

Background: In Alzheimer brain, I2PP2A is translocated from the neuronal nucleus to the cytoplasm and promotes abnormal hyperphosphorylation of Tau.
Results: Inactivation of nuclear localization signal (NLS) causes retention of I2PP2A in the cell cytoplasm, where it promotes Tau hyperphosphorylation by affecting PP2A signaling.
Conclusion: Retention of I2PP2A in cell cytoplasm results in Tau hyperphosphorylation.
Significance: The study provides potential tools for investigating Tau-based therapeutics.
Keywords: Alzheimer Disease, Ca2+/Calmodulin-dependent Protein Kinase (CaMK), Protein Serine/Threonine Phosphatase (PSP), Serine/Threonine Protein Kinase, Tau Protein (Tau)
Abstract
Abnormal hyperphosphorylation of Tau leads to the formation of neurofibrillary tangles, a hallmark of Alzheimer disease (AD), and related tauopathies. The phosphorylation of Tau is regulated by protein phosphatase 2A (PP2A), which in turn is modulated by endogenous inhibitor 2 (I2PP2A). In AD brain, I2PP2A is translocated from neuronal nucleus to cytoplasm, where it inhibits PP2A activity and promotes abnormal phosphorylation of Tau. Here we describe the identification of a potential nuclear localization signal (NLS) in the C-terminal region of I2PP2A containing a conserved basic motif, 179RKR181, which is sufficient for directing its nuclear localization. The current study further presents an inducible cell model (Tet-Off system) of AD-type abnormal hyperphosphorylation of Tau by expressing I2PP2A in which the NLS was inactivated by 179RKR181 → AAA along with 168KR169 → AA mutations. In this model, the mutant NLS (mNLS)-I2PP2A (I2PP2AAA-AAA) was retained in the cell cytoplasm, where it physically interacted with PP2A and inhibited its activity. Inhibition of PP2A was associated with the abnormal hyperphosphorylation of Tau, which resulted in microtubule network instability and neurite outgrowth impairment. Expression of mNLS-I2PP2A activated CAMKII and GSK-3β, which are Tau kinases regulated by PP2A. The immunoprecipitation experiments showed the direct interaction of I2PP2A with PP2A and GSK-3β but not with CAMKII. Thus, the cell model provides insights into the nature of the potential NLS and the mechanistic relationship between I2PP2A-induced inhibition of PP2A and hyperphosphorylation of Tau that can be utilized to develop drugs preventing Tau pathology.
Introduction
Abnormal hyperphosphorylation and aggregation of microtubule-associated protein Tau into paired helical filaments/neurofibrillary tangles is a hallmark of neurodegenerative tauopathies, including Alzheimer disease (AD),2 frontotemporal dementias, tangle-only dementia, Pick disease, argyrophilic grain disease, progressive supranuclear palsy, corticobasal degeneration, Guam parkinsonism dementia complex, dementia pugilistica, and traumatic brain injury/chronic traumatic encephalopathy (1, 2). Tau is a highly soluble and unfolded protein that stabilizes the assembly of microtubules. However, Tau abnormal hyperphosphorylation negatively regulates its microtubule binding, dissociation of Tau from microtubules, and sequestration of normal Tau and other microtubule-associated proteins, causing its aggregation, breakdown of the microtubule network, and eventually cell death (3–7). Studies have shown that Tau normally contains 2–3 mol of phosphate/mol of Tau, but it is 3–4-fold more phosphorylated in AD brain (8). The number of neurofibrillary tangles correlates with progressive neuronal dysfunction, synaptic loss, and functional decline in humans and transgenic mouse models (9–12). Although the triggering mechanism leading to Tau hyperphosphorylation is yet to be clarified, it is well recognized that an imbalanced regulation in Tau protein kinases and phosphatases can directly cause AD-like Tau hyperphosphorylation (1).
Protein phosphatase 2A (PP2A) is the major brain Tau phosphatase that regulates Tau phosphorylation, both directly and indirectly regulating the activities of several Tau kinases, which include glycogen synthase kinase-3β (GSK-3β), cyclin-dependent kinase 5, Ca2+/calmodulin-dependent protein kinase II (CaMKII), MAPK (MEK1/2), ERK1/2, and protein kinase A (1). PP2A accounts for ∼70% of Tau phosphatase activity in the human brain, regulating nearly all Tau phosphorylation sites (13, 14), and its activity is compromised in the AD brain (15, 16). The activity of PP2A is regulated by two endogenous inhibitors, I1PP2A and I2PP2A (17, 18), along with post-translational modifications, including phosphorylation at Tyr307 (19), which inactivates PP2A, and methylation at Leu309 (20), which activates PP2A. I2PP2A, also known as SETα, TAF-1β, and PHAPII, is a nuclear protein that regulates cell cycle (21), cell proliferation (22), and cell motility (23). Moreover, I2PP2A controls gene transcription (24) by regulating histone acetylation (25) and is possibly involved in neuronal apoptotic pathways in AD brain (26). We have shown previously that both the mRNA and protein expressions of I2PP2A are up-regulated, and I2PP2A is selectively cleaved at Asn175 into two fragments, N-terminal and C-terminal fragments (I2NTF and I2CTF), by asparaginyl endopeptidase and is translocated from its primary localization in the nucleus to the cytoplasm (27–29). Because PP2A and Tau are localized in the cytoplasm, the increased neuronal cytoplasmic residing of I2PP2A in the AD brain leads to the inhibition of PP2A, Tau hyperphosphorylation, and formation of neurofibrillary tangles (28, 30, 31). More recently, we have shown that I2CTF alone sufficiently induces Tau pathology and cognitive impairment in a non-transgenic rat model of AD (32). As a nuclear protein, I2PP2A requires a specific sequence called the nuclear localization signal (NLS) to be targeted to the nucleus. Indeed, the NLS of I2PP2A has been reported at 168KRSSQTQNKASRKR181, and targeted expression of I2PP2A is found to be associated with neuronal death (33). Therefore, cytoplasmic withholding of I2PP2A regulated by NLS and its association with PP2A appear to be the key upstream molecular mechanism controlling the abnormal phosphorylation of Tau, which is considered one of the earliest signs of neuronal degeneration that precede Tau aggregation/neurofibrillary tangles in AD and related tauopathies.
In this study, we first show that the functional NLS of I2PP2A is localized at 179RKR181, which controls the shuttle of I2PP2A between the cellular nucleus and cytoplasm. Second, we report a PC12 stably expressing human Tau441 cell line that allows the inducible expression of mNLS-I2PP2A (168KR169 → AA/179RKR181 → AAA) based on the Tet-Off system. Employing this cell line, we found that cytoplasmic accumulation of I2PP2A is associated with inhibition of PP2A activity and activation of Tau kinases regulated by PP2A, hyperphosphorylation of Tau, and promotion of microtubule instability. The mNLS-I2PP2A cell model can be useful in screening of therapeutic drugs for AD and other tauopathies.
EXPERIMENTAL PROCEDURES
Construction of Expression Vectors
To create a wild type I2PP2A response vector in the Tet-Off system, PCR was performed using primers based on the WT human I2PP2A sequence (GenBankTM number AY349172): sense primer, 5′-ACATCGGATCCATGTCGGCGCCGGCGGCCAAAGTC-3′ (BamHI-I2PP2A, N-terminal positions 1–24 of human I2PP2A and a BamHI site); antisense primer, 5′-ATAAGAATGCGGCCGCCTAAGCGTAATCTGGAACATCGTATGGGTAGTCATCTTCTCCTTCATCCTCCTCTCC-3′ (C-terminal 805–834 of human I2PP2A and an HA tag plus NotI site). The PCR product was digested with BamHI and NotI and ligated into a pTRE2hyg response vector (Clontech) previously digested with BamHI and NotI to generate pTRE2hyg HA tag I2PP2AWT. I2PP2A mutant 179RKR181 → 179AAA181 cDNA was generated using a two-step PCR strategy. Briefly, two sets of primer pairs (sense primer 5′-ACATCGGATCCATGTCGGCGCCGGCGGCCAAAGTC-3′ (BamHI-I2PP2A, N-terminal positions 1–24 of human I2PP2A and a BamHI site) and antisense primer 5′-GGTAAAGAAGCTCTCTGGTTCCTCATGCTGCGCCGCCGCGCTGGCTTTATTCTGCGTTTGACTC-3′ (reverse sequence 510–573 of human I2PP2A mutant); sense primer 5′-GAGTCAAACGCAGAATAAAGCCAGCGCGGCGGCGCAGCATGAGGAACCAGAGAGCTTCTTTACC-3′ (sense sequence 510–573 of human I2PP2A mutant) and antisense primer 5′-ATAAGAATGCGGCCGCCTAAGCGTAATCTGGAACATCGTATGGGTAGTCATCTTCTCCTTCATCCTCCTCTCC-3′ (C-terminal 805–834 of human I2PP2A and an HA tag plus NotI site) were individually incubated with pGEX-6P-1-I2PP2A as a template in the first PCR. The PCR products were gel-purified, combined, and incubated with BamHI-N-I2PP2A and NotI-C-I2PP2A primers to synthesize full-length I2PP2A RKR179 → AAA cDNA in the second PCR. The resulting product was digested with NotI and BamHI and inserted into NotI and BamHI sites of pTRE2hyg to generate the pTRE2hyg I2PP2A RKR179 → AAA mutation. pTRE2hyg I2PP2A mutants were generated by the same procedure.
