

Background: 7,8-DHF (7,8-dihydroxyflavone) is a TrkB agonist, but its receptor activation mechanism is not well understood.
Results: 7,8-DHF and BDNF display differential receptor binding affinity, receptor activation kinetics, receptor phosphorylation pattern, and ligand-induced receptor degradation.
Conclusion: 7,8-DHF and BDNF utilize different mechanisms to activate TrkB.
Significance: This report provides mechanistic insights into how 7,8-DHF induces TrkB activation.
Keywords: Brain-Derived Neurotrophic Factor (BDNF); Cell Signaling; Cell Surface Receptor; Endocytosis; Phosphorylation; 7,8-Dihydroxyflavone; TrkB
Abstract
7,8-dihydroxyflavone (7,8-DHF), a newly identified small molecular TrkB receptor agonist, rapidly activates TrkB in both primary neurons and the rodent brain and mimics the physiological functions of the cognate ligand BDNF. Accumulating evidence supports that 7,8-DHF exerts neurotrophic effects in a TrkB-dependent manner. Nonetheless, the differences between 7,8-DHF and BDNF in activating TrkB remain incompletely understood. Here we show that 7,8-DHF and BDNF exhibit different TrkB activation kinetics in which TrkB maturation may be implicated. Employing two independent biophysical approaches, we confirm that 7,8-DHF interacts robustly with the TrkB extracellular domain, with a Kd of ∼10 nm. Although BDNF transiently activates TrkB, leading to receptor internalization and ubiquitination/degradation, in contrast, 7,8-DHF-triggered TrkB phosphorylation lasts for hours, and the internalized receptors are not degraded. Notably, primary neuronal maturation may be required for 7,8-DHF but not for BDNF to elicit the full spectrum of TrkB signaling cascades. Hence, 7,8-DHF interacts robustly with the TrkB receptor, and its agonistic effect may be mediated by neuronal development and maturation.
Introduction
Trk receptor-mediated neurotrophic signaling regulates various neuronal events, including cell survival, proliferation, axon and dendrite growth and patterning, and synaptic plasticity. BDNF is a member of the neurotrophin family, which also includes NGF, NT-3, and NT-4/5. BDNF, like the other neurotrophins, exerts its biological functions on neurons through two transmembrane receptors: p75NTR and the TrkB receptor (NGF binds to TrkA, BDNF and NT-4/5 bind to TrkB, and NT-3 binds preferentially to TrkC) (1). BDNF triggers TrkB dimerization and autophosphorylation of tyrosine residues in its intracellular domain, resulting in activation of the downstream signaling pathways. Autophosphorylation of some tyrosine residues in the intracellular domain of the receptor provides the docking sites for the binding and activation of signaling intermediates that initiate the signal transduction cascade. Known intermediaries include PLC-γ1, SHC, and PI3K (2). Accumulating evidence supports the “signaling endosome hypothesis” that NGF-TrkA complexes are internalized at the axon terminal and transported retrogradely to the cell body. Following NGF treatment, clathrin-coated vesicles containing NGF-bound TrkA and activated signaling proteins of the Ras-MAP kinase pathway were formed. This model has been extrapolated to retrograde signaling by all neurotrophins (3–5). Indeed, binding of BDNF to the TrkB receptor triggers internalization of the ligand-receptor signal complex, which is transported from the nerve terminal to the cell body in the form of signaling endosomes to mediate numerous signaling events (6). In neurons, the endosomal TrkB receptor remains tyrosine-phosphorylated and bound to BDNF when they are transported retrogradely (7, 8). Interestingly, BDNF treatment elicits TrkB receptor ubiquitination and degradation (9–12). Inhibition of tyrosine phosphorylation by K252a prevents TrkB receptor internalization and ubiquitination (9, 13).
To search for a small compound that mimics the biological functions of BDNF, we developed a cell-based high throughput screening assay and identified small molecular TrkB agonists with different structural backbones, including 7,8-dihydroxyflavone (7,8-DHF)3 and deoxygedunin (14, 15). Using 3H-labeled 7,8-DHF and recombinant TrkB extracellular domain (ECD) recombinant proteins, we demonstrated that these small agonists directly bind TrkB-ECD in a filter binding assay and trigger its dimerization and autophosphorylation, leading to downstream PI3K/Akt and MAPK signaling activation (14, 15). Mounting evidence also supports that these small agonists mimic the physiological actions of BDNF and demonstrate promising therapeutic efficacy in various cellular and animal models. For instance, excessive sensory ganglion, including the vestibular ganglion, degeneration occurs in mutant mice lacking BDNF (16). Strikingly, small TrkB agonists (7,8-DHF and deoxygedunin) simulate BDNF and prevent the degeneration of the vestibular ganglion in BDNF−/− pups (15). Moreover, 7,8-DHF promotes neuronal survival in various models, including dopaminergic neurons in Parkinson disease (14), retinal ganglion cells (17), and motoneurons (18). Noticeably, 7,8-DHF protects the spiral ganglion from degeneration and promotes axon regeneration in a TrkB-dependent manner (19, 20). It also enhances neuromuscular transmission via activation of TrkB in the diaphragm muscle, which is TrkB-dependent as well (21). BDNF/TrkB signaling plays a critical role in synaptic plasticity. Accordingly, 7,8-DHF rescues synaptic plasticity in aged animals (22–24). Moreover, 7,8-DHF induces neurogenesis and dampens the development of the “depressive” phenotypes in chronic social defeat and binge-like alcohol consumption mouse models (25, 26). Remarkably, 7,8-DHF imitates BDNF and regulates the consolidation of emotional learning (27–29), rescues the BDNF deficiency-induced learning and memory phenotypes in proprotein convertase PC7 knockout mice (30), and exhibits therapeutic efficacy in a mouse model of Rett syndrome (MeCP2 mutant mice that have reduced levels of BDNF) (31). 7,8-DHF also penetrates the blood-brain barrier and exerts potent neurotrophic effects in various neurological disease models, including stroke (14, 32–34), and neurodegenerative diseases, including Huntington disease (35) and Alzheimer disease (36, 37). Hence, 7,8-DHF fully mimics the biological and physiological effects of BDNF and exerts neurotrophic effects by activating the TrkB receptors in various neurological diseases.
Accumulating evidence strongly supports that 7,8-DHF imitates the cognate ligand BDNF and acts as a TrkB receptor agonist. However, 7,8-DHF is a small compound with a molecular mass of only 254 Da, which is ∼1% of the size of the active BDNF dimer. Conceivably, these two ligands may utilize distinct mechanisms to activate TrkB receptors. In this report, we show that 7,8-DHF binds strongly to TrkB-ECD, as detected by surface plasmon resonance and fluorescent quenching biophysical assays. It swiftly activates TrkB receptors, which lasts for hours, and the activated receptors are internalized but not degraded. Additionally, TrkB receptor activation by the small molecular ligand is much more sensitive to primary neuronal maturation in vitro than BDNF.
EXPERIMENTAL PROCEDURES
Reagents and Cells
Recombinant human TrkB-Fc proteins purified from mouse myeloma cell lines were from R&D Systems (catalog no. 688-TK). Anti-p-TrkB 817 was from Epitomics. It specifically recognizes human and rat phospho-TrkB with a 1:5000∼20,000 dilution. Anti-p-TrkB 706 was from Santa Cruz Biotechnology, and it recognizes rat and mouse phospho-TrkB (1:200 or 500 dilution). The phospho-TrkB Tyr816 antibody has been described before. It recognizes human, mouse, and rat TrkB (1: 2000 dilution for immunoblotting) (38). Anti-TrkB (Cell Signaling Technology; it recognizes both full-length and truncated TrkB) was used for immunoblotting. P-Akt 473 Sandwich ELISA was from Cell Signaling Technology. BDNF was from Peprotech. Anti-phospho-TrkB515, phospho-Akt-473, anti-Akt, and Anti-p-Erk1/2 antibodies were from Cell Signaling. Anti-p-PKC and anti-PKC were from Cell Signaling Technology. Mouse monoclonal anti-EEA was from BD Biosciences. FITC-conjugated anti-avidin and Alexa Fluor 555-conjugated goat anti-mouse secondary antibody were from Invitrogen. 7,8-Dihydroxyflavone was purchased from TCI. All chemicals not included above were purchased from Sigma. T48 cells, stably transfected with rat HA-TrkB, were cultured in the DMEM containing 1 mm pyruvate and 10% FBS with 300 μg/ml G418.
Determination of Binding Parameters by Surface Plasmon Resonance
A Biacore T200 was used to determine the binding specificity and dissociation constant, Kd, via surface plasmon resonance (SPR). Human His-TrkB-ECD or His-TrkA-ECD purified from human cell lines (Sino Biological) was immobilized to an NTA-coated sensor ship at an average surface density of 11,000 resonance units (RU) for TrkB or 12,000 RU for TrkA when 25 μl was captured at 10 μg/ml in PBS (pH 7.4). The analytes, 7,8-DHF and 5,7-DHF (Sigma), were injected over the receptor-bound surface at 25 °C at a flow rate of 30 μl/min for 1 min in 100 mm Tris (pH 6.5). BDNF or NGF (Peprotech) was injected under similar conditions in PBS (pH 7.4). Following analysis of the interactions between the small molecule and the receptor, the surface was regenerated with 350 mm EDTA at a flow rate of 30 μl/min for 1 min. All analyte solutions were run simultaneously over a control flow cell that contained a blank surface with no immobilized protein. The control flow cell signal was subtracted to give response values, and the kinetic rate constants were calculated by using BIAevaluation kinetic software 1.0 (BIAcore) using the formula, KD = koff/kon.
