

Background: Metformin inhibits pancreatic cancer cell and tumor growth and down-regulated Sp transcription factors.
Results: Inhibition of mTOR and Ras signaling by metformin is due to decreased expression of Sp-regulated insulin-like growth factor-1 receptor (IGF-1R) and epidermal growth factor receptor (EGFR), respectively.
Conclusion: Metformin-induced down-regulation of Sp proteins affects multiple pro-oncogenic pathways.
Significance: These results identify important metformin-induced anticancer activities.
Keywords: Epidermal Growth Factor Receptor (EGFR), Gene Expression, Insulin-like Growth Factor (IGF), Pancreatic Cancer, Specificity Protein 1 (Sp1), IGFR Down-regulation, Ras Inhibition, Sp Transcription Factors, mTOR Down-regulation, Metformin
Abstract
The antidiabetic drug metformin exhibits both chemopreventive and chemotherapeutic activity for multiple cancers including pancreatic cancer; however, the underlying mechanism of action of metformin is unclear. A recent study showed that metformin down-regulated specificity protein (Sp) transcription factors (TFs) Sp1, Sp3, and Sp4 in pancreatic cancer cells and tumors, and this was accompanied by down-regulation of several pro-oncogenic Sp-regulated genes. Treatment with metformin or down-regulation of Sp TFs by RNAi also inhibits two major pro-oncogenic pathways in pancreatic cancer cells, namely mammalian target of rapamycin (mTOR) signaling and epidermal growth factor (EGFR)-dependent activation of Ras. Metformin and Sp knockdown by RNAi decreased expression of the insulin-like growth factor-1 receptor (IGF-1R), resulting in inhibition of mTOR signaling. Ras activity was also decreased by metformin and Sp knockdown of EGFR, another Sp-regulated gene. Thus, the antineoplastic activities of metformin in pancreatic cancer are due, in part, to down-regulation of Sp TFs and Sp-regulated IGF-1R and EGFR, which in turn results in inhibition of mTOR and Ras signaling, respectively.
Introduction
The antidiabetic drug metformin has been successfully used for treatment of type II diabetes, and there is increasing evidence that metformin is an anticancer agent that exhibits both chemopreventive and chemotherapeutic activities (1–4). It has been shown that cancer rates in diabetics using metformin are lower than in patients using other insulin-sensitizing agents (2, 5–12). In one study, the overall survival of type II diabetic patients with colorectal cancer was 76.9 months for individuals treated with metformin as compared with 56.9 months for those receiving other diabetic medications, and this represented a 30% improvement in overall survival (6). A comparable study in pancreatic cancer patients showed that diabetics using metformin had a 32% lower risk of death and longer overall survival than diabetics using other drugs (10). This latter study recommended using metformin as a supplemental therapy for treatment of pancreatic cancer patients (10).
The potential clinical applications for metformin in cancer chemotherapy also stems from studies on the anticancer activities of this drug in cancer cells in culture and in in vivo models (1–4, 13–28). Metformin inhibits growth and induces apoptosis and other antineoplastic responses in multiple cancer cell lines, and this is accompanied by modulation of different pathways and genes. Several studies demonstrate that metformin activates AMP-activated protein kinaseα (AMPKα), which results in the inhibition of the mTOR2 signaling pathway and downstream effects (15–20, 22–25), and this compliments one of the proposed mechanisms of action of metformin as an antidiabetic drug (29, 30).
Studies in this laboratory reported a novel mechanism of action for metformin in pancreatic cancer cells. This involved down-regulation of specificity protein (Sp) transcription factors Sp1, Sp3, and Sp4 and pro-oncogenic Sp-regulated genes such as bcl2, fatty acid synthase (FAS), survivin, vascular endothelial growth factor (VEGF), and VEGF receptor 1 (VEGFR1) (31). The anticancer activities of metformin are also similar to that observed after knockdown of Sp1 or all three Sp proteins by RNA interference in cancer cells, and this includes growth inhibition, induction of apoptosis, reversal of epithelial to mesenchymal transition, and decreased migration/invasion (32–36). Metformin also inhibits NFκB and decreases cyclin D1 and ErbB2 in cancer cell lines (13, 20, 27, 28), and these gene products are also decreased after Sp1, Sp3, and Sp4 silencing by RNAi or by other drugs that down-regulate Sp proteins (32–36). However, the relationship between metformin-induced down-regulation of Sp1, Sp3, and Sp4 and modulation of mTOR signaling has not been reported, except that total mTOR protein expression was unaffected by silencing of Sp transcription factors in pancreatic cancer cells (31).
Results of this study demonstrate novel findings indicating that metformin-induced antineoplastic activities are primarily due to down-regulation of Sp1, Sp3, and Sp4 transcription factors in pancreatic cancer cells. Firstly, treatment with metformin or silencing Sp transcription factors by RNAi down-regulated insulin-like growth factor-1 receptor (IGF-1R), which in turn inhibited activation of mTOR. Secondly, treatment with metformin or silencing Sp transcription factors by RNAi decreased epidermal growth factor receptor (EGFR) expression, resulting in inhibition of Ras activity, which is regulated by EGFR in pancreatic cancer cells (37, 38). Thus, we know show for the first time that metformin-dependent inhibition of both mTOR signaling and Ras activity is due to down-regulation of Sp transcription factors in pancreatic cancer.