Cell Culture, Transfection, and Differentiation
COS-7 cells (obtained from ATCC (Manassas, VA) were grown in 25-cm2 flasks at 37 °C, containing 5% CO2 in Dulbecco's modified Eagle's medium supplemented with 10% bovine calf serum. To investigate the localization I2PP2A and its mutants, cells were plated on 4-well Lab-Tek II Chamber Slides coated with poly-l-lysine (Nalge Nunc International, Naperville, IL) and transfected with expression plasmids using FuGENE 6 (Roche Applied Science).
In order to generate a stable cell line of Tau441/I2PP2A or its mutants, PC12 pheochromocytoma cells were grown at 37 °C in Dulbecco's modified Eagle's medium (high glucose) supplemented with 10% heat-inactivated horse serum plus 5% fetal calf serum. Cells were co-transfected with pCI-neoTau441, pTet-Off, and pTRE2hyg (vector) or pTRE2hyg containing human I2PP2AWT, I2PP2AAA, and I2PP2AAA-AAA tagged with HA using Lipofectamine 2000 (Invitrogen). The pTet-Off vector contains the neomycin resistance gene as a selectable marker. Drug selection was carried out for 2 weeks by seeding 1 × 104 cells/well in 96-well tissue culture plates (Nunc, Roskilde, Denmark) containing the same media supplemented with 400 μg/ml G418 (Sigma) and 100 μg/ml hygromycin B (EMD Biosciences). Among the G418-hygromycin-resistant clones, 20 clones were selected randomly and transferred into 6-well tissue culture plates (Nunc) for cell expansion in the presence of 200 μg/ml G418 and 100 μg/ml hygromycin. Overexpression of I2PP2A has been reported to be associated with cellular death (26, 30). We therefore maintained cells at a low dose of doxycycline (Dox; Sigma) (100 ng/ml) to keep a low level of I2PP2A expression. For the present study, cells were grown without Dox for 72–96 h to allow the expression of I2PP2A. The Tau441/I2PP2A cell lines were screened by Western blot analysis of HA expression in the presence or absence of 1 μg/ml Dox. For studying the effect of mNLS-I2PP2A-induced hyperphosphorylation of tau on the number and the length of neurites, stably transfected PC12 cells were differentiated into neurons by adding 100 ng/ml NGF to the culture medium for 5 days.
Subcellular Fractionation
Cytoplasmic and nuclear fractions were prepared from cells as described previously with minor modifications (29). Briefly, the cells were scraped and washed with cold PBS, and centrifuged at 200 × g for 7 min. The pellet was resuspended in lysis buffer comprising 50 mm Tris-HCl, pH 7.4, 0.32 m sucrose, 150 mm NaCl, 10% glycerol, 1 mm sodium vanadate, 50 mm sodium fluoride, 5 mm EDTA, 1 mm PMSF, 10 μg/ml aprotinin, 10 μg/ml leupeptin and homogenized for 1 min on ice using a Potter-Elvehjem homogenizer set to 600 rpm with 5–10 gentle strokes. The lysate was then inspected, and if intact cells were still evident, the homogenization was repeated. The cell lysate was centrifuged at 750 × g for 10 min in a swinging bucket rotor. The supernatant was kept, and the pellet was resuspended with half of the initial volume of the lysis buffer with 3–4 strokes and centrifuged as above. The second supernatant was combined with the previous one, and the pellet was resuspended with the same buffer and saved as the nuclear fraction. The pooled supernatant was centrifuged at 100,000 × g for 1 h, and the resulting supernatant was saved as the cytosolic fraction. After protein measurement, the samples were boiled in Laemmli's buffer and used for Western blots.
PP2A Activity Assay
PP2A activity was assayed in cell lysates using the phosphatase ELISA described previously (29). Briefly, 96-well plates were precoated with a 17-aa phosphopeptide corresponding to Tau aa 194–207 in which Ser199 was replaced with phosphoserine and to which KKK were added at the C terminus (coating buffer, 35 mm NaHCO3). After overnight blocking with a protein-free blocking solution (Pierce), the enzymatic reaction was performed by adding 2 μg of cell lysate protein in 60 μl of reaction buffer (50 mm Tris-HCl, pH 7.0, 2 mm MnCl2, 0.1 mg/ml BSA, 20 mm β-mercaptoethanol) at 30 °C for 30 min (in the presence or absence of 15 nm of okadaic acid) in a moist chamber. The reaction was stopped by adding 150 μl of Pierce blocking solution containing 50 mm NaF, followed by an overnight incubation with monoclonal antibody Tau-1, which recognizes Tau unphosphorylated at Ser198, Ser199, or Ser202 (30). HRP-conjugated secondary antibody treatment was for 60 min at room temperature. Finally 75 μl of tetramethylbenzidine substrate reagent (Sigma) was added, and the colorimetric development was measured using a microtiter plate reader at 650-nm wavelength.
Methylation Sensitivity Assay of PP2A Antibodies
In order to characterize the methylation sensitivity of PP2Ac antibodies used in the present study, PC12/Tau cells were incubated with 0.5 m NaOH for 5 min on ice, followed by neutralization with an equal volume of HCl and 0.5 volume of 1 m Tris-HCl, pH 6.8 (34). Control was treated with preneutralized base solution followed by 0.5 volume of Tris-HCl, pH 6.8. The samples were then analyzed by Western blots using three anti-PP2Ac antibodies: 1D6, R123d, and BD.
Co-immunoprecipitation
PC12 cells stably transfected with HA-tagged Tau441/I2PP2A or its mutants were lysed in co-immunoprecipitation lysis buffer (50 mm HEPES, pH 7.5, 150 mm NaCl, 1 mm EGTA, 10% glycerol, 1.5 mm magnesium chloride, 0.1% Triton X-100, 1 mm PMSF, 1 μg/ml leupeptin, and 50 units/ml aprotinin). After centrifugation at 16,000 × g for 15 min, the supernatants were then used for immunoprecipitation with rabbit anti-PP2Ac (R123d) (35), rabbit anti-GST (Cell Signaling), or rabbit anti-HA (Cell Signaling), followed by incubation with protein G-Sepharose (Thermo Scientific, Rockford, IL). The Western blots of immunoprecipitates were probed with mouse anti-HA (1:8000; Millipore, Billerica, MA), mouse anti-PP2Ac (ID6, 1:1500; Millipore), mouse anti-GFP (1:1000; Cell Signaling), mouse anti-I2PP2A (10E7, 1:1500) (27), mouse anti-c-Myc (1:1000; Cell Signaling), and mouse anti-PP1 (1:2000; BD Biosciences).
SDS-PAGE, Western Blots, and Quantification by Densitometry
Proteins were separated by 10% SDS-PAGE and transferred to PVDF membrane, and then the membrane was blocked with 5% skim milk for 1 h at room temperature. The membrane was probed with anti-I2PP2A (10E7, 1:1500) (27), anti-Tau (43D, 1:1500) (36), anti-Tau (R134d, 1:5000) (37), anti-Tau Ser(P)262/356 (12E8, 1:500) (38), anti-Tau Ser(P)396/404 (PHF1, 1:500; Peter Davis), anti-β-actin (1:3000; Sigma), anti-histone 3 (1:1000; Santa Cruz Biotechnology, Inc.), anti-PP1 (1:2000; BD Biosciences), anti-PP2Ac (1:5000; BD Biosciences), anti-GAPDH (1:2000; Santa Cruz Biotechnology), anti-GSK-3β, anti-Ser(P)9-GSK-3β, anti-ERK1/2, anti-Thr(P)202/Tyr(P)204-ERK1/2, anti-MEK1/2, anti-Ser(P)217/221-MEK1/2, anti-CAMKII, anti-Thr(P)286-CAMKII, anti-p70 S6 kinase, anti-Thr(P)389-p70 S6 kinase, anti-GFP, and anti-c-Myc (1:1000; Cell Signaling Technology, Danvers, MA). The membrane was then incubated with HRP-conjugated secondary antibody (Jackson ImmunoResearch, West Grove, PA). The protein bands were detected with the enhanced chemiluminescence reagents (Thermo Scientific). Band intensity was measured by Multi-Gauge version 3 software (Fuji Photo Film, Tokyo, Japan).
Immunofluorescent Staining
Histopathologically confirmed AD and age-matched control brain paraffin sections (Sun Health Research Institute, Brain Donation Program, Sun City, AZ) were treated with antigen retrieval buffer containing 7 mm citric acid and 10 mm sodium citrate in microwave for 2 min followed by primary antibody treatment. On the other hand, PC12 cells were grown on 4-chamber glass slides, fixed in cold 4% paraformaldehyde for 15 min, and permeabilized in 0.1% Triton X-100 for 15 min, followed by incubation in 5% goat serum to block the nonspecific staining for 1 h. The primary antibodies used at the indicated dilutions were as follows: anti-HA (1:3000, Sigma-Aldrich), anti-PP2Ac (R123d, 1:300), anti-I2PP2A (1483, 1:200) (29), anti-α-tubulin (DM1A, 1:2000; Sigma), and anti-GSK-3β (1:300; Cell Signaling) overnight at 4 °C. An Alexa-488/555-conjugated appropriate combination of anti-mouse/rabbit secondary antibody (1:500; Jackson Laboratory) was used for fluorescent labeling. The cells on slides were rinsed with PBS, mounted with Fluorogel (Electron Microscopy Sciences, Hartfield, PA), and examined under a laser confocal microscope (Nikon Eclipse 90i, Melville, NY). For quantitative analysis, the images were captured using the ×40 objective, and the antibody staining was quantified by measuring mean pixel intensity with the software Image ProPlus version 5.0 (Media Cybernetics, Silver Spring, MD), as described previously (39, 40). Each experimental condition was calculated using the average number of 5 fields/well for 3–4 wells. The numbers of cells bearing neurites and the length of the neurites were analyzed by 25–30 randomly selected NGF-differentiated PC12 cells using ImageJ software, as described previously (41).