Intrinsic Fluorescence Quenching Assay
The interaction between TrkB-ECD recombinant proteins and 7,8-DHF and the dissociation constant (Kd) of 7,8-DHF to TrkB were determined by tryptophan fluorescence titration. Two different concentrations of TrkB-ECD proteins (50 and 250 nm) were prepared in PBS buffer (pH 7.4). The fluorescent quenching was conducted in a cuvette with a 1-cm path length cell. Intrinsic fluorescence spectra of TrkB were recorded by fluorescence spectrophotometer (PTI) at room temperature using excitation at 280 nm and emission between 300 and 400 nm. Tryptophan fluorescence spectra were collected before and after titration with different concentrations of 7,8-DHF. 7,8-DHF itself produced negligible fluorescence changes under the same experimental conditions. The dissociation constant of 7,8-DHF to TrkB was determined by fitting the normalized fluorescence intensity at 335 nm under different concentrations of 7,8-DHF using equation 1 as follows:
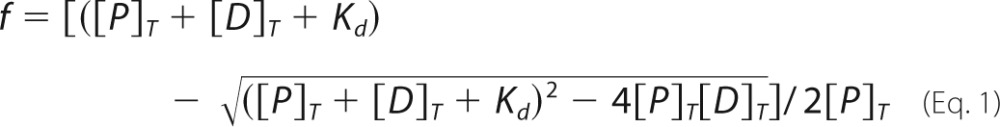 |
where f is the fractional change, Kd is the dissociation constant and [P]T and [D]T are the total concentration of protein and 7,8-DHF, respectively.
Organic Synthesis of Biotin-7,8-DHF
EDC (58 mg, 0.3 mmol) and DMAP (2 mg) were added to a mixture of 7,8-dihydroxyflavone (61 mg, 0.24 mmol) and biotin (50 mg, 0.2 mmol) in N,N-dimethylformamide (2 ml). The reaction mixture was stirred at room temperature for 24 h. The crude product was directly purified by flash chromatography on silica eluted with EtOAc/MeOH (10:1) to afford 7-biotinyl-8-hydroxyflavone as a white solid (15 mg, 16%): 1H NMR (dimethyl sulfoxide-d6, 400 MHz): δ 7.92 (m, 2H), 7.76 (d, J = 8.4 Hz, 1H), 7.57 (m, 3H), 7.05 (d, J = 8.2 Hz, 1H), 6.92 (s, 1H), 6.47 (s, 1H), 6.37 (s, 1H), 4.28 (m, 1H), 4.11 (m, 1H), 3.07 (m, 1H), 2.62–2.83 (m, 3H), 2.57 (d, J = 12.4 Hz, 1H), 1.4–1.7 (m, 6H); HRMS [M]+ Calcd for C25H24N2O6S: 480.1281, Found: 480.1316 (Structure 1).
STRUCTURE 1.
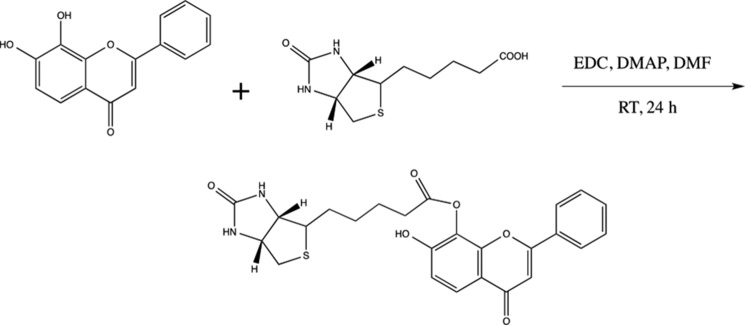
Synthesis of Bion-7,8-DHF.
Biotinylation of BDNF and Immunofluorescent Staining of TrkB Receptor Internalization
BDNF (100 μg, Peprotech, Inc.) was incubated with 2 mg NHS-LC-Biotin (Pierce) in 100 μl of PBS with Ca2+ and Mg2+ for 2 h at 4 °C. Biotinylated BDNF and unbound biotin were separated with a Zeba Spin desalting column (Thermo Scientific). Biotinylated BDNF (20 nm) or 500 nm biotin-7,8-DHF were mixed with or without unlabeled BDNF or unlabeled 7,8-DHF (200-fold excess) and applied to the primary neuron slides in DME containing 0.5% protamine and 10 mm Hepes for 30 min on ice. Unbound BDNF-biotin was washed out with culture medium. Internalization was initiated by applying warm culture medium (37 °C) to the cells. For the acid wash, after 30 min of incubation on ice, the primary cultures were washed with ice-cold acid (0.2 m acetic acid) for 20 min to remove the surface-bound BDNF. After 30 min of incubation, the cultures were fixed in 4% paraformaldehyde in PBS and permeabilized, and the BDNF-biotin or 7,8-DHF-biotin was visualized by FITC-conjugated avidin (1:500, Invitrogen) in 0.4% Triton X-100, 5% goat serum in PBS. The slides with the cells were mounted with mounting medium and visualized by confocal microscopy.
Biotinylation Assay of TrkB Internalization in Primary Neurons
The primary cultured DIV 13 cortical neurons were treated with 8 nm BDNF and 200 nm 7,8-DHF for 30 min on ice. Unbound BDNF and 7,8-DHF were washed off three times with ice-cold PBS. The cell surface proteins were labeled with 0.5 mg/ml EZ-Link Sulfo-NHS-SS-Biotin (Pierce) in PBS with Ca2+ and Mg2+ for 2.5 min at 37 °C and then washed extensively with ice-cold PBS. Internalization was initiated by switching to warm medium (37 °C) for 30 min. The remaining, biotinylated surface proteins were debiotinylated by washing with glutathione buffer (50 mm reduced glutathione, 100 mm NaCl, 1 mg/ml BSA, 1 mg/ml glucose, and 50 mm Tris (pH 8.6)) for 3 × 30 min at 4 °C. The cells were washed for an additional two times with PBS and harvested with lysis buffer (the same buffer as that used for the Western blot analysis). The internalized, biotinylated proteins were precipitated by immobilized streptavidin, separated by SDS-PAGE, and subjected to Western blot analysis using an anti-TrkB antibody.
Primary Rat Cortical Neuron Cultures
Cortical primary neurons were dissected from an embryonic day 18 rat embryo, and the cortex was extirpated, cross-chopped, and suspended by pipetting for separation in DMEM containing 5% FCS, and 5% horse serum. The cell suspension was then centrifuged at 250 × g for 5 min. This operation was repeated again. Cells were seeded into polyethyleneimine-coated 10-cm dishes and 12-well plates, including coated coverslips, and incubated at 37 °C in 5% CO2/95% air. After 3 h, the culture medium was changed to Neurobasal containing a B-27 supplement (Invitrogen) and incubated for 4 days. For maintenance, a half medium was changed to fresh Neurobasal/B-27 every 4 days. After 1 week or at different DIV, the cultured neurons were used for experiments.
RESULTS
The Specific Binding of 7,8-DHF to the TrkB Receptors Using SPR
To study the interactions between TrkB-ECD and 7,8-DHF, SPR was performed using the Biacore T200 system. A sensor chip coated with NTA was used to capture the His-tagged TrkB-ECD recombinant proteins to the Ni2+-chelated sensor chip surface. Initially, the optimal analyte buffer was found by measuring the RU response for 250 μm 7,8-DHF in various buffers at different pH levels (Fig. 1A). The strongest signal was observed when 7,8-DHF was dissolved in Tris (pH 6.5) (TrkB-ECD has a pI of 5.3), so this buffer was used to perform the rest of the binding experiments. 7,8-DHF was able to specifically bind to TrkB-ECD because only a minimal signal was observed when the molecule was injected over the TrkA-ECD-bound chip surface (Fig. 1, B and C). The natural ligands NGF and BDNF yielded robust binding signals for their specific receptors, TrkA and TrkB, respectively. Because the protein ligands have significantly larger molecular masses compared with 7,8-DHF (molecular mass of 5,7-DHF or 7,8-DHF, 254 Da; molecular mass of the NGF homodimer, 26,000 Da; molecular mass of BDNF, 28,000 Da), the absolute response associated with protein binding was significantly greater than that of the small molecule. Therefore, the response signal was normalized to the molecular weights of the compounds (Fig. 1, B and C). The small-molecule 7,8-DHF clearly bound to TrkB-ECD in a dose-dependent manner, whereas the inactive analog, 5,7-DHF, did not bind at significant levels to either receptor (Fig. 1D). A kinetic SPR analysis was performed to measure the binding constant, KD, for the physiological TrkB ligand BDNF and for 7,8-DHF. Fig. 1, E and F, displays the binding curves for both analytes, and the sensorgrams obtained for both analytes were used to calculate the KD as a function of their rates of association and dissociation (Fig. 1, G and H). The high TrkB binding affinities (Kd = 1.7 nm for BDNF and Kd = 15.4 nm for 7,8-DHF) observed for both ligands demonstrate their specificity for the receptor.