EXPERIMENTAL PROCEDURES
Cell Lines, Antibodies, and Reagents
Human Panc1 pancreatic cancer cells were purchased from American Type Culture Collection (Manassas, VA). Panc28 and L3.6pL pancreatic cancer cells that express Ras mutation were provided by The University of Texas M.D. Anderson Cancer Center. All three cell lines were maintained in DMEM/F-12 (Sigma) supplemented with 0.22% sodium bicarbonate, 0.022% bovine serum albumin, 5% fetal bovine serum, and 10 ml/liter 100× antibiotic, antimycotic solution (Sigma) at 37 °C in the presence of 5% CO2. Sp1 antibody was purchased from Millipore (Temecula, CA); Sp3 and Sp4 antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). FAS, Ras, p-mTOR, mTOR, p-4EBP, 4EBP, S6 ribosomal protein, and phospho-S6 ribosomal protein were purchased from Cell Signaling Technology (Danvers, MA). Metformin was purchased from Calbiochem (EMD Millipore, Billerica, MA). Chemiluminescence reagents (Immobilon Western) for Western blot imaging were purchased from Millipore. Active Ras detection assay kit was purchased from Cell Signaling Technology. Nuclear and cytoplasmic extraction kit was purchased from Thermo Scientific.
Cell Proliferation Assay
Panc28 and L3.6pL pancreatic cancer cells (3 × 104/well) were seeded in 12-well plates with 2.5% charcoal-stripped FBS, allowed to attach for 24 h, and treated with different concentrations of NVP-BEZ235, a dual PI3K/mTOR inhibitor. Cells were then trypsinized and counted after 24 and 48 h using a Coulter Z1 cell counter. Each experiment was determined in triplicate, and results are expressed as mean ± S.E. for each set of experiments.
Nuclear and Cytoplasmic Extraction and Western Blot
Panc28 and L3.6pL cells (3 × 105/well) were seeded in DMEM/Ham's F-12 medium in 6-well plates. After 24 h, cells were treated with different concentrations of metformin. Nuclear and cytoplasmic contents were extracted using nuclear and cytoplasmic extraction kit (Thermo Scientific) according to the manufacturer's protocol. Cells were lysed using high salt lysis buffer containing 50 mmol/liter HEPES, 0.5 mol/liter sodium chloride, 1.5 mmol/liter magnesium chloride, 1 mmol/liter EGTA, 10% (v/v) glycerol, 1% Triton X-100, and protease inhibitor mixture, 1:1000 (Sigma). Lysates were collected and vortexed every 15 min for 1 h, centrifuged at 20,000 × g for 10 min at 4 °C, and quantified with Bradford reagent. Western blot analysis was carried out by separating the proteins by SDS-PAGE at 120 V for 4 h. Proteins were then transferred onto PVDF membranes (Bio-Rad) by wet electroblotting, and membranes were blocked with 5% milk in TBST buffer containing 1.576 g/liter Tris, 8.776 g/liter sodium chloride, and 0.5 ml/liter Tween 20. The PVDF membranes were then probed with primary antibodies followed by incubation with horseradish peroxidase-conjugated secondary antibodies. Immobilon Western chemiluminescence substrates (Millipore) were used to develop the membrane, and images were captured on a Kodak 4000 MM Pro image station.
siRNA Interference Assay
Panc28, Panc1, and L3.6pL pancreatic cancer cells were seeded (1 × 105/well) in 6-well plates in DMEM/Ham's F-12 medium supplemented with 2.5% charcoal-stripped FBS without antibiotic and left to attach for 24 h. Knockdown of Sp1, Sp3, and Sp4 along with iLamin as control was carried out using Lipofectamine 2000 reagent according to the manufacturer's instructions and as described previously (35). Small inhibitory RNAs were prepared by (Sigma-Aldrich).
Active Ras Detection Assay
Pancreatic cancer cells (Panc28 and L3.6pL, 3 × 105/well) were seeded in Dulbecco's modified Eagle's medium/Ham's F-12 medium in 6-well plates. After 24 h, cells were treated with or without metformin (15 mm) for 36 h. Cells were harvested under nondenaturing conditions and rinsed with ice-cold PBS. Cells were lysed using lysis buffer. Affinity precipitation of active Ras was performed using active Ras detection assay kit according to the manufacturer's protocol. Cell lysates (500 μg) were treated with GTPγS or GDP to activate or inactivate Ras, which acts as a positive or a negative control, respectively. The lysates were then incubated with GST-Raf1-RBD fusion protein in glutathione resin (where RBD is Ras-binding domain). The eluted samples were electrophoresed and immunoblotted using Ras mouse monoclonal antibody.
Orthotopic Nude Mice Study and Immunohistochemical Staining
Male athymic nude mice (NCI-nu) (at least four per treatment group) were injected with suspensions of L3.6pL cells consisting of single cells with >90% viability directly into the pancreas as described previously (31, 32). Mice were treated (orally) with vehicle (control) or 250 mg/kg of metformin daily and sacrificed 4–5 weeks after injection, and body weights were recorded. Primary tumors in the pancreas were excised, measured, and weighed. For immunohistochemistry, tumor tissue specimens were fixed in 10% formaldehyde embedded in paraffin and sectioned into 3–5-mm-thick slices. In this study, we used slides generated from an orthotopic tumor model (31). Slides were deparaffinized with xylene, dehydrated with alcohol, and rinsed with distilled water. After antigen retrieval, sections were incubated with the following primary antibodies: anti-p-mTOR antibody (1:500, Cell Signaling, 2971), anti-IGF-1R antibody (1:100, Cell Signaling, 3027), overnight at 4 °C. Slides were then incubated with horseradish peroxidase-conjugated secondary antibody (30 min) and large volume HRP polymer (45 min) for sections incubated with p-mTOR and IGF-1R primary antibody, respectively. The substrate diaminobenzidine was added followed by hematoxylin counterstaining. After dehydration, the slides were soaked in xylene for 3–5 min. Immunostaining of IGF1 receptor was carried out using Lab Vision Autostainer (Thermo Scientific). Sections from normal pancreas were used as positive control. The negative control sections were incubated with antibody dilution buffer without primary antibody (31).