Statistical Analysis
The data were analyzed with Student's t test when comparing means between two groups or with one-way analysis of variance plus Bonferroni post hoc multiple comparison test to compare mean values across multiple treatment groups. In all cases, a probability statistic <0.05 was taken to indicate significance. All data are expressed throughout as means ± S.E.
RESULTS
Mutant NLS-I2PP2A Translocates to the Cell Cytoplasm from the Nucleus and Inhibits PP2A Activity
We previously reported that I2PP2A is translocated from neuronal nucleus to the cytoplasm, co-localizes with PP2A, and promotes abnormal hyperphosphorylation of Tau by inhibiting PP2A in AD brain (27, 28), suggesting the critical role of I2PP2A translocation and its association with PP2A in the pathogenesis of AD. Because the NLS of proteins plays an important role in facilitating the nuclear translocation, we focused on the 168KRSSQTQNKASRKR181 region of I2PP2A, as described by Qu et al. (33). We further carried out multiple alignments of I2PP2A sequence among different species and found that 178RKR181 emerged as the most conserved sequence, suggesting its important functional implication (Fig. 1A). To identify the critical region of I2PP2A NLS, we generated various mutants of this protein (Fig. 1B) and overexpressed them in COS7 cells transiently. Confocal microscopy analysis of their subcellular localization revealed that the mutants I2PP2AAAA and I2PP2AAA-AAA were present in a diffuse manner throughout the cells, whereas the wild type I2PP2A and I2PP2A 168AA169 were localized mainly in the nuclei (Fig. 1C), but low cytoplasmic staining can also be observed at higher microscopic gain (data not shown), suggesting that the 178RKR181 motif is required for the nuclear localization of I2PP2A. We further observed that mutation of Lys180 to Ala alone confined the I2PP2A into the cytoplasm of COS7 cells (Fig. 1D).
FIGURE 1.

Identification of nuclear localization signal of I2PP2A. A, the structural similarities between human, chimpanzee, pig, bovine, rat, mouse, and Drosophila were analyzed to determine the conservation and homology of potential I2PP2A NLS among various species. B, schematic diagram of the constructs of I2PP2A and its NLS mutants employed to study their intracellular localization. C, photomicrographs of COS7 cells transiently transfected with vector (vec), HA-tagged human I2PP2A (I2PP2AWT), and its NLS mutants (I2PP2AAA, I2PP2AAAA, and I2PP2AAA-AAA) for the identification of critical regions of I2PP2A required for its translocation from the cell nucleus to the cytoplasm. Following 48 h of transfection with HA-tagged I2PP2A (WT and NLS mutants), COS7 cells were subjected to immunocytochemical staining with anti-HA, and TOPRO-3 was used for nuclear staining. Mutations at amino acid residues 178–181 markedly increased the cytoplasmic retention of I2PP2A. D, COS7 cells transiently transfected with R179A-I2PP2A and K180A-I2PP2A, followed by immunostaining with anti-HA (I2PP2A). Immunofluorescence images indicate that lysine 180 is required for I2PP2A nuclear localization (i.e. NLS). Scale bar, 50 μm.
Next we evaluated whether translocation of I2PP2A is associated with inhibition of PP2A activity. As expected, total cell lysates from all transfections of I2PP2A constructs inhibited PP2A activity significantly, independent of their cellular localization (Fig. 2A). We found similar expressions of endogenous and various constructs of HA-tagged exogenous I2PP2A (Fig. 2, B and C). In order to investigate the effect of the cytoplasmic localization of I2PP2A on PP2A activity, we fractionated the total cell lysate into cytosolic and nuclear fractions where histone-3 and β-actin were used as markers for nuclear and cytosolic fractions, respectively (Fig. 2, D--G). When we assayed PP2A activity with cytosolic fractions, only NLS mutants (I2PP2AAAA and I2PP2AAA-AAA) significantly inhibited the activity (Fig. 2H). These results suggest that the translocation of I2PP2A from neuronal nucleus to cytosol is the critical step in inhibition of PP2A activity.
FIGURE 2.

NLS mutant I2PP2A translocates from the cell nucleus to the cytoplasm and inhibits PP2A activity. A, following 48 h of transient transfection with human I2PP2AWT and its NLS mutants (I2PP2AAA, I2PP2AAAA, and I2PP2AAA-AAA), COS7 cells were lysed, and PP2A activity was measured by ELISA using total cell lysate. I2PP2AWT along with all of its NLS mutants inhibited PP2A activity. B, the expression of endogenous and exogenous I2PP2A-HA was determined by Western blots. C, the quantitation of blots in B. The exogenous I2PP2A expression did not show any significant difference among the groups. D and F, nuclear and cytoplasmic fractions were prepared from COS7 cells transiently transfected (48 h), and the expression of I2PP2A in each fraction was analyzed by Western blots. Histone-3 and β-actin were used as markers of nuclear and cytoplasmic fractions, respectively. E and G, quantitative analysis of blots in D and F, respectively. The cytoplasmic retention of I2PP2A is associated with inactivation of NLS. H, PP2A activity assayed in the cytosolic fraction showed a significant decrease in NLS-I2PP2A mutants (I2PP2AAAA and I2PP2AAA-AAA). Data are expressed as mean ± S.E. (error bars) for three separate experiments. *, p < 0.05; **, p < 0.01.
Direct Interaction of PP2Ac with Translocated mNLS-I2PP2A Leads to Inhibition of PP2A Activity
Abnormal hyperphosphorylation of Tau is considered a key event leading to neurofibrillary tangle formation, which correlates with dementia in AD (42, 43). Based on the present finding that translocated mutant NLS-I2PP2A inhibits PP2A activity, the major phosphatase in the brain, we developed a Tet-Off cellular model of AD-type abnormal hyperphosphorylation of Tau by stably overexpressing HA-tagged mNLS-I2PP2A (I2PP2AAA-AAA) along with human Tau441 in PC12 cells. We also developed stably transfected cell lines for vector and I2PP2AWT in an identical manner. To validate this model, we first determined the PP2A activity in the cytosolic fraction of the cell lines. PP2A activity was significantly decreased in mNLS-I2PP2A cells as compared with controls (vec) (Fig. 3A). PP2A activity was also decreased by I2PP2AWT but did not reach statistical significance (Fig. 3A). We then characterized the methylation sensitivity of PP2Ac antibodies used in the present study by treating PC12 cell lysates with NaOH, which causes the demethylation of the PP2Ac pool (34). We found that R123d and BD PP2Ac antibodies recognize total PP2Ac, whereas 1D6 recognizes the demethylated PP2Ac pool (Fig. 3B). Next, we examined whether translocated mNLS-I2PP2A can bind to PP2Ac and thus inhibit its activity. To study the interaction between PP2Ac and mNLS-I2PP2A, the cytosolic fraction of PC12 cell lysates was also used to immunoprecipitate endogenous PP2Ac, using rabbit polyclonal antibody R123d to PP2Ac. Western blots developed with HA antibody clearly showed significantly higher interaction between I2PP2AAA-AAA and PP2Ac as compared with I2PP2AWT (Fig. 3, C and D). It is also evident that I2PP2AWT also co-immunoprecipitated with cytosolic PP2Ac, possibly caused by the overexpression of the transgene. To further confirm these findings, we carried out immunofluorescence analysis with stably transfected cell lines using antibodies to PP2Ac (R123d) and I2PP2A (HA). A diffuse pattern of I2PP2A staining predominantly localized to the nucleus was observed in I2PP2AWT cells (Fig. 3E). In contrast, PP2Ac and I2PP2A were found to co-localize in the cytoplasmic compartment in mNLS-I2PP2A (I2PP2AAA-AAA) cells (Fig. 3E). These findings suggest that the direct physical interaction of NLS mutant-I2PP2A with PP2A catalytic subunit could be involved in PP2A inhibition.
FIGURE 3.

Increased interaction between I2PP2A and PP2A in NLS mutant stably transfected PC12/Tau cells. Cells from stably expressed vector (vec), HA-tagged I2PP2AWT, and mNLS-I2PP2A (I2PP2AAA-AAA) were lysed and used for PP2A activity and immunoprecipitation. A, PP2A activity using cytoplasmic fraction, which was significantly reduced in mNLS-I2PP2A cells. B, methylation sensitivity of PP2A antibodies. PC12/Tau cells were incubated with or without 0.5 m NaOH, and the samples were then analyzed by Western blots using three anti-PP2Ac antibodies: 1D6, R123d, and BD. 1D6 predominantly recognizes demethylated PP2Ac, whereas R123d and BD recognize the total PP2Ac pool. C and D, quantitative analysis of PP2Ac immunoprecipitates blotted for co-immunoprecipitating I2PP2A showed a marked increase in interaction between PP2A and I2PP2A in the cytoplasm. E, immunocytochemical staining of I2PP2A-HA and PP2Ac in stably expressed vector, I2PP2AWT, and mNLS-I2PP2A in PC12/Tau cells. mNLS-I2PP2A co-localizes with PP2Ac in the cell cytoplasm, whereas I2PP2AWT stays mostly in the nucleus. Error bars, S.E. Scale bar, 50 μm.