FIGURE 1.

7,8-DHF binds specifically to the extracellular domain of the TrkB receptor. A, optimization of buffer conditions for the SPR binding assay. SPR was used to measure the binding of 7,8-DHF to human His-TrkB-ECD, which was immobilized to an NTA-coated sensor chip. Various buffers with different pH values were tested to determine the optimal buffer for the analyte. Tris buffer (pH 6.5) was found to have the most optimal binding signal. B, the selectivity of 7,8-DHF binding was determined by measuring the binding between PBS, 250 μm 5,7-DHF, 250 μm 7,8-DHF, or 0.2 μm NGF to His-TrkA-ECD. The response (RU) was normalized by molecular weight (MW) to account for the difference in molecular weight between the small molecule and protein analytes (molecular mass of 5,7-DHF and 7,8-DHF, 254 Da; molecular mass of the NGF homodimer, 26,000 Da). The inset displays the absolute binding signal observed for each analyte. Error bars represent mean ± S.D. of two independent experiments. C, the binding of PBS, 250 μm 5,7-DHF, 250 μm 7,8-DHF, or 0.2 μm BDNF was measured for His-TrkB-ECD. The response (RU) was normalized by molecular weight to account for the difference in molecular weight between the small molecule and protein analytes (molecular mass of BDNF, 28,000 Da). The inset displays the absolute binding signal observed for each analyte. Error bars represent mean ± S.D. of two independent experiments. B and C, bottom panel, schematic of the human TrkB-ECD with a C-terminal His6 tag. LRRD, leucine-rich repeat domain. D, the specific binding of 7,8-DHF to TrkB is concentration-dependent as the signal for 100 and 250 μm 7,8-DHF increases, whereas no signal is observed for 5,7-DHF binding. E, binding curve for BDNF binding to TrkB showing the RUs obtained when 0, 0.1, 0.5, 1, 5, or 10 nm BDNF was injected over TrkB. Error bars represent mean ± S.D. of two independent experiments. F, binding curve for 7,8-DHF binding to TrkB showing the RUs obtained when 0, 1, 5, 10, 25, 50, 100, 250, or 500 μm 7,8-DHF was injected over TrkB. Error bars represent mean ± S.D. of two independent experiments. G, sensorgrams obtained from the SPR analysis of the binding affinity of BDNF, which was injected over the TrkB receptor-bound chip surface. The KD was 1.7 nm and was obtained by fitting the data in the BIAevaluation kinetic analysis software. H, sensorgrams obtained from the SPR analysis of the binding affinity of 7,8-DHF, which was injected over the TrkB receptor-bound chip surface. The KD was 15.4 nm and was obtained by fitting the data in the BIAevaluation kinetic analysis software.
7,8-DHF Binds to TrkB-ECD in the Fluorescence Quenching Assay
To further test whether 7,8-DHF interacts directly with TrkB-ECD, we examined the tryptophan fluorescence differences of TrkB-ECD in the presence of different concentrations of 7,8-DHF because binding of 7,8-DHF to TrkB induced a change of the tryptophan environment, which could be visualized by fluorescence spectroscopy. As shown in Fig. 2, tryptophan emission maximized at 335 nm (excited at 280 nm), suggesting that the tryptophan residues of the purified recombinant protein TrkB-ECD were largely buried, which is consistent with the reported partial structure of the ECD domain of the TrkB receptor and other well folded proteins (39, 40). The tryptophan fluorescence was quenched by 7,8-DHF in a concentration-dependent manner, and the fluorescence decrease was maximal at the highest concentration of 7,8-DHF. Addition of 7,8-DHF to TrkB-ECD resulted in about 10% fluorescence decrease, whereas the maximum emission wavelength remained unchanged. 7,8-DHF itself produced a negligible fluorescence change under the same experimental conditions (data not shown). On the basis of the 7,8-DHF-induced fluorescence change, quantitative analysis demonstrated that the Kd of 7,8-DHF binding to TrkB was 12.1 ± 1.6 nm, which was calculated by equation 1 (see “Experimental Procedures”. Hence, the two independent biophysical assays support that 7,8-DHF strongly binds to TrkB-ECD with an affinity of ∼10 nm.
FIGURE 2.
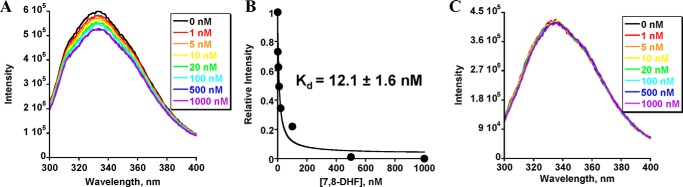
Tryptophan fluorescence quenching assay for the direct interaction between TrkB-ECD and 7,8-DHF. A, tryptophan fluorescence titration of TrkB-ECD recombinant protein with 7,8-DHF. The tryptophan fluorescence of TrkB excited at 280 nm showed decreased intensity after titrating different concentrations of 7,8-DHF, and this fluorescence decrease was saturated when 7,8-DHF concentrations increased to about 200 nm. B, the normalized tryptophan intensities at an emission of 335 nm were plotted as a function of total added concentration of 7,8-DHF. The dissociation constant between 7,8-DHF and TrkB was then calculated by fitting this plot with equation 1. C, tryptophan fluorescence titration of deglycosylated TrkB-ECD recombinant protein with 7,8-DHF. The tryptophan fluorescence of TrkB excited at 280 nm did not show any significant signal decrease after adding 0–1000 nm 7,8-DHF.
The TrkB receptor is highly glycosylated with at least 10 N-glycosylation sites on its ECD (50). To examine whether glycosylation mediates TrkB interaction with 7,8-DHF, we treated the purified recombinant TrkB-ECD proteins with N-glycanase (PNGase F, Promega) to obtain deglycosylated TrkB-ECD proteins and further tested the interaction between 7,8-DHF and deglycosylated TrkB-ECD using a fluorescence quenching assay. Titration of 0–1000 nm 7,8-DHF did not cause any significant change of the florescence in deglycosylated TrkB-ECD (Fig. 2C). This result indicates that glycosylation in TrkB-ECD is essential for the interaction between 7,8-DHF and TrkB-ECD.
Comparison of TrkB Tyrosine Phosphorylation by BDNF and 7,8-DHF
Neurotrophins bind to Trk receptors and elicit phosphorylation of a panel of well conserved tyrosine residues in the intracellular domain. For instance, BDNF potently induces TrkB Tyr515, Tyr706, and Tyr816 phosphorylation in neurons. To compare the TrkB phosphotyrosine signaling elicited by BDNF and 7,8-DHF, we treated primary neurons with vehicle, BDNF (100 ng/ml), and 7,8-DHF (500 nm) for 15 min and monitored the phosphorylation of each indicated tyrosine residue by immunoblotting with the specific antibodies. The signals were quantified and analyzed against total TrkB levels using ImageJ software. As expected, BDNF triggered robust phosphotyrosine signals on all of the analyzed residues. 7,8-DHF induced stronger tyrosine phosphorylation on Tyr515, Tyr706, and Tyr816 residues than control vehicle, but the signals were weaker than those elicited by BDNF. Similar activation patterns were also observed for the downstream effectors Akt and MAPK. Nonetheless, p-PKC, a downstream factor for PLC-γ1, was not substantially up-regulated by either BDNF or 7,8-DHF (Fig. 3, A and B). Because mouse/rat TrkB-ICD and human TrkB-ICD are nearly identical, the extreme C-terminal tail for the PLC-γ1 binding site is the same for these species. Hence, we also examined TrkB activation with phospho-Tyr817 antibody (human Tyr-817 is equivalent to mouse/rat Tyr-816). The rabbit monoclonal phospho-Tyr817 antibody that recognizes both human and rat TrkB receptors exhibited a more prominent TrkB activation effect for 7,8-DHF (Fig. 3, A and B). Therefore, this antibody was employed for analysis of TrkB activation in primary neurons from rat embryonic cultures (embryonic days 17–18).
FIGURE 3.

BDNF and 7,8-DHF exhibit different temporal patterns in triggering TrkB activation. A and B, immunoblotting (IB) and quantitative analysis of TrkB receptor and its downstream p-Akt. A, primary cortical neurons (DIV 13) were treated with BDNF (100 ng/ml), 7,8-DHF (500 nm), and vehicle control for 15 min, followed by Western blot analysis with antibodies against p-TrkB Tyr515, p-TrkB Tyr706, p-TrkB Tyr816, p-TrkB Tyr817, and total TrkB, p-Akt/Akt, p-MAPK/MAPK, and p-PKC/PKC. B, the signals were analyzed quantitatively and normalized. *, p < 0.05; **, p < 0.01 versus control, Student's t test, n = 2. M.W., molecular weight. C, BDNF, but not 7,8-DHF, induces TrkB receptor ubiquitination and degradation. Primary neurons were treated with BDNF or 7,8-DHF for different time points, and TrkB was immunoprecipitated (IP) with anti-Trk Cys14 and analyzed by immunoblotting with anti-TrkB (center row) or anti-ubiquitin (bottom row). The cell lysates were analyzed by immunoblotting with anti-p-TrkB Tyr817 or anti-TrkB (top row). Quantitative analysis of TrkB ubiquitination was performed with results from independent experiments. *, p < 0.05 versus 0 min, one-way analysis of variance, Tukey's multiple comparison test, n = 2.