RESULTS
Metformin and Sp Down-regulation Inhibit mTOR Signaling
Fig. 1A shows that NVP-BE235, an mTOR and PI3 kinase inhibitor, also decreases proliferation of Panc28 and L3.6pL cells. Similar results were observed after treatment with 15 mm metformin, and L3.6pL cells were more sensitive than Panc28 cells to the growth inhibitory effects of both compounds. Treatment of Panc28 and L3.6pL cells with 50 nm NVP-BE235 decreased activation (phosphorylation) of both mTOR and AKT, and 5–20 mm metformin also inhibited phosphorylation of mTOR and AKT (Fig. 1B), demonstrating that like NVP-BE235, metformin blocks mTOR signaling in pancreatic cancer cells. Similar results were observed in Panc1 cells; however, in this cell line, metformin also decreased total mTOR and AKT protein levels. Immunostaining of pancreatic tumors and normal pancreatic tissue from an orthotopic mouse model using L3.6pL cells (31) also showed that the enhanced staining of phospho-mTOR in tumors was decreased in pancreatic tumors from mice treated with metformin (Fig. 1C). Moreover, Western blot analysis showed that phospho-AKT and phospho-mTOR decreased in tumors from mice treated with metformin as compared with vehicle-treated mice (Fig. 1D). The lack of a significant decrease in phospho-mTOR was due to a single animal outlier.
FIGURE 1.

Effects of metformin and PI3 kinase inhibitor on mTOR signaling. A, inhibition of Panc28 and L3.6pL cell proliferation. Cells were treated with 50 and 100 nm NVP-BE235 (NVP, PI3 kinase inhibitor) or 15 mm metformin, and the effects on cell growth were determined after 24 and 48 h as described under “Experimental Procedures.” B, NVP-BE235- and metformin-mediated down-regulation of phosphorylated mTOR and AKT. Panc28 and L3.6pL cells were treated with 50 nm NVP-BE235 or metformin (5, 10 and 20 mm) for 36 h, and cell lysates were analyzed by Western blot analysis. C, metformin decreased expression of p-mTOR and p-AKT expression in pancreatic tumors. Panels a–f, immunostaining of phospho-mTOR in normal pancreas (panels a and b), orthotopic pancreatic tumor tissue (panels c and d), and tumor tissue treated with metformin (panels e and f). D, lysates from tumors of four mice were analyzed by Western blot analysis and quantitated (relative to β-actin; control values set at 100%) as outlined under “Experimental Procedures.” A significant (p < 0.05) decrease in protein expression in tumors from metformin-treated mice as compared with controls is indicated (*). Results (A and D) were quantitated and are given as mean ± S.E.
Knockdown of Sp transcription factors by RNAi did not decrease expression of mTOR protein in pancreatic cancer cells (31); however, there is evidence from RNAi studies that Sp transcription factors regulate activation of kinases such as AKT (39) and, therefore, we further investigated the effects of Sp silencing on inhibition of mTOR and downstream kinases. Fig. 2A shows that in Panc28 cells transfected with oligonucleotides targeting Sp1 (iSp1), Sp3 (iSp3), and Sp4 (iSp4), there was specific knockdown of the individual Sp proteins as described previously (35), and this was accompanied by decreased expression of phospho-mTOR and AKT but not total mTOR and AKT proteins. A similar approach was used for L3.6pL cells (Fig. 2B), and silencing of Sp1, Sp3, and Sp4 also decreased phosphorylated mTOR and AKT, indicating that all three Sp transcription factors regulated activation of both kinases. There was some variability in these responses and this was due, in part, to the efficiency of Sp1, Sp3, and Sp4 knockdown. A comparison of the effects of metformin with Sp silencing showed that both treatments decreased activation (phosphorylation) of mTOR-regulated S6RP and 4EBP gene products in Panc28 (Fig. 2C), L3.6pL (Fig. 2D), and Panc1 (Fig. 2E) cells. Metformin decreased activation of mTOR and mTOR-regulated genes in all three cell lines; however, there were some concentration-dependent differences in these effects. These results suggest that metformin-induced down-regulation of Sp1, Sp3, and Sp4 plays a role in inhibiting activation of the mTOR pathway.
FIGURE 2.

Effects of metformin and Sp knockdown on mTOR signaling. A–E, knockdown of Sp proteins (iSp1–iSpi4) or treatment with metformin (Met) decreased activation/phosphorylation of mTOR and AKT. Panc28 and L3.6pL cells were transfected with siRNA against Sp1, Sp3, and Sp4 or treatment with metformin (5, 10, 20 mm), and whole cell lysates were analyzed by Western blot analysis. iLamin (iL) was used as control oligonucleotide. E, results of treatment of Panc1 cells with metformin are also shown. Results (A and B) were quantitated and are given as mean ± S.E. for three replicate determinations for each treatment group, and a significant (p < 0.05) decrease in expression of phospho-mTOR and phospho-AKT is indicated (*).
Metformin and Sp Down-regulation Inhibit Lipogenic Genes
mTOR activation is also important for lipogenesis and enhances cleavage of sterol regulatory element-binding protein 1 (SREBP-1) to give the cleaved (active) transcription factor SREBP-1c, which in turn regulates expression of FAS (40–42). Treatment of Panc28 cells with metformin decreases SREBP-1c expression in both nuclear and cytosolic fractions (Fig. 3A). Similar results were observed in L3.6pL cells (Fig. 3B); however, the concentration-dependent decrease in p-SREBP-1c was also cell context-dependent. It has been previously reported that both SREBP-1c and FAS are Sp1-regulated genes in breast and colon cancer cells (43), and silencing of Sp1, Sp3, and Sp4 or all three proteins combined (iSp1/3/4) in Panc28 and L3.6pL decreased expression of phosphorylated SREBP-1c, whereas total SREBP-1c expression was not significantly decreased by the treatments (Fig. 3C). The results suggest that in pancreatic cancer cells, SREBP-1c is not directly regulated by Sp transcription factors, and the observed decrease in phospho-SREBP-1c is due to inactivation of mTOR by Sp silencing (Fig. 2, A and B). FAS protein expression is also decreased by metformin in pancreatic and other cancer cell lines (43), and we observed that silencing of Sp transcription factors in Panc28 and L3.6pL (Fig. 3D) cells also decreased FAS expression. Thus, the decreased expression of FAS by metformin is due to both direct effects from the loss of Sp proteins and also decreased activation of SREBP due to inhibition of mTOR. Knockdown of Sp1, Sp3, or Sp4 alone or in combination also induced poly(ADP-ribose) polymerase-2 (PARP) cleavage, a marker of apoptosis.