Effect of mNLS-I2PP2A on Tau Phosphorylation and Neurite Outgrowth
PP2A is the most implicated protein phosphatase in regulating Tau phosphorylation because it accounts for more than ∼70% of Tau dephosphorylation activity (13). Inhibition of PP2A activity has been reported to be associated with abnormal hyperphosphorylation of Tau in cultured cells and in the brain (16, 29, 32, 40, 44–46). In the current study, we therefore investigated the functional consequences of the inhibition of PP2A activity due to direct interaction of PP2Ac with mNLS-I2PP2A in the I2PP2AAA-AAA cells as compared with I2PP2AWT cells. Because both I2PP2AWT and I2PP2AAA-AAA were Tet-Off cell lines, we determined abnormal hyperphosphorylation of Tau at 12E8 (Ser262/Ser356) and PHF1 (Ser396/Ser404) sites in the presence or absence of 1 μg/ml Dox for 72 h. The level of the abnormal hyperphosphorylation of Tau at 12E8 and PHF1 sites, which are considered major sites in AD-type neurofibrillary degeneration, was increased significantly both in I2PP2AWT (p < 0.05) and mNLS-I2PP2A (p < 0.01) as compared with the vector cells (Fig. 4). However, the extent of Tau hyperphosphorylation at 12E8 (p = 0.054) and PHF1 (p = 0.046) sites in mNLS-I2PP2A was higher than in I2PP2AWT cells. Treatment with Dox normalized the level of I2PP2A transgene expression and the associated increase in abnormal hyperphosphorylation of Tau (Fig. 4). No significant change was detected in total Tau level as determined by human-specific anti-Tau, 43D, among the cell lines (Fig. 4A).
FIGURE 4.

mNLS-I2PP2A promotes abnormal hyperphosphorylation of Tau in Tau441-Tet-Off-mNLS-I2PP2A PC12 cells, an inducible cell model. A, cells were treated with or without Dox (1 μg/ml) for 72 h, and the cell lysates were used to measure Tau phosphorylation by Western blots developed with 12E8 (Ser(P)262/Ser(P)356) and PHF1 (Ser(P)396/Ser(P)404) Tau antibodies. B, quantitative analysis showing the suppression of the expressions of I2PP2AWT and I2PP2AAA-AAA with Dox. C, quantitative analysis showing Tau hyperphosphorylation at 12E8 and PHF1 sites expressed as mean ± S.E. (error bars) after normalization with total Tau (43D). The abnormal hyperphosphorylation of Tau at 12E8 and PHF1 sites seen in a Dox-dependent manner was higher in I2PP2AAA-AAA than in I2PP2AWT cells due to the cytoplasmic localization of the former. *, p < 0.05; **, p < 0.01.
The functional aftermath of abnormal hyperphosphorylation of Tau is the disruption of microtubule stability, which could reduce the number and the length of neurite outgrowth (41). To investigate this possibility, we differentiated the stable cell lines of I2PP2AWT and I2PP2AAA-AAA with 100 ng/ml NGF for 5 days and then carried out immunofluorescence using rabbit polyclonal antibody to HA and mouse monoclonal antibody DM1A to tubulin. The morphology of cells expressing I2PP2AAA-AAA was significantly altered, with marked decreases in the number and length of neurites. The neurite outgrowth was also reduced in I2PP2AWT cells but was not statistically significant when compared with controls (vec) (Fig. 5, A–C). The number of neurite-bearing cells was reduced to ∼22 and ∼39%, whereas the mean neurite length was reduced to ∼17 and ∼49% in PC12 cells stably expressing I2PP2AWT and mNLS-I2PP2A, respectively (Fig. 5). These data imply that the abnormal hyperphosphorylation of Tau by mNLS-I2PP2A suppresses its microtubule binding and assembly, which eventually impairs the neurite outgrowth and neuronal morphology.
FIGURE 5.

Effect of I2PP2A on neuronal morphology and neurite outgrowth in PC12 cell. PC12/Tau stably transfected cells expressing vector (Vec), I2PP2AWT, or mNLS-I2PP2A (I2PP2AAA-AAA) were differentiated with 100 ng/ml NGF for 5 days and then processed for double immunofluorescence using anti-tubulin (DM1A) and anti-HA (I2PP2A). A, representative confocal images. B and C, quantitative analysis of cells bearing neurites and neurite length, respectively. The morphology of cells expressing mNLS-I2PP2A was significantly altered with a marked decrease in the number and length of neurites as compared with vector and I2PP2AWT cells. Scale bar, 50 μm. Error bars, S.E. *, p < 0.05; **, p < 0.01.
Effect of mNLS-I2PP2A on GSK-3β and CAMKII Signaling Pathways
The abnormal hyperphosphorylation of Tau that results from the inhibition of PP2A activity is probably due to not only a direct decrease in the dephosphorylation by PP2A but also an increase in phosphorylation of Tau by Tau kinases that are regulated by PP2A. Among the Tau kinases in the brain, GSK-3β, CAMKII, ERK1/2, MEK1/2, and P70S6 kinase are regulated by PP2A (47). To explore the involvement of PP2A-regulated Tau kinases, we measured the levels of total and activated/inactivated forms of these kinases by Western blots. The levels of phosphorylated/activated CaMKII increased, whereas phosphorylated/inactivated GSK-3β at Ser9 decreased noticeably in mNLS-I2PP2A cells (Fig. 6, A and B). However, we detected no significant change in the levels of phosphorylated ERK1/2, MEK1/2, and p70 S6 kinase among the cells. CAMKII phosphorylates Tau at Ser262/Ser356 (12E8 site), whereas GSK-3β is the responsible kinase phosphorylating Tau at both Ser396 and Ser404 (PHF1 site) (48). Treatment with KN-93 (CAMKII inhibitor) but not with LiCl (GSK-3β inhibitor) significantly (p < 0.05) inhibited the mNLS-I2PP2A-induced Tau hyperphosphorylation at 12E8 site (Fig. 6, C and D). On the contrary, LiCl treatment significantly (p < 0.05) prevented the Tau phosphorylation at PHF1 but not at the 12E8 site induced by mNLS-I2PP2A expression (Fig. 6, C and D). Combined treatment of both inhibitors (KN-93 and LiCl) showed a synergistic effect in reversing the hyperphosphorylation of Tau in mNLS-I2PP2A cells. These results suggested that mNLS-I2PP2A promotes abnormal hyperphosphorylation of Tau both by directly inhibiting PP2A and by up-regulating the activities of Tau kinases regulated by PP2A.
FIGURE 6.

Activation of CAMKII and GSK-3β in response to cytoplasmic retention of I2PP2A. Lysates from stably expressing vector (vec), I2PP2AWT, or mNLS-I2PP2A (I2PP2AAA-AAA) were used to detect the activation/inactivation of Tau protein kinases. A, Western blot patterns of total and phosphorylated kinases; B, quantitation of phosphorylated kinases after normalization with the level of corresponding kinase. C and D, cell lines were incubated with LiCl (1 mm) and/or KN-93 (10 μm) for 24 h, and the cell lysates were used to analyze the levels of hyperphosphorylation of Tau at 12E8 and PHF1 sites. C, representative Western blots. D, quantitation of blots from C showed that I2PP2A-induced Tau hyperphosphorylation at the 12E8 site was inhibited by KN93 and not by GSK-3β inhibitor LiCl, whereas the PHF1 site was inhibited by LiCl and not by KN93 alone. KN93 together with LiCl yielded the highest inhibition. Data are expressed as mean ± S.E. (error bars) from 3–4 separate experiments. *, p < 0.05; **, p < 0.01.
However, the question remains to be answered as to whether mNLS-I2PP2A has any direct interaction with CAMKII or GSK-3β, which might also contribute to the activation of these kinases. To address this issue, we performed co-immunoprecipitation studies with cell lysates from COS7 cells transiently transfected with a combination of I2PP2AAA-AAA-HA/CAMKII-GFP or I2PP2AAA-AAA-Myc/GSK-3β-HA. I2PP2A co-immunoprecipitated with GSK-3β along with PP2Ac, whereas no in vivo association of CAMKII with I2PP2A was detected (Fig. 7, A and B). Furthermore, protein phosphatase 1 (PP1) is reported to dephosphorylate GSK-3β at Ser9 and activate the kinase activity (49). Because mNLS-I2PP2A is found to be associated with GSK-3β with undefined functional implications, it is reasonable to speculate that I2PP2A might affect GSK-3β indirectly through PP1. Thus, we also evaluated the direct interaction between I2PP2A and PP1 by co-immunoprecipitation experiments. We did not find any association of I2PP2A with PP1 in our cell model (Fig. 7A). Furthermore, Western blot analysis of PP1 did not show any significant change between I2PP2AWT and mNLS-I2PP2A cells used in the study (Fig. 7, C and D). In addition, GSK-3β and I2PP2A were found to co-localize in cytoplasmic compartments of COS7 cells double-labeled with anti-GSK-3β and anti-HA (I2PP2AAA-AAA) (Fig. 7E). Next, we asked the question whether or not similar cross-talk between I2PP2A and GSK-3β is also evident in AD brain. Consistent with previous reports (27, 50), a significantly enhanced I2PP2A staining was detected in the cytoplasm of hippocampus of AD brains as compared with age-matched control brains, and the increased I2PP2A was co-localized with GSK-3β (Fig. 7F).