BDNF treatment elicits TrkB receptor ubiquitination and degradation (9, 11, 12, 41). To compare the TrkB activation kinetics by BDNF and 7,8-DHF, we treated primary neurons for various times, immunoprecipitated TrkB, and monitored its phosphorylation state and ubiquitination by immunoblotting. In neuronal lysates, the BDNF-triggered phospho-Tyr817 signal peaked at 10 min, decreased at 60 min, and faded away at 180 min. In alignment with this observation, TrkB was clearly ubiquitinated at 10 min. Its ubiquitination signals correlated tightly with its Tyr817 phosphorylation pattern (Fig. 3C, left panel). Accordingly, the total level of TrkB was discernibly reduced at 180 min, fitting with the previous findings (9, 11, 12, 41). In contrast, 7,8-DHF swiftly activated TrkB at 10 min, and the 7,8-DHF-elicited phospho-Tyr817 signal was sustained for 180 min, fitting with a previous report showing that TrkB activation by 7,8-DHF lasts more than 8 h in the retinal ganglion (17). Remarkably, no TrkB ubiquitination was detected. Consequently, total TrkB levels remained largely unchanged (Fig. 3C, left panel, center row). The quantitative analysis of TrkB ubiquitination from multiple independent experiments supported that BDNF, but not 7,8-DHF, elicited TrkB ubiquitination (Fig. 3C, right panel). Therefore, 7,8-DHF exhibits a different temporal pattern of inducing TrkB activation from BDNF, and the TrkB receptors activated by 7,8-DHF are not ubiquitinated or degraded.
The Agonistic Activity of 7.8-DHF Is Mediated by Neuronal Maturity and TrkB Glycosylation
Previous studies have demonstrate that BDNF/TrkB signaling is regulated by neuronal development in vitro and in vivo (42, 43). To explore whether 7,8-DHF-induced TrkB signaling is also modulated by developmental status, we treated primary neurons at different culture DIV with BDNF (100 ng/ml), 7,8-DHF, or deoxygedunin (500 nm) for 15 min. The neuronal lysates were analyzed by immunoblotting. As expected, BDNF elicited prominent TrkB tyrosine phosphorylation on both Tyr817 and Tyr706 residues as well as the downstream effectors Akt and MAPK in all cultured neurons. Notably, at DIV 5 and 7, the stimulatory effect by 7,8-DHF on TrkB Tyr817 phosphorylation was detectable but not very strong compared with the vehicle control (10% dimethyl sulfoxide/PBS). On the other hand, p-TrkB 706 was not detectable following treatment with 7,8-DHF in these neurons. Interestingly, Akt, but not MAPK, was activated by 7,8-DHF at both DIV 5 and 7. It is worth noting that 7,8-DHF-provoked TrkB Tyr817 and Tyr706 phosphorylation was evident in both DIV 13 and 15 compared with vehicle control. Consistently, the downstream effectors Akt and MAPK were activated prominently by 7,8-DHF (Fig. 4A). These observations support that the small molecular agonist 7,8-DHF triggers TrkB receptor activation and that this is regulated by neuronal development and maturation.
FIGURE 4.

Primary neuronal maturation and TrkB glycosylation mediate the agonistic effect of 7,8-DHF on TrkB signaling. A, immunoblot (IB) analysis of the TrkB receptor and its downstream p-Akt and p-MAPK. Primary cortical neurons at different DIV were treated with BDNF (100 ng/ml) in PBS, 7,8-DHF (500 nm) dissolved in 10% dimethyl sulfoxide/PBS, deoxygedunin (Deox., 500 nm) in dimethyl sulfoxide (DMSO, another small molecular TrkB agonist), and their own vehicle for 15 min, respectively, followed by Western blot analysis with antibodies against p-TrkB Tyr817 and total TrkB. M.W., molecular weight; Ct, control. B, neuronal development in vivo also regulates TrkB activation by 7,8-DHF. The mouse brain microslices were prepared from differently aged pups, followed by treatment with vehicle, BDNF (100 ng/ml), dopamine (DA) and norepinephrine (NE) (500 nm), and 7,8-DHF (500 nm) for 15 min at 37 °C. The brain lysates were prepared after treatment, followed by immunoblot analysis with anti-p-TrkB 706 and total TrkB antibody. C, rat cortical neurons, pretreated with or without tunicamycin (3 μg/ml, a glycosylation inhibitor) for 24 h, were harvested after 15 min of treatment with vehicle, BDNF, and 7,8-DHF, respectively. The cell lysates were prepared and analyzed by immunoblotting with pTrkB Tyr817 antibody and p-AKT antibody. Tunicamycin pretreatment abolished TrkB activation by 7,8-DHF and it also slightly reduced TrkB phosphorylation by BDNF.
Developmental regulation of the response of brain tissues to the TrkB ligands BDNF and NT-4/5 has been shown before. BDNF or NT-4/5-induced TrkB tyrosine phosphorylation in the cortex and hippocampus peak at embryonic day 19 through the first week after birth (P7), and there is a lack of a significant response to these neurotrophins in the adult brain (44). To further investigate whether the agonistic activity of 7,8-DHF toward TrkB is also regulated by development, we prepared microslices of cortex tissues from P4, P8, and P16 mouse brains and treated these tissues with BDNF (100 ng/ml) or 7,8-DHF (500 nm) for 5 min. As a control, we also employed two catechol group-containing chemicals, dopamine and norepinephrine, because our previous structure-activity relationship study demonstrated that the catechol group is essential for the agonistic effect of 7,8-DHF (45). Compared with vehicle control, BDNF triggered TrkB activation at both P4 and P8, and its stimulatory effect was reduced substantially at P16. In contrast, 7,8-DHF barely activated TrkB in P4 and P8 cortices but strongly elicited TrkB activation at P16. Notably, the two control compounds dopamine and norepinephrine induced TrkB activation at both P8 and P16 (Fig. 4B, top row). The kinase domain-truncated TrkB isoform or unglycosylated TrkB (full-length) was gradually up-regulated as the mouse brains matured (Fig. 4B, bottom row).
To test whether TrkB glycosylation plays a mediatory role in the agonistic effect of 7,8 DHF, we employed an N-linked glycosylation inhibitor. Preincubation of the primary cultures with tunicamycin markedly inhibited the TrkB agonistic effect of 7,8-DHF. In contrast, the TrkB stimulatory effect of BDNF was only reduced partially (Fig. 4C), supporting the notion that TrkB glycosylation indeed somehow mediates receptor activation by 7,8-DHF.
The TrkB Receptor Extracellular Domain Fusion Protein (TrkB-Fc) Neutralizes the Agonistic Activity of 7.8-DHF
TrkB-Fc is a His-tagged fusion protein of human TrkB-ECD (Cys32-His430) and human IgG1. It has been used extensively to specifically neutralize endogenous or exogenous BDNF (46–48). If 7,8-DHF activates the TrkB receptor specifically through binding to its ECD, one would expect that the TrkB-Fc inhibits this small molecular agonist as it does the cognate ligand BDNF. To explore this possibility, we pretreated primary neurons with different dosages of TrkB-Fc for 5 min, followed by a 10-min treatment of TrkB-Fc in combination with BDNF (500 pm) or 7,8-DHF (100 nm). The cell lysates were analyzed by immunoblotting. Compared with vehicle control, BDNF triggered TrkB phosphorylation, which was reduced by the addition of the receptor-based scavenger TrkB-Fc in a dose-dependent manner. We made a similar observation for the downstream signaling proteins p-Akt and p-MAPK (Fig. 5A, left column). Remarkably, 7,8-DHF-provoked TrkB phosphorylation, and its downstream p-Akt and p-MAPK signaling was gradually suppressed by progressively increasing TrkB-Fc concentrations (Fig. 5A, right column), indicating that 7,8-DHF exerts its agonistic effect through selectively interacting with the TrkB receptor on its ECD domain. A quantitative analysis of multiple independent experiments validated the observations and supported the conclusion (Fig. 5B).
FIGURE 5.

TrkB-Fc blocks the TrkB receptor agonistic activity of 7,8-DHF. Primary neurons (DIV 13) were preincubated with gradually increasing doses of TrkB-Fc for 15 min, followed by addition of BDNF (100 ng/ml) or 7,8-DHF (100 nm) for another 15 min. A, the cell lysates were analyzed by immunoblotting (IB) with various indicated antibodies. BDNF or 7,8-DHF-triggered TrkB phosphorylation and its downstream p-Akt and p-MAPK signals were inhibited progressively by TrkB-fc recombinant proteins. M.W., molecular weight; Ct, control. B, quantitative analysis of p-TrkB/Total TrkB from independent experiments. *, p < 0.05 versus control; #, p < 0.005 versus BDNF or 7,8-DHF treatment only; one-way analysis of variance, Tukey's multiple comparison test, n = 2.