FIGURE 3.
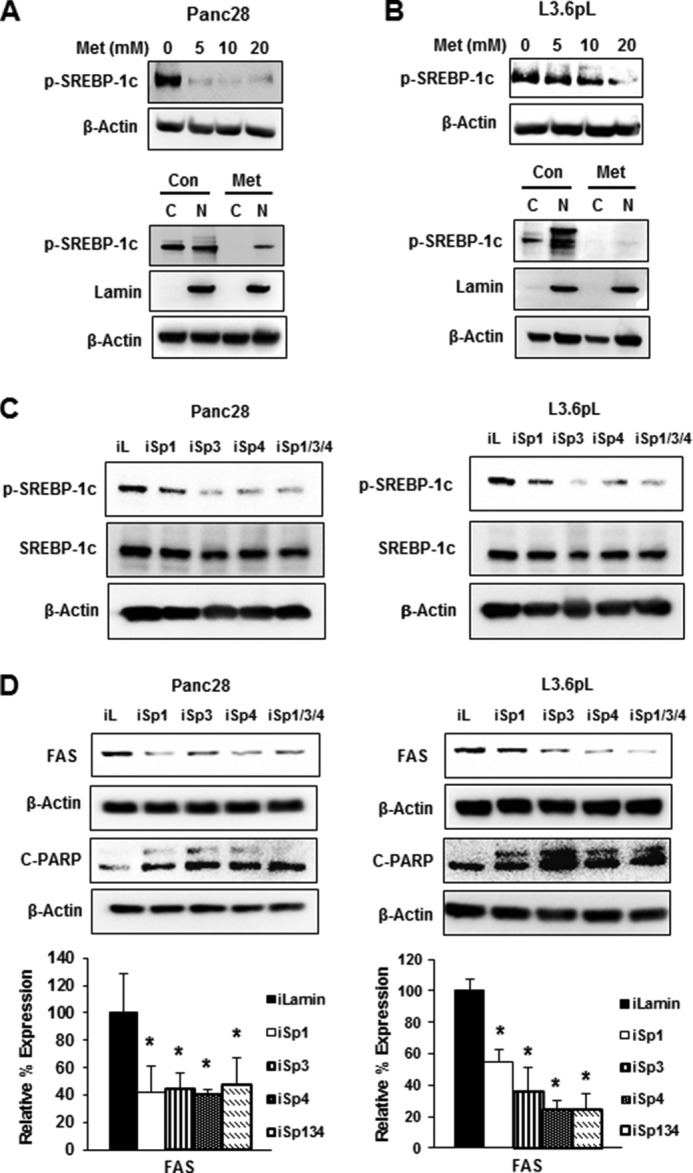
Effects of metformin and Sp knockdown on lipogenic gene products. Knockdown of Sp proteins or metformin (Met) down-regulated expression of phospho-SREBP-1c and FAS. A and B, Panc28 and L3.6pL cells were treated with 5, 10, and 20 mm metformin, and expression of phospho-SREBP-1c was analyzed in whole cell lysates and cytosolic (C) and nuclear (N) extracts. Con, control. C and D, cells were transfected with iLamin (iL), iSp1, iSp3,or iSp4, or a mixture of iSp1, iSp3, and iSp4, and expression of p-SREBP-1c and FAS was analyzed by Western blot analysis. Results (D) were quantitated and shown as mean ± S.E. for three replicate determinations. A significant (p < 0.05) decrease in FAS protein expression is indicated (*).
Metformin Inhibits Activation of mTOR through Down-regulation of IGF-1R
IGF-1R and other growth factor receptors are upstream activators of mTOR in pancreatic cancer cells (19, 22, 44), and IGF-1R is essential for proliferation of pancreatic cancer cells (44). It has been reported that IGF-1R expression is regulated by Sp1 in some cancer cell lines (37, 39, 45). Fig. 4A shows that metformin decreases IGF-1R expression in Panc28 and L3.6pL cells, and the role of IGF-1R in regulating the mTOR pathway was investigated by silencing of IGF-1R by RNAi in Panc28 and L3.6pL cells. Down-regulation of IGF-1R in these cells resulted in decreased phosphorylation of mTOR and AKT (Fig. 4B), and this was accompanied by decreased phosphorylation of S6RP and SREBP-1c and decreased expression of FAS protein (Fig. 4C). These results confirm the important role of IGF-1R in regulating mTOR and is consistent with inhibition of mTOR by metformin through down-regulation of IGF-1R. Transfection with iSp1, iSp3, iSp4, or iSp1/3/4 oligonucleotides also decreased IGF-1R expression in pancreatic cancer cells, confirming that IGF-1R is an Sp-regulated gene that is down-regulated by metformin (Fig. 4D). The overall contribution of IGF-1R to cell proliferation was confirmed by knockdown of IGF-1R by RNAi, which significantly decreased Panc28 and L3.6pL cell growth (Fig. 4E). Thus, inhibition of mTOR activation by metformin in pancreatic cancer cells is due to metformin-induced down-regulation of Sp1, Sp3, and Sp4 and the Sp-regulated IGF-1R gene.
FIGURE 4.