FIGURE 7.

In vivo association of I2PP2A with GSK-3β. A and B, COS7 cells were transiently transfected with a combination of I2PP2AAA-AAA-HA/CAMKII-GFP or I2PP2AAA-AAA-MYC/GSK-3β-HA. Cell lysates were subjected to immunoprecipitation (IP) with anti-HA, and the Western blots of immunoprecipitates were probed with anti-GFP for CAMKII, anti-HA/MYC for I2PP2A, anti-PP2Ac, and anti-PP1. I2PP2A was found to be associated with GSK-3β and with PP2Ac but not with CAMKII and PP1. C, the total level of PP1 was detected by Western blots in vector, I2PP2AWT, and mNLS-I2PP2A cell lines. D, quantitation of blots in C. Data are expressed as mean ± S.E. (error bars) from two separate experiments. E, COS7 cells were transiently co-transfected with GSK-3β-GFP and HA-tagged I2PP2AWT or mNLS-I2PP2A (I2PP2AAA-AAA), and cells were then stained with anti-HA (I2PP2A). The co-localization of the two is demonstrated by yellow color in the merged image, suggesting the cross-talk of translocated I2PP2A with GSK-3β in the cytoplasm. Scale bar, 20 μm. F, co-localization of I2PP2A and GSK-3β in the cytoplasm of hippocampal neurons in AD brain. Scale bar, 50 μm.
DISCUSSION
Tauopathies are age-associated neurodegenerative diseases characterized by neurofibrillary pathology made up of abnormally hyperphosphorylated Tau, and the severity of these lesions directly correlates with dementia. Given the magnitude and the impact of tauopathies, including AD, on public health, there is increasing interest in the field to decipher the molecular mechanism underlying the Tau pathology as well as identifying the therapeutic interventions where disease-relevant cellular models are of crucial importance. We have previously shown that the activity of PP2A, the major Tau phosphatase regulating abnormally hyperphosphorylated Tau, is decreased in AD brains due to the cleavage and translocation of I2PP2A from the neuronal nucleus to the cytoplasm by asparagine endopeptidase, possibly as a consequence of brain acidosis (27, 29). As observed in AD brain, we recently reported a selective increase in the cleavage of I2PP2A and inhibition of PP2A activity in the lateral sclerosis cases (46), suggesting I2PP2A-PP2A signaling as a common molecular target in different neurodegenerative diseases. To clarify whether cytoplasmic localization of I2PP2A is vital for its association and inhibition of PP2A and thus neurofibrillary degeneration, we identified the potential NLS of I2PP2A, and, employing the mutated (inactivated) NLS, we generated a cell model of the hyperphosphorylated Tau that exhibits the following characteristic features. 1) The system is developed on PC12 cells based on the mutation of NLS of I2PP2A, which restricts it to cytoplasmic localization where both PP2A and expressed human Tau441 are present. 2) The expression of mNLS-I2PP2A can be switched on and off in response to doxycycline (Tet-Off system) in order to control the inhibition of PP2A activity and resultant hyperphosphorylation of Tau. 3) The expression of mNLS-I2PP2A is associated with the down-regulation of PP2A activity along with up-regulation of PP2A-regulated Tau kinases, possibly synergistically increasing the phosphorylation of Tau and impairment in microtubule network and neurite outgrowth.
I2PP2A is predominantly a nuclear protein; hence, the NLS of I2PP2A is supposed to be the central element controlling its nuclear localization. Previous studies revealed two NLSs for I2PP2A; one is close to the N terminus, 6AKVSKK11 (51), and other one is in the area of the I2PP2A cleavage site at Asn175, 168KRSSQTQNKASRKR181 (33). Based on the sequence comparison analysis among different species, we found 168KRSSQTQNKASRKR181 as an apparent conserved region of I2PP2A and inferred that this region is a potential NLS. By using site-directed mutagenesis in combination with immunofluorescence analysis, we confirmed the NLS in the I2CTF in the current study and further narrowed it down to 179RKR181 because mutations of these three amino acid residues cause the I2PP2A to be diffusely distributed throughout the cytoplasm. The present study clearly suggests that NLS at amino acids 179–181 is sufficient to determine its subcellular localization. The most likely reason is that NLS at amino acids 6–11 is neutralized by phosphorylation at Ser9 in contrast to the NLS at 179–181 because the phosphorylation of Ser9 is considered to be critical for its interaction and thus inhibition of PP2A (23). However, one cannot rule out the existence of double NLS in a protein similar to that found in 5-lipoxygenase (52). In the present study, cell fractionation and subsequent analysis with subcellular markers followed by PP2A activity further confirmed that cytoplasmic localization of I2PP2A is associated with inhibition of PP2A activity, which is considered to be the major regulator of Tau hyperphosphorylation as seen in tauopathies. Thus, utilizing this observation, we developed an inducible cellular model of Tau hyperphosphorylation by overexpressing mNLS-I2PP2A (I2PP2AAA-AAA) along with human Tau441 in PC12 cells.
PP2A has been reported to interact physically with its endogenous inhibitors I1PP2A and I2PP2A and its cleavage fragments and thus inhibits its activity (31, 41). In the present study, we validated the newly developed cellular model in which cytoplasmic localization of I2PP2A leads to increased physical association with PP2Ac and inhibits its activity. Co-localization studies with PP2Ac and I2PP2A clearly demonstrated a significant cross-talk in the cytoplasmic compartment, which is consistent with the reduced PP2A activity. Co-immunoprecipitation experiments further confirmed that the increased physical interaction between PP2A and I2PP2A facilitates the inhibition of PP2A activity in our cell model.
Next, we addressed the question of whether or not PP2A inhibition promotes the abnormal hyperphosphorylation of Tau and microtubule instability as a functional consequence of cytoplasmic retention of I2PP2A in this cell model. We found a marked increase in the abnormal hyperphosphorylation of Tau in cells at Ser262/356 (12E8) and Ser396/404 (PHF1) sites, two prominent abnormal hyperphosphorylated sites found in PHF-Tau in AD brain (53). Ser262/356 is located in the microtubule (MT) binding domain of Tau, and phosphorylation at this site has been shown to induce MT disassembly, whereby the new unbound pool of Tau is susceptible to self-assembly into PHFs (14, 43, 54, 55). Phosphorylation of Tau at Ser262 in combination with Thr212 and Thr231 results not only in loss of its normal function but also gain of a toxic activity that causes disruption of microtubule networks and cell death (3, 14). With respect to Ser262, there is evidence that Tau phosphorylation at this site plays a critical role in Aβ42-induced Tau toxicity because Tau-induced toxicity was prevented by using a transgenic fly expressing a nonphosphorylatable form of Tau at Ser262 (56). The phosphorylation of Tau at Ser262 and Ser356 has another functional consequence: priming the phosphorylation at other sites, which increases further its microtubule inhibitory activity and self-aggregation into filaments, because we previously showed that a prior phosphorylation of Tau at Ser262/356 containing the Ser/Thr-X motif by CAMKII, a non-proline-dependent protein kinase (PDPK) was found to stimulate a subsequent phosphorylation catalyzed by GSK-3β, a PDPK (57). In contrast to Ser262, Ser396 and Ser404 are present in the C-terminal domain and have only a moderate influence on Tau-MT interaction but are up-regulated in AD and other tauopathies (58, 59). Moreover, phosphorylation of Tau at the PHF1 (Ser396/404) site in combination with the AT8 (Ser199/Ser202/Ser205) site has been shown to induce a compaction of the paper clip folding of Tau that generates a pathological conformation, which aggregates somewhat more readily (60). Thus, it appears that the missorting of I2PP2A into the cytoplasm probably contributes to the deficit in PP2A activity, which in turn promotes the hyperphosphorylation and loss of biological function of Tau in our cellular model.
There is significant evidence that abnormal hyperphosphorylation of Tau results in Tau dysfunction due to the disturbances in microtubule dynamics, axonal transport, and neurite outgrowth, which synergistically contribute to the pathogenic processes. In the current study, we found that mNLS-I2PP2A decreased not only the mean neurite length but also the average number of NGF-induced differentiated cells bearing neurites. Our data are consistent with our previous observation that I1PP2A, another endogenous inhibitor of PP2A, impairs MT network and neurite outgrowth via hyperphosphorylation of Tau (41). Although our data do not imply a direct role of I2PP2A in microtubule instability, it is well recognized that once Tau is abnormally hyperphosphorylated, it can disrupt microtubules by sequestering normal microtubule-associated proteins (61). There is indirect evidence that Tau hyperphosphorylation might facilitate neurite retraction (62). Pharmacological inhibition, dominant negative down-regulation, and siRNA knockdown of PP2Ac lead to neurite retraction and inhibition of axiogenesis (63). Thus, I2PP2A could contribute to the destabilization of microtubules indirectly through PP2A-dependent phosphorylation states of Tau in our cell model. However, we cannot rule out the possibility that the effects of PP2A observed in the present study on neurite outgrowth and microtubule stability are partially Tau-independent because PP2A is not only the major Tau phosphatase but also the major brain phosphatase, which plays a crucial role in regulating most cellular functions, including neurite outgrowth (64, 65).