7,8-DHF Triggers TrkB Receptor Internalization
Internalization of the neurotrophin-Trk complex plays a critical role in signal transduction that initiates cell body responses to target-derived neurotrophins. The neurotrophin-Trk complex is internalized through clathrin-mediated endocytosis, leading to the formation of signaling endosomes (3, 49). To determine whether the small molecular agonist could mimic BDNF in provoking TrkB receptor internalization, we performed a TrkB endocytosis assay with primary cultures. We incubated primary cortical neurons at 37 °C with anti-TrkB antibody (3 μg/ml) and FITC-conjugated secondary antibody in the presence of vehicle control, BDNF (20 nm), or 7,8-DHF (2.5 μm) for 10 or 60 min, respectively. The neurons were fixed and immunostained with anti-EEA1, an early endosome marker. BDNF or 7,8-DHF elicited TrkB/EEA1 colocalization, indicating that the internalized ligand-TrkB complex is sorted to the early endosomal compartment. Representative pictures from 60-min groups are shown in Fig. 6A. Quantification of the internalized TrkB/EEA1 colocalization indicated that BDNF was more potent than 7,8-DHF in stimulating TrkB internalization and delivering it to the early endosomes at 10 min. At 60 min, both BDNF and 7,8-DHF substantially elevated TrkB endocytosis, and the early endosomal residency and 7,8-DHF appeared more robust than BDNF (Fig. 6B).
FIGURE 6.

TrkB receptor internalization in primary neurons. A and B, TrkB receptor internalization and sorting to early endosomes. Primary cultured cortical neurons were incubated with anti-TrkB antibody and FITC-conjugated secondary antibody (3 μg/ml). The cells were incubated with 20 nm BDNF or 2.5 μm 7,8-DHF for 10 or 60 min at 37 °C. The cells were then fixed in 4% paraformaldehyde and stained with mouse anti-EEA1 antibody and Alexa Fluor 555-conjugated goat anti-mouse secondary antibody. Scale bar = 10 μm. Quantitative analysis of internalized TrkB receptors was performed with results from independent experiments. *, p < 0.05; **, p < 0.01; Student's t test; n = 3. C, the effect of BDNF and 7,8-DHF on the internalization of TrkB as determined by surface biotinylation. Cell surface proteins were labeled by Sulfo-NHS-SS-Biotin before initiation of TrkB internalization by BDNF or 7,8-DHF for 30 min at 37 °C. The remaining biotinylated proteins on the cell surface were removed by glutathione. The internalized biotin-modified TrkB receptors were precipitated by streptavidin, followed by Western blot analysis using TrkB antibody. 7,8-DHF and BDNF enhanced TrkB internalization (full-length (145-kDa) and truncated (95-kDa) TrkB receptors) in primary neurons. The quantitative data were mean ± S.D. from two independent experiments. *, p < 0.05; ***, p < 0.001; Student's t test. M.W., molecular weight; IP, immunoprecipitation; IB, immunoblot.
To further investigate the effect of 7,8-DHF on triggering TrkB receptor internalization, we performed surface biotinylation with primary neurons. The cell surface proteins were labeled with Sulfo-NHS-SS-Biotin on ice, followed by addition of BDNF or 7,8-DHF. The internalization was initiated by placing the neurons at 37 °C and incubating for 30 min. The internalization was terminated by placing the primary cultures on ice, and biotin was removed from the remaining biotinylated surface proteins by the reducing agent glutathione. The internalized biotinylated proteins were precipitated with streptavidin, resolved on SDS-PAGE, and analyzed by immunoblotting with anti-TrkB antibody against its ECD domain. Although spontaneous internalization of TrkB was detected in the vehicle-treated neurons, both BDNF and 7,8-DHF significantly escalated TrkB internalization (Fig. 6C).
Biotin-labeled Ligands Trigger TrkB Endocytosis
Conventional immunofluorescent staining with TrkB antibodies cannot distinguish the newly synthesized receptors inserted into the cell surface from the internalized receptors. To visualize the internalization of cell surface TrkB receptors after binding to BDNF or 7,8-DHF, we employed biotinylated BDNF (BDNF-biotin) and 7,8-DHF-biotin to detect ligand-induced TrkB internalization. The biotin group was conjugated to the 8-hydroxyl group on 7,8-DHF via medicinal chemistry (the organic synthesis route is described under “Experimental Procedures”). Notably, both 7,8-DHF and its biotin-conjugated derivative elicited comparable p-TrkB in primary cultures. As expected, the application of BDNF-biotin to primary neurons induced the same level of TrkB tyrosine phosphorylation as the recombinant BDNF, suggesting that both 7,8-DHF-biotin and BDNF-biotin are bioactive, and conjugation of a biotin group to the 8 position did not impair the agonistic activity of 7,8-DHF (Fig. 7A). Hippocampal cultures were first incubated with BDNF-biotin or 7,8-DHF-biotin on ice to achieve saturated surface binding without any internalization, followed by extensive washing to remove unbound BDNF-biotin or 7,8-DHF-biotin. The primary cultures were then transferred to 37 °C and incubated for 30 min to allow TrkB receptor internalization. This approach measured only the internalized TrkB receptors bound to biotinylated BDNF or 7,8-DHF, and the newly inserted receptors were not detected (Fig. 7B, Experiment). Numerous fluorescent puncta, typical of endocytotic particles, were observed inside the cells treated with BDNF-biotin and 7,8-DHF-biotin (Fig. 7B, left column). Immunofluorescence signals were decreased markedly when the cultures were coincubated in the presence of a 100-fold excess unlabeled BDNF or 7,8-DHF (data not shown). When the cultures were kept on ice without switching to 37 °C, internalization did not occur, and FITC-conjugated avidine immunofluorescent staining was only observed on the cell surface (Fig. 7B, center column, Ice incubation). An acid wash completely eliminated the surface BDNF-biotin or 7,8-DHF-biotin staining (Fig. 7B, right column). The quantitative analysis demonstrated that 7,8-DHF and BDNF strongly induced TrkB receptor internalization in ∼85–90% of primary neurons (Fig. 7C). Hence, like BDNF, 7,8-DHF strongly induces TrkB receptor internalization.
FIGURE 7.
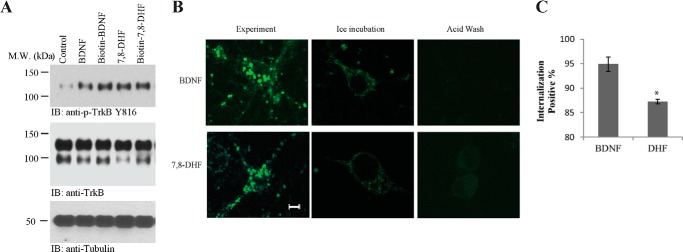
Biotin-labeled ligands trigger TrkB endocytosis. A, BDNF-biotin and 7,8-DHF-biotin induce TrkB phosphorylation. Primary neuronal cultures were treated with vehicle, BDNF, BDNF-biotin, 7,8-DHF, and 7,8-DHF-biotin. TrkB phosphorylation was detected by Western blot analysis using an anti-pTrkB antibody specific for phospho-Tyr816. M.W., molecular weight; IB, immunoblot. B, TrkB receptor internalization with BDNF-biotin and 7,8-DHF-biotin. The internalized receptor-BDNF-biotin complex and receptor-7,8-DHF-biotin complex by FITC-conjugated avidin were analyzed with confocal microscopy. Cortical cultures were first incubated with BDNF-biotin or 7,8-DHF-biotin on ice to achieve saturated binding and then switched to 37 °C for 30 min to allow receptor internalization. Many fluorescent puncta are seen in cultures incubated with BDNF-biotin and biotin-7,8-DHF (left column). Blockage of ligand-receptor internalization at low temperature on ice is seen (center column). The staining was completely eliminated after acid wash (0.2 m acetic acid) (right column). Scale bar = 5 μm. C, quantitative analysis of TrkB internalization by BDNF-biotin or 7,8-DHF-biotin. Fewer fluorescent puncta were observed in 7,8-DHF-biotin-incubated samples. The quantitative data were mean ± S.D. from two independent experiments. *, p < 0.05; Student t test.
DISCUSSION
In this study, we demonstrated that 7,8-DHF binds tightly to TrkB-ECD using two independent biophysical approaches: Biacore and fluorescent quenching. These two methods yield comparable binding affinities for 7,8-DHF to the human TrkB receptor (∼10 nm), which was purified from human CHO cells. The Kd is much stronger than our previous result (320 nm), for which we employed 3H-labeled 7,8-DHF and TrkB-ECD recombinant protein (purified from the BL21 bacterium) in the filter binding assay (14). TrkB-ECD is a highly glycosylated protein that contains 33.3% carbohydrate moieties. Among the 12 potential N-linked glycosylation sites in the extracellular domain of TrkB, 10 sites are actually glycosylated. Moreover, it contains two cysteine cluster domains in the N terminus and also possesses six disulfide linkages (Cys1-Cys7, Cys5-Cys14, Cys121-Cys145, Cys123-Cys163, Cys187-Cys235, and Cys271-Cys314) (50). Because much of the posttranslational modification machinery in eukaryotic cells is missing in prokaryotic systems, it is possible that our previously employed TrkB-ECD recombinant proteins are not folded or modified posttranslationally in the same manner as that purified from human CHO cells. This discrepancy might provide an explanation for the different binding dissociation constants. Molecular modeling also supports that 7,8-DHF might bind to the N-terminal LRR motif on TrkB-ECD (45). This approach may shed light on our future drug design for further improving the lead compound (45). Even though the cocrystal (7,8-DHF/TrkB-ECD) structure is not yet resolved experimentally, this independent biophysical evidence strongly supports that 7,8-DHF directly and strongly interacts with TrkB-ECD.