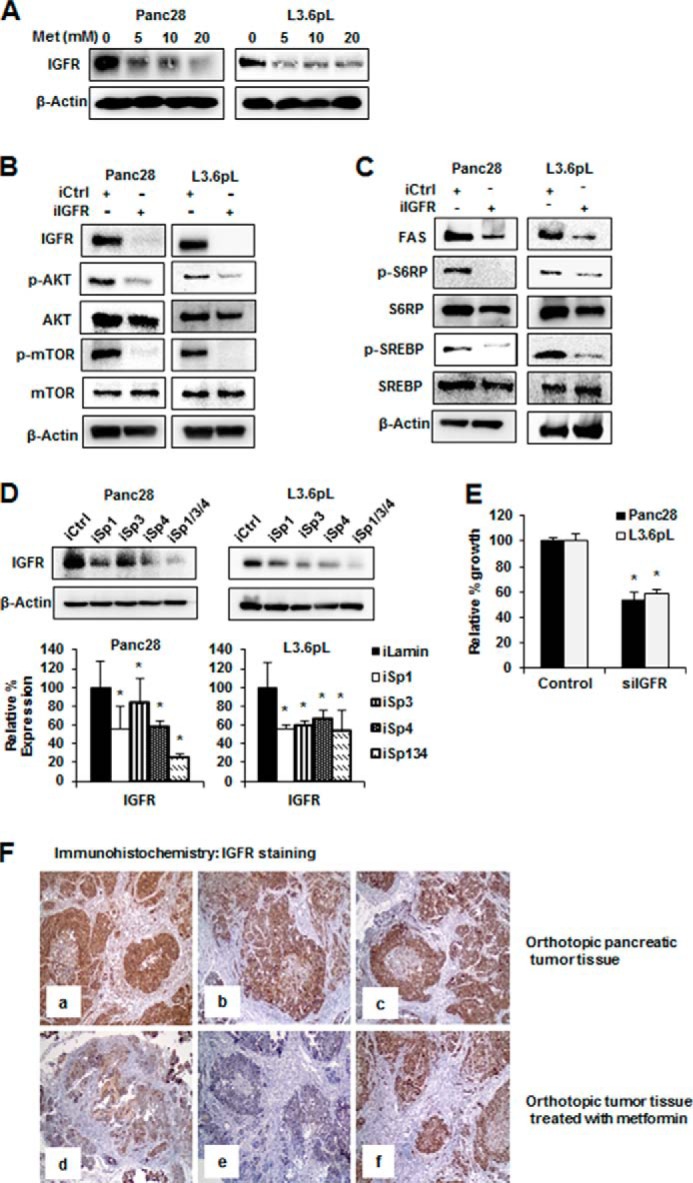
Effects of metformin and IGF-1R knockdown on mTOR signaling. A, metformin (Met) down-regulated IGF-1R expression. Panc28 and L3.6pL cells were treated with 5, 10, and 20 mm metformin, and expression of IGF-1R was analyzed by Western blot analysis. B and C, RNA interference with iIGF-1R (iIGFR) decreased mTOR signaling and cell proliferation. Panc28 and L3.6pL cells were transfected with siRNA against lamin or IGF-1R, and cell lysates were analyzed by Western blot analysis. iCtrl, control knockdown. D, knockdown of Sp proteins decreased expression of IGF-1R (IGFR). Cells were transfected with iLamin, iSp1, iSp3, or iSp4, or a mixture of iSp1, iSp3, and iSp4, and expression of IGF-1R was analyzed by Western blot analysis. E, the effects on cell proliferation were determined after 48 h as described under “Experimental Procedures.” F, metformin decreased IGF-1R expression in pancreatic tumor. Orthotopic pancreatic tumor tissue (panels a–c) and orthotopic tumor tissue treated with metformin (panels d–f) were probed for IGF-1R by immunohistochemistry analysis as described under “Experimental Procedures.” Results are expressed as the mean ± S.E. for at least three separate determinations, and a significant (p < 0.05) decrease in IGF-1R protein expression (D) and growth inhibition (E) is indicated (*).
Immunostaining and Western blot analysis of pancreatic tissue from orthotopic mouse model using L3.6pL cells (31) showed intense IGF-1R expression in tumor tissue sections from control animals (Fig. 4F, panels a–c). On the other hand, IGF-1R expression was reduced in the metformin-treated groups (Fig. 4F, panels d and e). However, the immunoreactivity was only minimally changed in tissues from one of the metformin-treated animals (Fig. 4F, panel f), and similar results were observed in Western blots of tumor lysates (data not shown). In normal pancreas, IGF-1R expression was moderate in ductal cells, and acinar cells were devoid of IGF-1R staining.
Metformin and Sp Down-regulation Target EGFR-Ras Signaling
The EGFR is essential for K-Ras signaling and subsequent Ras-dependent pancreatic cancer cell growth (37, 38). Like IGF-1R, knockdown of EGFR by RNAi also decreased proliferation of L3.6pL and Panc28 cells (Fig. 5A), and this is consistent with the role of the EGFR-Ras pathway in pancreatic cancer cell proliferation (37, 38). It has previously been reported that metformin decreases EGFR expression in pancreatic cancer cells (46), and similar results were observed in Panc28 and L3.6pL cells treated with metformin (Fig. 5B). The importance of metformin-mediated down-regulation of Sp transcription factors in decreasing EGFR was confirmed by RNAi where knockdown of Sp1 or all three Sp proteins (iSp1/3/4) significantly decreased EGFR protein levels in Panc28 and L3.6pL cells (Fig. 5C). Down-regulation of EGFR by Sp knockdown was not observed in cells transfected with iSp4, and silencing Sp3 decreased EGFR only in L3.6pL cells, showing that Sp1 is the major regulator of EGFR expression; this has been reported in other cancer cell lines (39, 47).
FIGURE 5.