In addition to the direct action of PP2A on Tau, the activation of Tau protein kinases can also induce Tau hyperphosphorylation. For instance, the proline-independent 12E8 site is phosphorylated by CaMKII, PKA, and MARK (66, 67). Ser396 is almost exclusively phosphorylated by GSK-3β, whereas both GSK-3β and Cdk5 phosphorylate Tau at Ser404. We found a significant activation of CAMKII, which is consistent with the report that the treatment of metabolically active rat brain slices by a PP2A inhibitor, okadaic acid, activated CAMKII activity and promoted Tau hyperphosphorylation at the 12E8 site (68). However, the levels of phosphorylated ERK1/2, MEK1/2, and p70 S6 kinase did not change among the cells. Overexpression of I2PP2A has been shown to increase the levels of ERK1 and ERK2 in HeLa cells (69), whereas Fukukawa (70) reported the opposite; overexpression of I2PP2A results in suppression of EGF-stimulated ERK activation, and knocking down I2PP2A by siRNA resulted in enhancement of the MEK/ERK pathway in HeLa cells. Phosphorylated ERK1/2 was reported to be reduced in head and neck squamous cell carcinoma stably expressing shRNA against I2PP2A (71). The exact reason for this discordance is not clear, but it is apparent that the regulation of the MEK/ERK pathway by I2PP2A is possibly a cell type-specific phenomenon; however, it demands further investigation. Additionally, we noticed a significant activation of GSK-3β, as detected by a decrease in inhibitory phosphorylation at Ser9. Our current finding is consistent with previous reports that showed that accumulation of I2PP2A or overexpression of the C-terminal cleaved fragment of I2PP2A, I2CTF, results in an increase in GSK-3β activity (32, 72). Co-treatment with LiCl (GSK-3β inhibitor) and KN-93 (CAMKII inhibitor) significantly reduced the I2PP2A-induced Tau hyperphosphorylation. The exact reason underlying the GSK-3β activation in our model is not clear. However, the direct physical interaction between I2PP2A and GSK-3β could possibly explain the observed phenomenon. Another possible reason could be that PP1 activation could lead to the dephosphorylation of Ser(P)9-GSK-3β associated with its activation. Consistent with this possibility, it has been reported that I1PP2A and I2PP2A can markedly increase PP1c activity in the presence of a nearly physiological concentration of Mn2+ in a substrate-specific manner (73). Although we did not measure PP1 activity, we could not detect any change in total PP1 protein level. On the other hand, co-immunoprecipitation experiments clearly suggest a physical association of I2PP2A with GSK-3β in contrast with CAMKII. Consistent with our finding, Liu et al. (72) showed that overactivation of GSK-3β inhibits PP2A through up-regulation of I2PP2A, indicating a positive correlation between I2PP2A and GSK-3β activity. We further confirmed the in vivo association of GSK-3β with I2PP2A in AD brain, where both are co-localized in the neuronal cytoplasm. Collectively, these results (Fig. 8) suggest 1) that inhibition of PP2A caused by cytoplasmic translocation of I2PP2A mimics the abnormal hyperphosphorylation of Tau, as observed in AD brain; 2) that the effect of mNLS-I2PP2A is modulated both directly by PP2A and indirectly by the Tau kinases that are regulated by PP2A; and 3) that the in vivo association of I2PP2A with GSK-3β could imply a direct role of I2PP2A in activation of GSK-3β.
FIGURE 8.
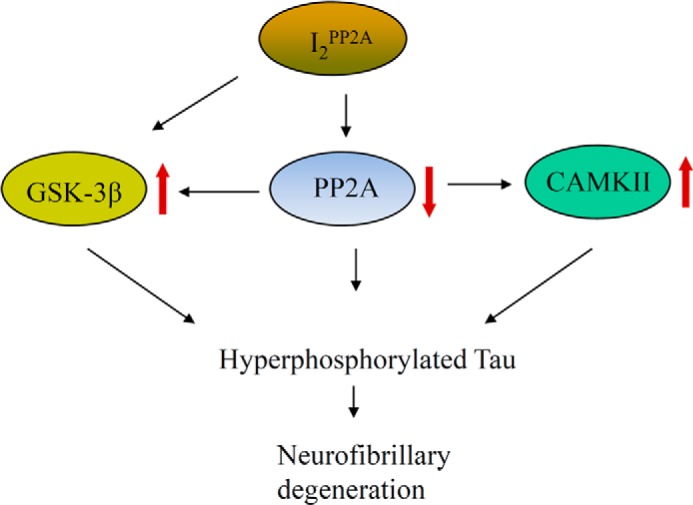
Proposed mechanism of I2PP2A-mediated Alzheimer-like abnormal hyperphosphorylation of Tau. Translocation of I2PP2A from the cell nucleus to the cytoplasm enables it to interact with PP2Ac and GSK-3β, leading to the inhibition of the phosphatase and activation of the kinase. Activation of CaMKII activity is secondary to the I2PP2A-dependent PP2A inactivation. Collectively, all of these events contribute to the hyperphosphorylation of Tau.
In conclusion, our study demonstrates a potential NLS at amino acids 179–181, which regulates the translocation of I2PP2A from neuronal cytoplasm to nucleus, and offers the development of a Tet-Off-inducible cell model of abnormal hyperphosphorylation of Tau in which the NLS of I2PP2A is mutated, and it is translocated to the cell cytoplasm. Cytoplasmic retention of I2PP2A results in inhibition of PP2A directly and indirectly by PP2A-regulated Tau kinases, all of which eventually lead to the hyperphosphorylation of Tau. The restoration of PP2A activity through the inhibition of I2PP2A-PP2A interaction offers a promising therapeutic target for preventing the neurofibrillary degeneration of the abnormally phosphorylated Tau. Our cellular model of tauopathies is suitable for screening drugs/compounds that can attenuate the abnormal hyperphosphorylation of Tau.
Acknowledgments
We thank Dr. Zane Martin Jones for helpful comments on the manuscript, Dr. George Merz for assistance with confocal microscopy, and Janet Murphy for secretarial assistance.
This work was supported, in whole or in part, by National Institutes of Health, NIA, Grant AG019158. This work was also supported by the New York State Office of People with Developmental Disabilities.
- AD
- Alzheimer disease
- PP2A
- protein phosphatase 2A
- PP1
- protein phosphatase 1
- NLS
- nuclear localization sequence
- mNLS
- mutant NLS
- MT
- microtubule
- CAMKII
- Ca2+/calmodulin-dependent protein kinase
- PDPK
- proline-directed protein kinase
- GSK-3β
- glycogen synthase kinase-3β
- PHF
- paired helical filament
- Tet-Off
- tetracycline-Off
- Dox
- doxycycline
- PC12
- pheochromocytoma.