Previous studies demonstrate that neurotrophins interact with the leucine-rich repeat (LRR) motif on the N termini of Trk receptors in addition to the Ig2 domain (51). Our prior truncation experiments also indicate that 7,8-DHF associates with the N-terminal LRM-CC2 region (amino acids 36–176) (14). Molecular modeling with a maximum entropy optimization-based docking method suggests that there is a large cavity between the N-terminal cap and the first repeat of the LRR domain, located on the back or the convex surface of the LRR domain. 7,8-DHF inserts into this pocket, with the carbonyl group pointing to the deepest recess. In this model, the rotatable benzene ring of 7,8-DHF would form hydrophobic interactions with TrkB Phe55 and Pro56 at the edge of the pocket (45). Hence, these findings suggest that 7,8-DHF might share the same binding motif on TrkB hot spots that are essential for triggering TrkB receptor autophosphorylation and activation compared with BDNF. In alignment with this finding, most recently, Bernard-Gauthier et al. (52) showed that BDNF competes dose-dependently with 18F-labeled 7,8-DHF for TrkB binding in the rodent brain, strongly supporting the observation that 7,8-DHF exerts its agonistic effect on TrkB phosphorylation via direct TrkB-ECD binding. This conclusion is also supported by our TrkB-Fc inhibition assay. Fusion proteins comprising the Fc domain of human IgG and extracellular domains of receptor tyrosine kinases can neutralize the activity of their cognate ligands when administered in molar excess (46). Accumulating evidence supports that TrkB-ECD-Fc specifically blocks the actions of BDNF in activating TrkB (47, 48). Accordingly, we observe that TrkB-ECD-Fc recombinant proteins also suppress the agonistic effect of 7,8-DHF on TrkB (Fig. 5), underscoring that the TrkB receptor, and not any other target, is the specific receptor for 7,8-DHF to induce TrkB signaling cascades in neurons.
The dissociated primary cultures of neurons undergo dynamic developmental changes that may recapitulate some aspects of in vivo development, such as synapse formation and maturation as well as establishment of functional synaptic connections. Interestingly, BDNF-induced p-MAPK and p-cAMP response element-binding protein signaling in primary neuronal cultures are regulated by developmental maturity. Neurons at DIV 7 displayed the highest sensitivity to exogenous BDNF. The sensitivity was reduced progressively along with neuronal maturation. Although p75NTR remained constant at different developmental stages, TrkB expression was relatively low at DIV 3, increased gradually, reached the highest level at DIV 13, and then declined at DIV 22 (42). Using an in vitro maturation model, Zhou et al. (42) reported that neurons at DIV 7, when functional synapses start to form and GABAergic inhibition emerges, display the most dynamic activation of ERK1/2 and cAMP response element-binding protein by exogenous BDNF. Neurons at DIV 13 and 22 display less dynamic responses to BDNF (42). Interestingly, we also find that 7,8-DHF-induced TrkB signaling cascade activation is mediated by neuronal maturation. We show that 7,8-DHF-triggered TrkB autophosphorylation in primary neurons at DIV 5 or 7 is not very high compared with vehicle control. However, the signals elicited by 7,8-DHF versus vehicle control are obviously elevated at DIV 11 and 13 (Fig. 4A). This observation fits with the findings for BDNF, suggesting that neuronal maturation indeed mediates TrkB signaling, regardless of whether it is triggered by the cognate ligand, BDNF, or small molecular agonists.
In the rat forebrain, adult mRNA levels for full-length TrkB are reached by birth, whereas the truncated TrkB message does not peak until postnatal days 10–15. Western blot analysis indicates that full-length TrkB protein is the major form during early development, whereas truncated TrkB protein predominates in all forebrain regions of late postnatal and adult rats. These data also suggest that the glycosylation state of these receptors changes during postnatal maturation (43, 53). In alignment with these observations, we find that the agonistic effect of 7,8-DHF in triggering TrkB activation in mouse brain tissues is also regulated by development. The TrkB activation pattern observed by 7,8-DHF is different from BDNF. Knusel et al. (44) showed that BDNF displays an agonistic effect from E19 to P7 and that the activity is lost at P14 in the adult brain, whereas 7,8-DHF agonistic activity escalates in brain microslices from P8 to P16 (Fig. 4B). During their maturation, cortical neurons show increased or stable protein expression of glycolytic enzymes, synaptophysin, synapsin IIa, α and β synucleins, and glutamate receptors. Synaptogenesis is prominent during the first 15 days, and then synaptic markers remain stable through DIV 60 (54). Previously, Segal and co-workers showed that TrkA glycosylation regulates the receptor subcellular localization and activity (55). Although only four N-glycosylation sites are conserved in the Trk family (Asn26, Asn90, Asn174, and Asn223), differences in N-glycosylation may also affect the specificity of ligand-receptor interaction (50). Our observation that neuronal maturation regulates the agonistic effect of 7,8-DHF toward TrkB suggests that TrkB glycosylation might play some role in mediating this event. Indeed, preincubation of the primary cultures with tunicamycin suppresses the TrkB agonistic effect of 7,8-DHF, suggesting that TrkB glycosylation indeed mediates receptor activation by 7,8-DHF. It remains unknown why the glycosylation inhibitor tunicamycin reduces TrkB expression (Fig. 4C). Tunicamycin blocks N-linked glycosylation (N-glycans). Presumably, glycosylation on TrkB-ECD is required to stabilize the protein expression levels, and inhibition of this posttranslational modification may lead to its reduction in primary neurons. Previously, it has been shown that tunicamycin reduces 140-kDa TrkA and increases unglycosylated 80-kDa TrkA core protein in PC12 cells (55). Nonetheless, both the 140- and 90-kDa forms of TrkB in primary cortical cultures are reduced in response to tunicamycin. It appears that TrkA and TrkB might display different responses to tunicamycin treatment.
It has been well documented that the internalized Trk receptor remains tyrosine-phosphorylated and activated, with its extracellular domain bound to the neurotrophic ligand inside the signaling endosomes, and that the intracellular domain remains tightly associated with a number of signaling molecules such as PLC-γ1, PI3K, and proteins of the Ras/MAPK pathway in the cytoplasm of the responsive neurons (3, 4). BDNF elicits numerous tyrosine residue phosphorylations in TrkB-ICD. The phosphorylated mouse/rat TrkB Tyr515 provides a docking site for Shc, eliciting the Ras/Raf/MAPK signaling cascade, whereas phosphorylated Tyr816 associates with PLC-γ1. Tyr706 localizes in the activation loop, well conserved in the kinase domain, which is responsible for initiating Trk autophosphorylation (56). Because BDNF is 100-fold larger than 7,8-DHF in molecular weight, it has multiple binding sites on the TrkB receptor. Accordingly, the phosphotyrosine signals on these sites are much stronger by BDNF than 7,8-DHF in primary neurons (Fig. 3, A and B). Moreover, it is possible that BDNF may trigger the phosphorylation of a large panel of tyrosine residues on its ICD, and the small molecular agonist might only induce the phosphorylation of a portion of these residues, leading to partial imitation of the biological actions of BDNF by the small molecule. Conceivably, in some of the BDNF-mediated biological assays, this small agonist might be unable to elicit the same effects as BDNF. These hypotheses might apply to many of the most recently identified small TrkB agonists (14, 15, 57). It is worth noting that 7,8-DHF fails to trigger Tyr706 activation in primary neurons at DIV 5 or 7, although it induces robust phospho-Tyr706 in the matured cultures at DIV 13 and 15. Interestingly, Akt is readily activated at DIV 5 and 7, whereas p-MAPK is not activated in these neurons (Fig. 4A). These findings suggest that the small molecular agonists behave very differently from BDNF because the latter always simultaneously activates both the PI3K/Akt and Raf/Raf/MAPK pathways. It has been suggested before that residue Tyr751 on human TrkA (corresponding to human Tyr783 and rat TrkB Tyr782) is the major docking site for PI3K and TrkA Tyr490 (human TrkB Tyr516 and rat TrkB Tyr515) is responsible for Shc binding, leading to Ras/Raf/MAPK signaling activation (58). Conceivably, PI3K/Akt signaling is readily activated by 7,8-DHF when a portion of TrkB-ICD tyrosine residues are phosphorylated, whereas p-MAPK might require numerous key tyrosine residues including Tyr515, Tyr706, and Tyr785 phosphorylation to elicit the full activation of MAPK pathway (56, 59). Nonetheless, BDNF-elicited TrkB signals are transient and fade away within 1 h. In contrast, the 7,8-DHF-induced TrkB activation signal is sustained for hours. Furthermore, BDNF provokes activated TrkB receptors to be ubiquitinated and degraded. Conversely, the small agonist does not induce TrkB ubiquitination or degradation (Fig. 3C). These are the clear differences between the cognate ligand and the small molecular agonist. However, we observed that both BDNF and 7,8-DHF robustly induced TrkB internalization and subsequent shuttling to the early endosomes (Fig. 6). Clearly, further investigation of these observations is required to elucidate the precise molecular mechanism accounting for these events. Our study suggests that 7,8-DHF is a specific TrkB agonist that selectively binds to the TrkB receptor ECD with ∼10 nm affinity. It shares many biological and physiological functions with the cognate ligand BDNF in vitro and in vivo. Most importantly, it is orally bioactive, passes the blood-brain barrier and exhibits promising therapeutic efficacy in a variety of neurological and psychological diseases. Notably, it displays different kinetic and temporal activities in activating TrkB and its downstream effectors from BDNF, although both strongly trigger ligand-bound TrkB receptor internalization.