Effects of metformin and Sp knockdown on EGFR expression and Ras activation. A, RNA interference with siRNA against EGFR (siEGFR) decreased cell proliferation. Cells were transfected with siRNA against lamin or EGFR, and cells were counted after 48 h as described under “Experimental Procedures.” B, metformin (Met) decreased EGFR expression. Panc28 and L3.6pL cells were treated with 5, 10, and 20 mm metformin, and expression of EGFR was analyzed by Western blot analysis. C, knockdown of Sp proteins decreased expression of EGFR. Cells were transfected with iLamin, iSp1, iSp3, or iSp4, or a mixture of iSp1, iSp3, and iSp4, and expression of EGFR was analyzed by Western blot analysis. iCtrl, control knockdown. D and E, metformin or knockdown of Sp proteins and EGFR decreased levels of active Ras (Ras-GTP). Panc28 and L3.6pL cells were treated with or without metformin or transfected with small inhibitory RNA for lamin or Sp (a mixture of iSp1, iSp3, and iSp4) (D) or knockdown of EGFR (iEGFR) (E). Levels of Ras-GTP were determined using active Ras detection assay. GTPγS and GDP act as positive and negative controls, respectively. Activated Ras was quantified and normalized to total Ras levels. Results are expressed as the mean ± S.E. for at least three separate determinations, and a significant (p < 0.05) growth inhibition (A), decrease in EGFR expression (C), and decreased Ras activity (D) are indicated (*).
The effects of metformin and Sp silencing on Ras activity were determined using an active Ras detection assay. Treatment of Panc28 and L3.6pL cells with metformin or transfection of these cells with iSp1/3/4 (combined) decreased active Ras-GTP levels (Fig. 5D), and similar results were observed after silencing EGFR (iEGFR) by RNAi in Panc28 and L3.6pL cells (Fig. 5E). Thus, metformin-induced down-regulation of Sp1, Sp3, and Sp4 and Sp-regulated IGF-1R and EGFR genes resulted in inhibition of both the mTOR and the Ras pathways in pancreatic cancer cells, and metformin also decreased IGF-1R and EGFR expression in pancreatic tumors from an orthotopic mouse model.
Metformin-induced down-regulation of Sp transcription factors in pancreatic cancer cells was dependent on induction of mitogen-activated protein kinase phosphatase 1 (MKP1) and MKP5, and this response was blocked by the phosphatase inhibitor sodium orthovanadate (SOV) (31). Results in Fig. 6A show that SOV also blocks metformin-mediated inhibition of mTOR and AKT phosphorylation and also blocks phosphorylation of 4EBP and S6RP (Fig. 6B) in Panc28 and L3.6pL cells. These results confirm that an important underlying mechanism of action of metformin in pancreatic cancer cells was due to down-regulation of Sp transcription factors and the Sp-regulated IGF-1R and EGFR genes, which results in the inhibition of mTOR and Ras pathways as illustrated in Fig. 6C.
FIGURE 6.

Role of phosphatases in metformin mediated down-regulation of mTOR signaling. A and B, phosphatase inhibitor reversed metformin (Met)-mediated down-regulation of mTOR signaling. Panc28 and L3.6pL cells were pretreated with phosphatase inhibitor SOV (20 μm) for 45 min followed by treatment with 15 mm metformin for 36 h, and whole cell lysates were analyzed by Western blots. C, proposed mechanism of action of metformin in pancreatic cancer. IGFR, IGF-1R. P, phosphorylation.
DISCUSSION
Pancreatic cancer is a highly aggressive disease that is not readily detected in its early stages, and the 1- and 5-year overall survival rates are 26 and 6%, respectively (48). Improvements in pancreatic cancer patient survival will depend on development of reliable biomarkers for early stage disease and on improved therapies for treating patients with early and late stage disease. Pancreatic tumors are complex and heterogeneous and typically express activated pro-oncogenic factors including Ras and receptor tyrosine kinases and mutations of tumor suppressor genes. Recent studies report that diabetic cancer patients who take metformin exhibit improved outcomes as compared with patients taking other antidiabetic drugs (10), and this has spurred interest in possible clinical applications of metformin for cancer therapy. One of the hallmarks of metformin action is associated with inhibition of the mTOR signaling in both cancer and non-cancer tissues and cells (15–20, 22–25). For example, metformin inhibited constitutive and induced activation of mTOR in several pancreatic cancer cell lines, and the inhibitory effects were higher in cells grown in normal 5 mm glucose as compared with cells cultured in 25 mm glucose (19, 23, 24). It has also been reported that metformin suppresses the IGF-1R and mTOR signaling in pancreatic cancer cells, and this contributes to the antineoplastic activity of this agent (19).
Treatment of pancreatic cancer cells with metformin down-regulates expression of Sp1, Sp3, and Sp4 and pro-oncogenic Sp-regulated genes including cyclin D1, bcl2, survivin, and VEGF and its receptor 1 (31). The importance of targeting Sp transcription factors in pancreatic cancer was confirmed by knockdown of Sp1, which resulted in inhibition of growth and invasion and induction of apoptosis (49). Moreover, high Sp1 expression in pancreatic tumors is a prognostic factor for decreased pancreatic cancer patient survival (50). It has also been reported that knockdown of Sp1, Sp3, and Sp4 also decreased expression of receptor tyrosine kinases and phosphorylation of other kinases such as AKT (39), and in this study, we initially investigated the role of metformin-induced down-regulation of Sp transcription factors on the mTOR pathway. In this study and others, a range of relatively high concentrations (5–20 mm) of metformin is often used; however, many of the same responses are observed at lower concentrations when treatment times are extended (data not shown).