REFERENCES
- 1. Iqbal K., Liu F., Gong C. X., Alonso Adel C., Grundke-Iqbal I. (2009) Mechanisms of tau-induced neurodegeneration. Acta Neuropathol. 118, 53–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Goedert M., Clavaguera F., Tolnay M. (2010) The propagation of prion-like protein inclusions in neurodegenerative diseases. Trends Neurosci. 33, 317–325 [DOI] [PubMed] [Google Scholar]
- 3. Alonso A. D., Di Clerico J., Li B., Corbo C. P., Alaniz M. E., Grundke-Iqbal I., Iqbal K. (2010) Phosphorylation of Tau at Thr212, Thr231, and Ser262 combined causes neurodegeneration. J. Biol. Chem. 285, 30851–30860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Alonso A. D., Grundke-Iqbal I., Barra H. S., Iqbal K. (1997) Abnormal phosphorylation of tau and the mechanism of Alzheimer neurofibrillary degeneration: sequestration of microtubule-associated proteins 1 and 2 and the disassembly of microtubules by the abnormal tau. Proc. Natl. Acad. Sci. U.S.A. 94, 298–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Alonso A. C., Grundke-Iqbal I., Iqbal K. (1996) Alzheimer's disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat. Med. 2, 783–787 [DOI] [PubMed] [Google Scholar]
- 6. Alonso A. C., Zaidi T., Grundke-Iqbal I., Iqbal K. (1994) Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 91, 5562–5566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li B., Chohan M. O., Grundke-Iqbal I., Iqbal K. (2007) Disruption of microtubule network by Alzheimer abnormally hyperphosphorylated tau. Acta Neuropathol. 113, 501–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Köpke E., Tung Y. C., Shaikh S., Alonso A. C., Iqbal K., Grundke-Iqbal I. (1993) Microtubule-associated protein tau. Abnormal phosphorylation of a non-paired helical filament pool in Alzheimer disease. J. Biol. Chem. 268, 24374–24384 [PubMed] [Google Scholar]
- 9. Alafuzoff I., Iqbal K., Friden H., Adolfsson R., Winblad B. (1987) Histopathological criteria for progressive dementia disorders: clinical-pathological correlation and classification by multivariate data analysis. Acta Neuropathol. 74, 209–225 [DOI] [PubMed] [Google Scholar]
- 10. Arriagada P. V., Growdon J. H., Hedley-Whyte E. T., Hyman B. T. (1992) Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology 42, 631–639 [DOI] [PubMed] [Google Scholar]
- 11. Polydoro M., Acker C. M., Duff K., Castillo P. E., Davies P. (2009) Age-dependent impairment of cognitive and synaptic function in the htau mouse model of tau pathology. J. Neurosci. 29, 10741–10749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Small S. A., Duff K. (2008) Linking Aβ and tau in late-onset Alzheimer's disease: a dual pathway hypothesis. Neuron 60, 534–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu F., Grundke-Iqbal I., Iqbal K., Gong C. X. (2005) Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur. J. Neurosci. 22, 1942–1950 [DOI] [PubMed] [Google Scholar]
- 14. Wang J. Z., Grundke-Iqbal I., Iqbal K. (2007) Kinases and phosphatases and tau sites involved in Alzheimer neurofibrillary degeneration. Eur. J. Neurosci. 25, 59–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gong C. X., Shaikh S., Wang J. Z., Zaidi T., Grundke-Iqbal I., Iqbal K. (1995) Phosphatase activity toward abnormally phosphorylated tau: decrease in Alzheimer disease brain. J. Neurochem. 65, 732–738 [DOI] [PubMed] [Google Scholar]
- 16. Gong C. X., Singh T. J., Grundke-Iqbal I., Iqbal K. (1993) Phosphoprotein phosphatase activities in Alzheimer disease brain. J. Neurochem. 61, 921–927 [DOI] [PubMed] [Google Scholar]
- 17. Li M., Guo H., Damuni Z. (1995) Purification and characterization of two potent heat-stable protein inhibitors of protein phosphatase 2A from bovine kidney. Biochemistry 34, 1988–1996 [DOI] [PubMed] [Google Scholar]
- 18. Li M., Makkinje A., Damuni Z. (1996) The myeloid leukemia-associated protein SET is a potent inhibitor of protein phosphatase 2A. J. Biol. Chem. 271, 11059–11062 [DOI] [PubMed] [Google Scholar]
- 19. Chen J., Martin B. L., Brautigan D. L. (1992) Regulation of protein serine-threonine phosphatase type-2A by tyrosine phosphorylation. Science 257, 1261–1264 [DOI] [PubMed] [Google Scholar]
- 20. Favre B., Zolnierowicz S., Turowski P., Hemmings B. A. (1994) The catalytic subunit of protein phosphatase 2A is carboxyl-methylated in vivo. J. Biol. Chem. 269, 16311–16317 [PubMed] [Google Scholar]
- 21. Canela N., Rodriguez-Vilarrupla A., Estanyol J. M., Diaz C., Pujol M. J., Agell N., Bachs O. (2003) The SET protein regulates G2/M transition by modulating cyclin B-cyclin-dependent kinase 1 activity. J. Biol. Chem. 278, 1158–1164 [DOI] [PubMed] [Google Scholar]
- 22. Shin K. S., Shin E. Y., Bae S. C., Kim S. R., Jeong G. B., Kwak S. J., Ballermann B. J., Kim E. G. (1999) Expression of SET is modulated as a function of cell proliferation. J. Cell Biochem. 74, 119–126 [PubMed] [Google Scholar]
- 23. ten Klooster J. P., Leeuwen I., Scheres N., Anthony E. C., Hordijk P. L. (2007) Rac1-induced cell migration requires membrane recruitment of the nuclear oncogene SET. EMBO J. 26, 336–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Compagnone N. A., Zhang P., Vigne J. L., Mellon S. H. (2000) Novel role for the nuclear phosphoprotein SET in transcriptional activation of P450c17 and initiation of neurosteroidogenesis. Mol. Endocrinol. 14, 875–888 [DOI] [PubMed] [Google Scholar]
- 25. Seo S. B., McNamara P., Heo S., Turner A., Lane W. S., Chakravarti D. (2001) Regulation of histone acetylation and transcription by INHAT, a human cellular complex containing the set oncoprotein. Cell 104, 119–130 [DOI] [PubMed] [Google Scholar]
- 26. Madeira A., Pommet J. M., Prochiantz A., Allinquant B. (2005) SET protein (TAF1β, I2PP2A) is involved in neuronal apoptosis induced by an amyloid precursor protein cytoplasmic subdomain. FASEB J. 19, 1905–1907 [DOI] [PubMed] [Google Scholar]
- 27. Tanimukai H., Grundke-Iqbal I., Iqbal K. (2005) Up-regulation of inhibitors of protein phosphatase-2A in Alzheimer's disease. Am. J. Pathol. 166, 1761–1771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tsujio I., Zaidi T., Xu J., Kotula L., Grundke-Iqbal I., Iqbal K. (2005) Inhibitors of protein phosphatase-2A from human brain structures, immunocytological localization and activities towards dephosphorylation of the Alzheimer type hyperphosphorylated tau. FEBS Lett. 579, 363–372 [DOI] [PubMed] [Google Scholar]
- 29. Basurto-Islas G., Grundke-Iqbal I., Tung Y. C., Liu F., Iqbal K. (2013) Activation of asparaginyl endopeptidase leads to Tau hyperphosphorylation in Alzheimer disease. J. Biol. Chem. 288, 17495–17507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chohan M. O., Khatoon S., Iqbal I. G., Iqbal K. (2006) Involvement of I2PP2A in the abnormal hyperphosphorylation of tau and its reversal by Memantine. FEBS Lett. 580, 3973–3979 [DOI] [PubMed] [Google Scholar]
- 31. Arnaud L., Chen S., Liu F., Li B., Khatoon S., Grundke-Iqbal I., Iqbal K. (2011) Mechanism of inhibition of PP2A activity and abnormal hyperphosphorylation of tau by I2PP2A/SET. FEBS Lett. 585, 2653–2659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang X., Blanchard J., Kohlbrenner E., Clement N., Linden R. M., Radu A., Grundke-Iqbal I., Iqbal K. (2010) The carboxy-terminal fragment of inhibitor-2 of protein phosphatase-2A induces Alzheimer disease pathology and cognitive impairment. FASEB J. 24, 4420–4432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Qu D., Zhang Y., Ma J., Guo K., Li R., Yin Y., Cao X., Park D. S. (2007) The nuclear localization of SET mediated by impα3/impβ attenuates its cytosolic toxicity in neurons. J. Neurochem. 103, 408–422 [DOI] [PubMed] [Google Scholar]
- 34. Yu X. X., Du X., Moreno C. S., Green R. E., Ogris E., Feng Q., Chou L., McQuoid M. J., Pallas D. C. (2001) Methylation of the protein phosphatase 2A catalytic subunit is essential for association of Bα regulatory subunit but not SG2NA, striatin, or polyomavirus middle tumor antigen. Mol. Biol. Cell 12, 185–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tatebayashi Y., Iqbal K., Grundke-Iqbal I. (1999) Dynamic regulation of expression and phosphorylation of tau by fibroblast growth factor-2 in neural progenitor cells from adult rat hippocampus. J. Neurosci. 19, 5245–5254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liu F., Iqbal K., Grundke-Iqbal I., Hart G. W., Gong C. X. (2004) O-GlcNAcylation regulates phosphorylation of tau: a mechanism involved in Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 101, 10804–10809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shi J., Zhang T., Zhou C., Chohan M. O., Gu X., Wegiel J., Zhou J., Hwang Y. W., Iqbal K., Grundke-Iqbal I., Gong C. X., Liu F. (2008) Increased dosage of Dyrk1A alters alternative splicing factor (ASF)-regulated alternative splicing of tau in Down syndrome. J. Biol. Chem. 283, 28660–28669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Seubert P., Mawal-Dewan M., Barbour R., Jakes R., Goedert M., Johnson G. V., Litersky J. M., Schenk D., Lieberburg I., Trojanowski J. Q., Lee V. M.-Y. (1995) Detection of phosphorylated Ser262 in fetal tau, adult tau, and paired helical filament Tau. J. Biol. Chem. 270, 18917–18922 [DOI] [PubMed] [Google Scholar]