This work was supported, in whole or in part, by National Institutes of Health Grants RO1, DC010204 and NS045627 (to K. Y.).
- 7,8-DHF
- 7,8-dihydroxyflavone
- ECD
- extracellular domain
- SPR
- surface plasmon resonance
- RU
- resonance unit
- DIV
- days in vitro
- P
- postnatal day
- LRR
- leucine-rich region
- SHC
- Src homology 2 domain containing adapter protein
- EEA
- early endosome antigen 1
- NTA
- nickel-nitriloacetic acid
- EDC
- 1-ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride
- DMAP
- 4-dimethylaminopyridine
- HRMS
- high-resolution mass spectrum
- ICD
- intracellular domain.
REFERENCES
- 1. Kaplan D. R., Miller F. D. (2000) Neurotrophin signal transduction in the nervous system. Curr. Opin. Neurobiol. 10, 381–391 [DOI] [PubMed] [Google Scholar]
- 2. Kaplan D. R., Stephens R. M. (1994) Neurotrophin signal transduction by the Trk receptor. J. Neurobiol. 25, 1404–1417 [DOI] [PubMed] [Google Scholar]
- 3. Grimes M. L., Zhou J., Beattie E. C., Yuen E. C., Hall D. E., Valletta J. S., Topp K. S., LaVail J. H., Bunnett N. W., Mobley W. C. (1996) Endocytosis of activated TrkA: evidence that nerve growth factor induces formation of signaling endosomes. J. Neurosci. 16, 7950–7964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Howe C. L., Valletta J. S., Rusnak A. S., Mobley W. C. (2001) NGF signaling from clathrin-coated vesicles: evidence that signaling endosomes serve as a platform for the Ras-MAPK pathway. Neuron 32, 801–814 [DOI] [PubMed] [Google Scholar]
- 5. Campenot R. B., MacInnis B. L. (2004) Retrograde transport of neurotrophins: fact and function. J. Neurobiol. 58, 217–229 [DOI] [PubMed] [Google Scholar]
- 6. Philippidou P., Valdez G., Akmentin W., Bowers W. J., Federoff H. J., Halegoua S. (2011) Trk retrograde signaling requires persistent, Pincher-directed endosomes. Proc. Natl. Acad. Sci. U.S.A. 108, 852–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Delcroix J. D., Valletta J. S., Wu C., Hunt S. J., Kowal A. S., Mobley W. C. (2003) NGF signaling in sensory neurons: evidence that early endosomes carry NGF retrograde signals. Neuron 39, 69–84 [DOI] [PubMed] [Google Scholar]
- 8. Ye H., Kuruvilla R., Zweifel L. S., Ginty D. D. (2003) Evidence in support of signaling endosome-based retrograde survival of sympathetic neurons. Neuron 39, 57–68 [DOI] [PubMed] [Google Scholar]
- 9. Makkerh J. P., Ceni C., Auld D. S., Vaillancourt F., Dorval G., Barker P. A. (2005) p75 neurotrophin receptor reduces ligand-induced Trk receptor ubiquitination and delays Trk receptor internalization and degradation. EMBO Rep. 6, 936–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Geetha T., Jiang J., Wooten M. W. (2005) Lysine 63 polyubiquitination of the nerve growth factor receptor TrkA directs internalization and signaling. Mol. Cell 20, 301–312 [DOI] [PubMed] [Google Scholar]
- 11. Arévalo J. C., Waite J., Rajagopal R., Beyna M., Chen Z. Y., Lee F. S., Chao M. V. (2006) Cell survival through Trk neurotrophin receptors is differentially regulated by ubiquitination. Neuron 50, 549–559 [DOI] [PubMed] [Google Scholar]
- 12. Jadhav T., Geetha T., Jiang J., Wooten M. W. (2008) Identification of a consensus site for TRAF6/p62 polyubiquitination. Biochem. Biophys. Res. Commun. 371, 521–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Du J., Feng L., Zaitsev E., Je H. S., Liu X. W., Lu B. (2003) Regulation of TrkB receptor tyrosine kinase and its internalization by neuronal activity and Ca2+ influx. J. Cell Biol. 163, 385–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jang S. W., Liu X., Yepes M., Shepherd K. R., Miller G. W., Liu Y., Wilson W. D., Xiao G., Blanchi B., Sun Y. E., Ye K. (2010) A selective TrkB agonist with potent neurotrophic activities by 7,8-dihydroxyflavone. Proc. Natl. Acad. Sci. U.S.A. 107, 2687–2692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jang S. W., Liu X., Chan C. B., France S. A., Sayeed I., Tang W., Lin X., Xiao G., Andero R., Chang Q., Ressler K. J., Ye K. (2010) Deoxygedunin, a natural product with potent neurotrophic activity in mice. PLoS ONE 5, e11528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ernfors P., Lee K. F., Jaenisch R. (1994) Mice lacking brain-derived neurotrophic factor develop with sensory deficits. Nature 368, 147–150 [DOI] [PubMed] [Google Scholar]
- 17. Gupta V. K., You Y., Gupta V. B., Klistorner A., Graham S. L. (2013) TrkB receptor signalling: implications in neurodegenerative, psychiatric and proliferative disorders. Int. J. Mol. Sci. 14, 10122–10142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tsai T., Klausmeyer A., Conrad R., Gottschling C., Leo M., Faissner A., Wiese S. (2013) 7,8-Dihydroxyflavone leads to survival of cultured embryonic motoneurons by activating intracellular signaling pathways. Mol. Cell Neurosci. 56, 18–28 [DOI] [PubMed] [Google Scholar]
- 19. Yu Q., Chang Q., Liu X., Wang Y., Li H., Gong S., Ye K., Lin X. (2013) Protection of spiral ganglion neurons from degeneration using small-molecule TrkB receptor agonists. J. Neurosci. 33, 13042–13052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. English A. W., Liu K., Nicolini J. M., Mulligan A. M., Ye K. (2013) Small-molecule trkB agonists promote axon regeneration in cut peripheral nerves. Proc. Natl. Acad. Sci. U.S.A. 110, 16217–16222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mantilla C. B., Ermilov L. G. (2012) The novel TrkB receptor agonist 7,8-dihydroxyflavone enhances neuromuscular transmission. Muscle Nerve 45, 274–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zeng Y., Tan M., Kohyama J., Sneddon M., Watson J. B., Sun Y. E., Xie C. W. (2011) Epigenetic enhancement of BDNF signaling rescues synaptic plasticity in aging. J. Neurosci. 31, 17800–17810 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 23. Zeng Y., Liu Y., Wu M., Liu J., Hu Q. (2012) Activation of TrkB by 7,8-dihydroxyflavone prevents fear memory defects and facilitates amygdalar synaptic plasticity in aging. J. Alzheimers Dis. 31, 765–778 [DOI] [PubMed] [Google Scholar]
- 24. Zeng Y., Lv F., Li L., Yu H., Dong M., Fu Q. (2012) 7,8-dihydroxyflavone rescues spatial memory and synaptic plasticity in cognitively impaired aged rats. J. Neurochem. 122, 800–811 [DOI] [PubMed] [Google Scholar]
- 25. Blugeot A., Rivat C., Bouvier E., Molet J., Mouchard A., Zeau B., Bernard C., Benoliel J. J., Becker C. (2011) Vulnerability to depression: from brain neuroplasticity to identification of biomarkers. J. Neurosci. 31, 12889–12899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Briones T. L., Woods J. (2013) Chronic binge-like alcohol consumption in adolescence causes depression-like symptoms possibly mediated by the effects of BDNF on neurogenesis. Neuroscience 254, 324–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Choi D. C., Maguschak K. A., Ye K., Jang S. W., Myers K. M., Ressler K. J. (2010) Prelimbic cortical BDNF is required for memory of learned fear but not extinction or innate fear. Proc. Natl. Acad. Sci. U.S.A. 107, 2675–2680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Choi D. C., Gourley S. L., Ressler K. J. (2012) Prelimbic BDNF and TrkB signaling regulates consolidation of both appetitive and aversive emotional learning. Transl. Psychiatry 2, e205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Andero R., Daviu N., Escorihuela R. M., Nadal R., Armario A. (2012) 7,8-dihydroxyflavone, a TrkB receptor agonist, blocks long-term spatial memory impairment caused by immobilization stress in rats. Hippocampus 22, 399–408 [DOI] [PubMed] [Google Scholar]