Metformin inhibited phosphorylation of mTOR and AKT in Panc28 and L3.6pL cells (Fig. 1B), and this was accompanied by decreased activation of downstream kinases (S6RP and 4EBP) (Fig. 2, C and D) and decreased formation of the cleaved (and activated) form of SREBP (Fig. 3, A and B). These results confirm that metformin inhibits mTOR signaling as observed previously in other studies (40–42). However, knockdown of Sp1, Sp3, and Sp4 also decreased activation of mTOR and mTOR-regulated kinases/genes, suggesting that inhibition of mTOR by metformin is due, in part, to Sp down-regulation. Metformin-induced down-regulation of Sp1, Sp3, and Sp4 was phosphatase-dependent in Panc1 cells (31), and similar results were observed in colon cancer cell lines that were treated with a synthetic cannabinoid (WIN 55,212-2) that also decreases expression of Sp transcription factors (36). Moreover the effects of both metformin and WIN 55,212-2 on expression of Sp1, Sp3, and Sp4 were inhibited in cells cotreated with the phosphatase inhibitor SOV, and similar results were observed in Panc28 and L3.6pL cells treated with SOV (data not shown). SOV also reversed the inhibitory effects of metformin on mTOR signaling (Fig. 6, A and B), further confirming a role for metformin-dependent down-regulation of Sp transcription factors as an important pathway for mTOR inhibition.
Rescue experiments of metformin-induced Sp down-regulation and Sp-dependent genes/responses by overexpression of Sp1 and other Sp transcription factors are problematic because Sp1 induces apoptosis (51), although it regulates survival genes (survivin) and responses. Therefore, we further investigated selected Sp-regulated genes (and their knockdowns) that significantly contribute to pancreatic cancer growth and survival. Receptor tyrosine kinases play a particularly important role in the pancreatic cancer/tumor phenotype because IGF-1R is an upstream activator of mTOR (19, 22–24), and the EGFR is required for Ras activation (37, 38). Both the IGF-1R and the EGFR contain GC-rich promoters and are regulated by Sp1 in some cancer cell lines (39, 45, 47). Knockdown of Sp transcription factors (alone or combined) by RNAi in Panc28 and L3.6pL cells clearly demonstrates that both receptors are Sp-regulated genes in pancreatic cancer cells (Figs. 4D and 5B). Thus, metformin-induced down-regulation of the Sp-regulated genes IGF-1R and EGFR in L3.6pL and Panc28 cells (Figs. 4A and 5A) is critical for inhibition of mTOR and Ras activity, respectively, and the role of these receptors in regulating these pathways was also confirmed by RNAi (Figs. 4 and 5).
The antineoplastic activities of metformin in cancer cell lines includes the inhibition of several pathways and genes that are important for cancer cell proliferation, survival, migration, and invasion (13–28). Metformin down-regulates expression of Sp1, Sp3, and Sp4 and several pro-oncogenic Sp-regulated genes, and this study shows that inhibition of mTOR signaling and RAS activation by metformin in pancreatic cancer cells is also due to decreased expression of the Sp-regulated upstream receptor tyrosine kinases (RTKs) IGF-1R and EGFR, respectively. Thus, Sp transcription factors are not only important as prognostic factors for pancreatic cancer patients but also regulate multiple pro-oncogenic pathways/genes in pancreatic cancer cells. Moreover, knockdown of Sp1 in pancreatic cancer cells decreases growth and invasion and induces apoptosis, confirming the pro-oncogenic functions of this factor (49). These results suggest that drugs such as metformin and other agents (31–36, 39) that target Sp1, Sp3, and Sp4 represent a class of new mechanism-based drugs that can be used in combination therapies for treating this deadly disease.
This work was supported, in whole or in part, by National Institutes of Health Grants R01CA136571 and P30ES023512 (to S. S.). This work was also supported by Texas AgriLife Research.
- mTOR
- mammalian target of rapamycin
- Sp
- specificity protein
- EGFR
- epidermal growth factor receptor
- IGF-1R
- insulin-like growth factor-1 receptor
- FAS
- fatty acid synthase
- SREBP
- sterol regulatory element-binding protein
- SOV
- sodium orthovanadate
- GTPγS
- guanosine 5′-3-O-(thio)triphosphate
- p
- phosphorylated.
REFERENCES
- 1. Pollak M. (2010) Metformin and other biguanides in oncology: advancing the research agenda. Cancer Prev. Res. (Phila.) 3, 1060–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ben Sahra I., Le Marchand-Brustel Y., Tanti J. F., Bost F. (2010) Metformin in cancer therapy: a new perspective for an old antidiabetic drug? Mol. Cancer Ther. 9, 1092–1099 [DOI] [PubMed] [Google Scholar]
- 3. Engelman J. A., Cantley L. C. (2010) Chemoprevention meets glucose control. Cancer Prev. Res. (Phila.) 3, 1049–1052 [DOI] [PubMed] [Google Scholar]
- 4. Dowling R. J., Niraula S., Stambolic V., Goodwin P. J. (2012) Metformin in cancer: translational challenges. J. Mol. Endocrinol. 48, R31–R43 [DOI] [PubMed] [Google Scholar]
- 5. Evans J. M., Donnelly L. A., Emslie-Smith A. M., Alessi D. R., Morris A. D. (2005) Metformin and reduced risk of cancer in diabetic patients. BMJ 330, 1304–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Garrett C. R., Hassabo H. M., Bhadkamkar N. A., Wen S., Baladandayuthapani V., Kee B. K., Eng C., Hassan M. M. (2012) Survival advantage observed with the use of metformin in patients with type II diabetes and colorectal cancer. Br. J. Cancer 106, 1374–1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jiralerspong S., Palla S. L., Giordano S. H., Meric-Bernstam F., Liedtke C., Barnett C. M., Hsu L., Hung M. C., Hortobagyi G. N., Gonzalez-Angulo A. M. (2009) Metformin and pathologic complete responses to neoadjuvant chemotherapy in diabetic patients with breast cancer. J. Clin. Oncol. 27, 3297–3302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Libby G., Donnelly L. A., Donnan P. T., Alessi D. R., Morris A. D., Evans J. M. (2009) New users of metformin are at low risk of incident cancer: a cohort study among people with type 2 diabetes. Diabetes Care 32, 1620–1625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Murtola T. J., Tammela T. L., Lahtela J., Auvinen A. (2008) Antidiabetic medication and prostate cancer risk: a population-based case-control study. Am. J. Epidemiol. 168, 925–931 [DOI] [PubMed] [Google Scholar]