- 39. Blanchard J., Wanka L., Tung Y. C., Cárdenas-Aguayo Mdel C., LaFerla F. M., Iqbal K., Grundke-Iqbal I. (2010) Pharmacologic reversal of neurogenic and neuroplastic abnormalities and cognitive impairments without affecting Aβ and Tau pathologies in 3xTg-AD mice. Acta Neuropathol. 120, 605–621 [DOI] [PubMed] [Google Scholar]
- 40. Bolognin S., Blanchard J., Wang X., Basurto-Islas G., Tung Y. C., Kohlbrenner E., Grundke-Iqbal I., Iqbal K. (2012) An experimental rat model of sporadic Alzheimer's disease and rescue of cognitive impairment with a neurotrophic peptide. Acta Neuropathol. 123, 133–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chen S., Li B., Grundke-Iqbal I., Iqbal K. (2008) I1PP2A affects Tau phosphorylation via association with the catalytic subunit of protein phosphatase 2A. J. Biol. Chem. 283, 10513–10521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Grundke-Iqbal I., Iqbal K., Tung Y. C., Quinlan M., Wisniewski H. M., Binder L. I. (1986) Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. U.S.A. 83, 4913–4917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Alonso A., Zaidi T., Novak M., Grundke-Iqbal I., Iqbal K. (2001) Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments. Proc. Natl. Acad. Sci. U.S.A. 98, 6923–6928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gong C. X., Shaikh S., Grundke-Iqbal I., Iqbal K. (1996) Inhibition of protein phosphatase-2B (calcineurin) activity towards Alzheimer abnormally phosphorylated tau by neuroleptics. Brain Res. 741, 95–102 [DOI] [PubMed] [Google Scholar]
- 45. Sun X. Y., Wei Y. P., Xiong Y., Wang X. C., Xie A. J., Wang X. L., Yang Y., Wang Q., Lu Y. M., Liu R., Wang J. Z. (2012) Synaptic released zinc promotes Tau hyperphosphorylation by inhibition of protein phosphatase 2A (PP2A). J. Biol. Chem. 287, 11174–11182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang X., Blanchard J., Grundke-Iqbal I., Wegiel J., Deng H. X., Siddique T., Iqbal K. (2014) Alzheimer disease and amyotrophic lateral sclerosis: an etiopathogenic connection. Acta Neuropathol. 127, 243–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Iqbal K., Grundke-Iqbal I. (2005) Metabolic/signal transduction hypothesis of Alzheimer's disease and other tauopathies. Acta Neuropathol. 109, 25–31 [DOI] [PubMed] [Google Scholar]
- 48. Qian W., Shi J., Yin X., Iqbal K., Grundke-Iqbal I., Gong C. X., Liu F. (2010) PP2A regulates tau phosphorylation directly and also indirectly via activating GSK-3β. J. Alzheimers Dis. 19, 1221–1229 [DOI] [PubMed] [Google Scholar]
- 49. Hernández F., Langa E., Cuadros R., Avila J., Villanueva N. (2010) Regulation of GSK3 isoforms by phosphatases PP1 and PP2A. Mol. Cell Biochem. 344, 211–215 [DOI] [PubMed] [Google Scholar]
- 50. Facchinetti P., Dorard E., Contremoulins V., Gaillard M. C., Déglon N., Sazdovitch V., Guihenneuc-Jouyaux C., Brouillet E., Duyckaerts C., Allinquant B. (2014) SET translocation is associated with increase in caspase cleaved amyloid precursor protein in CA1 of Alzheimer and Down syndrome patients. Neurobiol. Aging 35, 958–968 [DOI] [PubMed] [Google Scholar]
- 51. Yu G., Yan T., Feng Y., Liu X., Xia Y., Luo H., Wang J. Z., Wang X. (2013) Ser9 phosphorylation causes cytoplasmic detention of I2PP2A/SET in Alzheimer disease. Neurobiol. Aging 34, 1748–1758 [DOI] [PubMed] [Google Scholar]
- 52. Luo M., Pang C. W., Gerken A. E., Brock T. G. (2004) Multiple nuclear localization sequences allow modulation of 5-lipoxygenase nuclear import. Traffic 5, 847–854 [DOI] [PubMed] [Google Scholar]
- 53. Pei J. J., Gong C. X., An W. L., Winblad B., Cowburn R. F., Grundke-Iqbal I., Iqbal K. (2003) Okadaic-acid-induced inhibition of protein phosphatase 2A produces activation of mitogen-activated protein kinases ERK1/2, MEK1/2, and p70 S6, similar to that in Alzheimer's disease. Am. J. Pathol. 163, 845–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Singh T. J., Wang J. Z., Novak M., Kontzekova E., Grundke-Iqbal I., Iqbal K. (1996) Calcium/calmodulin-dependent protein kinase II phosphorylates tau at Ser-262 but only partially inhibits its binding to microtubules. FEBS Lett. 387, 145–148 [DOI] [PubMed] [Google Scholar]
- 55. Sengupta A., Kabat J., Novak M., Wu Q., Grundke-Iqba I., Iqbal K. (1998) Maximal inhibition of tau binding to microtubules requires the phosphorylation of tau at both Thr 231 and Ser 262. Neurobiol. Aging 19, S124–S524 [DOI] [PubMed] [Google Scholar]
- 56. Iijima K., Gatt A., Iijima-Ando K. (2010) Tau Ser262 phosphorylation is critical for Aβ42-induced tau toxicity in a transgenic Drosophila model of Alzheimer's disease. Hum. Mol. Genet. 19, 2947–2957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Singh T. J., Zaidi T., Grundke-Iqbal I., Iqbal K. (1995) Modulation of GSK-3-catalyzed phosphorylation of microtubule-associated protein tau by non-proline-dependent protein kinases. FEBS Lett. 358, 4–8 [DOI] [PubMed] [Google Scholar]
- 58. Gong C. X., Liu F., Grundke-Iqbal I., Iqbal K. (2005) Post-translational modifications of tau protein in Alzheimer's disease. J. Neural Transm. 112, 813–838 [DOI] [PubMed] [Google Scholar]
- 59. Mandelkow E., von Bergen M., Biernat J., Mandelkow E. M. (2007) Structural principles of tau and the paired helical filaments of Alzheimer's disease. Brain Pathol. 17, 83–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Jeganathan S., Hascher A., Chinnathambi S., Biernat J., Mandelkow E. M., Mandelkow E. (2008) Proline-directed pseudo-phosphorylation at AT8 and PHF1 epitopes induces a compaction of the paperclip folding of Tau and generates a pathological (MC-1) conformation. J. Biol. Chem. 283, 32066–32076 [DOI] [PubMed] [Google Scholar]
- 61. Alonso Adel C., Li B., Grundke-Iqbal I., Iqbal K. (2006) Polymerization of hyperphosphorylated tau into filaments eliminates its inhibitory activity. Proc. Natl. Acad. Sci. U.S.A. 103, 8864–8869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sayas C. L., Avila J., Wandosell F. (2002) Regulation of neuronal cytoskeleton by lysophosphatidic acid: role of GSK-3. Biochim. Biophys. Acta 1582, 144–153 [DOI] [PubMed] [Google Scholar]
- 63. Zhu L. Q., Zheng H. Y., Peng C. X., Liu D., Li H. L., Wang Q., Wang J. Z. (2010) Protein phosphatase 2A facilitates axonogenesis by dephosphorylating CRMP2. J. Neurosci. 30, 3839–3848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Virshup D. M., Shenolikar S. (2009) From promiscuity to precision: protein phosphatases get a makeover. Mol. Cell 33, 537–545 [DOI] [PubMed] [Google Scholar]
- 65. Sontag J. M., Sontag E. (2014) Protein phosphatase 2A dysfunction in Alzheimer's disease. Front. Mol. Neurosci. 7, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Drewes G., Lichtenberg-Kraag B., Döring F., Mandelkow E. M., Biernat J., Goris J., Dorée M., Mandelkow E. (1992) Mitogen activated protein (MAP) kinase transforms tau protein into an Alzheimer-like state. EMBO J. 11, 2131–2138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Sironi J. J., Yen S. H., Gondal J. A., Wu Q., Grundke-Iqbal I., Iqbal K. (1998) Ser-262 in human recombinant tau protein is a markedly more favorable site for phosphorylation by CaMKII than PKA or PhK. FEBS Lett. 436, 471–475 [DOI] [PubMed] [Google Scholar]
- 68. Bennecib M., Gong C. X., Grundke-Iqbal I., Iqbal K. (2001) Inhibition of PP-2A upregulates CaMKII in rat forebrain and induces hyperphosphorylation of tau at Ser 262/356. FEBS Lett. 490, 15–22 [DOI] [PubMed] [Google Scholar]
- 69. Lam B. D., Anthony E. C., Hordijk P. L. (2013) Cytoplasmic targeting of the proto-oncogene SET promotes cell spreading and migration. FEBS Lett. 587, 111–119 [DOI] [PubMed] [Google Scholar]
- 70. Fukukawa C., Shima H., Tanuma N., Okada T., Kato N., Adachi Y., Kikuchi K. (2005) The oncoprotein I-2PP2A/SET negatively regulates the MEK/ERK pathway and cell proliferation. Int. J. Oncol. 26, 751–756 [PubMed] [Google Scholar]
- 71. Sobral L. M., Sousa L. O., Coletta R. D., Cabral H., Greene L. J., Tajara E. H., Gutkind J. S., Curti C., Leopoldino A. M. (2014) Stable SET knockdown in head and neck squamous cell carcinoma promotes cell invasion and the mesenchymal-like phenotype in vitro, as well as necrosis, cisplatin sensitivity and lymph node metastasis in xenograft tumor models. Mol. Cancer 13, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Liu G. P., Zhang Y., Yao X. Q., Zhang C. E., Fang J., Wang Q., Wang J. Z. (2008) Activation of glycogen synthase kinase-3 inhibits protein phosphatase-2A and the underlying mechanisms. Neurobiol. Aging 29, 1348–1358 [DOI] [PubMed] [Google Scholar]
- 73. Katayose Y., Li M., Al-Murrani S. W., Shenolikar S., Damuni Z. (2000) Protein phosphatase 2A inhibitors, I1PP2A and I2PP2A, associate with and modify the substrate specificity of protein phosphatase 1. J. Biol. Chem. 275, 9209–9214 [DOI] [PubMed] [Google Scholar]