- 30. Wetsel W. C., Rodriguiz R. M., Guillemot J., Rousselet E., Essalmani R., Kim I. H., Bryant J. C., Marcinkiewicz J., Desjardins R., Day R., Constam D. B., Prat A., Seidah N. G. (2013) Disruption of the expression of the proprotein convertase PC7 reduces BDNF production and affects learning and memory in mice. Proc. Natl. Acad. Sci. U.S.A. 110, 17362–17367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Johnson R. A., Lam M., Punzo A. M., Li H., Lin B. R., Ye K., Mitchell G. S., Chang Q. (2012) 7,8-Dihydroxyflavone exhibits therapeutic efficacy in a mouse model of Rett syndrome. J. Appl. Physiol. 112, 704–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang B., Wu N., Liang F., Zhang S., Ni W., Cao Y., Xia D., Xi H. (2013) 7,8-Dihydroxyflavone, a small-molecule tropomyosin-related kinase B (TrkB) agonist, attenuates cerebral ischemia and reperfusion injury in rats. J. Mol. Histol. 45, 129–140 [DOI] [PubMed] [Google Scholar]
- 33. Hung P. L., Huang C. C., Huang H. M., Tu D. G., Chang Y. C. (2013) Thyroxin treatment protects against white matter injury in the immature brain via brain-derived neurotrophic factor. Stroke 44, 2275–2283 [DOI] [PubMed] [Google Scholar]
- 34. Uluc K., Kendigelen P., Fidan E., Zhang L., Chanana V., Kintner D., Akture E., Song C., Ye K., Sun D., Ferrazzano P., Cengiz P. (2013) TrkB receptor agonist 7,8-dihydroxyflavone triggers profound gender-dependent neuroprotection in mice after perinatal hypoxia and ischemia. CNS Neurol. Disord. Drug Targets 12, 360–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jiang M., Peng Q., Liu X., Jin J., Hou Z., Zhang J., Mori S., Ross C. A., Ye K., Duan W. (2013) Small-molecule TrkB receptor agonists improve motor function and extend survival in a mouse model of Huntington's disease. Hum. Mol. Genet. 22, 2462–2470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Devi L., Ohno M. (2012) 7,8-Dihydroxyflavone, a small-molecule TrkB agonist, reverses memory deficits and BACE1 elevation in a mouse model of Alzheimer's disease. Neuropsychopharmacology 37, 434–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang Z., Liu X., Schroeder J. P., Chan C. B., Song M., Yu S. P., Weinshenker D., Ye K. (2014) 7,8-Dihydroxyflavone prevents synaptic loss and memory deficits in a mouse model of Alzheimer's disease. Neuropsychopharmacology 39, 638–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liu X., Chan C. B., Jang S. W., Pradoldej S., Huang J., He K., Phun L. H., France S., Xiao G., Jia Y., Luo H. R., Ye K. (2010) A synthetic 7,8-dihydroxyflavone derivative promotes neurogenesis and exhibits potent antidepressant effect. J. Med. Chem. 53, 8274–8286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ultsch M. H., Wiesmann C., Simmons L. C., Henrich J., Yang M., Reilly D., Bass S. H., de Vos A. M. (1999) Crystal structures of the neurotrophin-binding domain of TrkA, TrkB and TrkC. J. Mol. Biol. 290, 149–159 [DOI] [PubMed] [Google Scholar]
- 40. Wiesmann C., Ultsch M. H., Bass S. H., de Vos A. M. (1999) Crystal structure of nerve growth factor in complex with the ligand-binding domain of the TrkA receptor. Nature 401, 184–188 [DOI] [PubMed] [Google Scholar]
- 41. Geetha T., Kenchappa R. S., Wooten M. W., Carter B. D. (2005) TRAF6-mediated ubiquitination regulates nuclear translocation of NRIF, the p75 receptor interactor. EMBO J. 24, 3859–3868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhou X., Xiao H., Wang H. (2011) Developmental changes of TrkB signaling in response to exogenous brain-derived neurotrophic factor in primary cortical neurons. J. Neurochem. 119, 1205–1216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fryer R. H., Kaplan D. R., Feinstein S. C., Radeke M. J., Grayson D. R., Kromer L. F. (1996) Developmental and mature expression of full-length and truncated TrkB receptors in the rat forebrain. J. Comp. Neurol. 374, 21–40 [DOI] [PubMed] [Google Scholar]
- 44. Knüsel B., Rabin S. J., Hefti F., Kaplan D. R. (1994) Regulated neurotrophin receptor responsiveness during neuronal migration and early differentiation. J. Neurosci. 14, 1542–1554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Liu X., Chan C. B., Qi Q., Xiao G., Luo H. R., He X., Ye K. (2012) Optimization of a small tropomyosin-related kinase B (TrkB) agonist 7,8-dihydroxyflavone active in mouse models of depression. J. Med. Chem. 55, 8524–8537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Croll S. D., Chesnutt C. R., Rudge J. S., Acheson A., Ryan T. E., Siuciak J. A., DiStefano P. S., Wiegand S. J., Lindsay R. M. (1998) Co-infusion with a TrkB-Fc receptor body carrier enhances BDNF distribution in the adult rat brain. Exp. Neurol. 152, 20–33 [DOI] [PubMed] [Google Scholar]
- 47. Mannion R. J., Costigan M., Decosterd I., Amaya F., Ma Q. P., Holstege J. C., Ji R. R., Acheson A., Lindsay R. M., Wilkinson G. A., Woolf C. J. (1999) Neurotrophins: peripherally and centrally acting modulators of tactile stimulus-induced inflammatory pain hypersensitivity. Proc. Natl. Acad. Sci. U.S.A. 96, 9385–9390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chan J. R., Cosgaya J. M., Wu Y. J., Shooter E. M. (2001) Neurotrophins are key mediators of the myelination program in the peripheral nervous system. Proc. Natl. Acad. Sci. U.S.A. 98, 14661–14668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Beattie E. C., Howe C. L., Wilde A., Brodsky F. M., Mobley W. C. (2000) NGF signals through TrkA to increase clathrin at the plasma membrane and enhance clathrin-mediated membrane trafficking. J. Neurosci. 20, 7325–7333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Haniu M., Talvenheimo J., Le J., Katta V., Welcher A., Rohde M. F. (1995) Extracellular domain of neurotrophin receptor trkB: disulfide structure, N-glycosylation sites, and ligand binding. Arch. Biochem. Biophys. 322, 256–264 [DOI] [PubMed] [Google Scholar]
- 51. Windisch J. M., Marksteiner R., Lang M. E., Auer B., Schneider R. (1995) Brain-derived neurotrophic factor, neurotrophin-3, and neurotrophin-4 bind to a single leucine-rich motif of TrkB. Biochemistry 34, 11256–11263 [DOI] [PubMed] [Google Scholar]
- 52. Bernard-Gauthier V., Boudjemeline M., Rosa-Neto P., Thiel A., Schirrmacher R. (2013) Toward tropomyosin-related kinase B (TrkB) receptor ligands for brain imaging with PET: radiosynthesis and evaluation of 2-(4-[(18)F]fluorophenyl)-7,8-dihydroxy-4H-chromen-4-one and 2-(4-([N-methyl-(11)C]-dimethylamino)phenyl)-7,8-dihydroxy-4H-chromen-4-one. Bioorg Med. Chem. 21, 7816–7829 [DOI] [PubMed] [Google Scholar]
- 53. Escandón E., Soppet D., Rosenthal A., Mendoza-Ramírez J. L., Szönyi E., Burton L. E., Henderson C. E., Parada L. F., Nikolics K. (1994) Regulation of neurotrophin receptor expression during embryonic and postnatal development. J. Neurosci. 14, 2054–2068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lesuisse C., Martin L. J. (2002) Long-term culture of mouse cortical neurons as a model for neuronal development, aging, and death. J. Neurobiol. 51, 9–23 [DOI] [PubMed] [Google Scholar]
- 55. Watson F. L., Porcionatto M. A., Bhattacharyya A., Stiles C. D., Segal R. A. (1999) TrkA glycosylation regulates receptor localization and activity. J. Neurobiol. 39, 323–336 [DOI] [PubMed] [Google Scholar]
- 56. Cunningham M. E., Stephens R. M., Kaplan D. R., Greene L. A. (1997) Autophosphorylation of activation loop tyrosines regulates signaling by the TRK nerve growth factor receptor. J. Biol. Chem. 272, 10957–10967 [DOI] [PubMed] [Google Scholar]
- 57. Massa S. M., Yang T., Xie Y., Shi J., Bilgen M., Joyce J. N., Nehama D., Rajadas J., Longo F. M. (2010) Small molecule BDNF mimetics activate TrkB signaling and prevent neuronal degeneration in rodents. J. Clin. Invest. 120, 1774–1785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Obermeier A., Lammers R., Wiesmüller K. H., Jung G., Schlessinger J., Ullrich A. (1993) Identification of Trk binding sites for SHC and phosphatidylinositol 3′-kinase and formation of a multimeric signaling complex. J. Biol. Chem. 268, 22963–22966 [PubMed] [Google Scholar]
- 59. Guiton M., Gunn-Moore F. J., Tavare J. M. (1995) Critical role for the phosphorylation of tyrosines 674 and 675 in signalling by the Trk tyrosine kinase. Biochem. Soc. Trans. 23, 176S. [DOI] [PubMed] [Google Scholar]