- 10. Sadeghi N., Abbruzzese J. L., Yeung S. C., Hassan M., Li D. (2012) Metformin use is associated with better survival of diabetic patients with pancreatic cancer. Clin. Cancer Res. 18, 2905–2912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Spillane S., Bennett K., Sharp L., Barron T. I. (2013) A cohort study of metformin exposure and survival in patients with stage I–III colorectal cancer. Cancer Epidemiol. Biomarkers Prev. 22, 1364–1373 [DOI] [PubMed] [Google Scholar]
- 12. Wright J. L., Stanford J. L. (2009) Metformin use and prostate cancer in Caucasian men: results from a population-based case-control study. Cancer Causes Control 20, 1617–1622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ben Sahra I., Laurent K., Loubat A., Giorgetti-Peraldi S., Colosetti P., Auberger P., Tanti J. F., Le Marchand-Brustel Y., Bost F. (2008) The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene 27, 3576–3586 [DOI] [PubMed] [Google Scholar]
- 14. Ben Sahra I., Regazzetti C., Robert G., Laurent K., Le Marchand-Brustel Y., Auberger P., Tanti J. F., Giorgetti-Peraldi S., Bost F. (2011) Metformin, independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer Res. 71, 4366–4372 [DOI] [PubMed] [Google Scholar]
- 15. Buzzai M., Jones R. G., Amaravadi R. K., Lum J. J., DeBerardinis R. J., Zhao F., Viollet B., Thompson C. B. (2007) Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 67, 6745–6752 [DOI] [PubMed] [Google Scholar]
- 16. Cerezo M., Tichet M., Abbe P., Ohanna M., Lehraiki A., Rouaud F., Allegra M., Giacchero D., Bahadoran P., Bertolotto C., Tartare-Deckert S., Ballotti R., Rocchi S. (2013) Metformin blocks melanoma invasion and metastasis development in AMPK/p53-dependent manner. Mol. Cancer Ther. 12, 1605–1615 [DOI] [PubMed] [Google Scholar]
- 17. Gotlieb W. H., Saumet J., Beauchamp M. C., Gu J., Lau S., Pollak M. N., Bruchim I. (2008) In vitro metformin anti-neoplastic activity in epithelial ovarian cancer. Gynecol. Oncol. 110, 246–250 [DOI] [PubMed] [Google Scholar]
- 18. Gou S., Cui P., Li X., Shi P., Liu T., Wang C. (2013) Low concentrations of metformin selectively inhibit CD133+ cell proliferation in pancreatic cancer and have anticancer action. PLoS One 8, e63969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Karnevi E., Said K., Andersson R., Rosendahl A. H. (2013) Metformin-mediated growth inhibition involves suppression of the IGF-I receptor signalling pathway in human pancreatic cancer cells. BMC Cancer 13, 235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kim H. G., Hien T. T., Han E. H., Hwang Y. P., Choi J. H., Kang K. W., Kwon K. I., Kim B. H., Kim S. K., Song G. Y., Jeong T. C., Jeong H. G. (2011) Metformin inhibits P-glycoprotein expression via the NF-κB pathway and CRE transcriptional activity through AMPK activation. Br. J. Pharmacol. 162, 1096–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kisfalvi K., Eibl G., Sinnett-Smith J., Rozengurt E. (2009) Metformin disrupts crosstalk between G protein-coupled receptor and insulin receptor signaling systems and inhibits pancreatic cancer growth. Cancer Res. 69, 6539–6545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rozengurt E., Sinnett-Smith J., Kisfalvi K. (2010) Crosstalk between insulin/insulin-like growth factor-1 receptors and G protein-coupled receptor signaling systems: a novel target for the antidiabetic drug metformin in pancreatic cancer. Clin. Cancer Res. 16, 2505–2511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sinnett-Smith J., Kisfalvi K., Kui R., Rozengurt E. (2013) Metformin inhibition of mTORC1 activation, DNA synthesis and proliferation in pancreatic cancer cells: dependence on glucose concentration and role of AMPK. Biochem. Biophys. Res. Commun. 430, 352–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Soares H. P., Ni Y., Kisfalvi K., Sinnett-Smith J., Rozengurt E. (2013) Different patterns of Akt and ERK feedback activation in response to rapamycin, active-site mTOR inhibitors and metformin in pancreatic cancer cells. PLoS One 8, e57289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Storozhuk Y., Hopmans S. N., Sanli T., Barron C., Tsiani E., Cutz J. C., Pond G., Wright J., Singh G., Tsakiridis T. (2013) Metformin inhibits growth and enhances radiation response of non-small cell lung cancer (NSCLC) through ATM and AMPK. Br. J. Cancer 108, 2021–2032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tan B. K., Adya R., Chen J., Lehnert H., Sant Cassia L. J., Randeva H. S. (2011) Metformin treatment exerts antiinvasive and antimetastatic effects in human endometrial carcinoma cells. J. Clin. Endocrinol. Metab. 96, 808–816 [DOI] [PubMed] [Google Scholar]
- 27. Yasmeen A., Beauchamp M. C., Piura E., Segal E., Pollak M., Gotlieb W. H. (2011) Induction of apoptosis by metformin in epithelial ovarian cancer: involvement of the Bcl-2 family proteins. Gynecol. Oncol. 121, 492–498 [DOI] [PubMed] [Google Scholar]
- 28. Zhuang Y., Miskimins W. K. (2011) Metformin induces both caspase-dependent and poly(ADP-ribose) polymerase-dependent cell death in breast cancer cells. Mol. Cancer Res. 9, 603–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shaw R. J., Lamia K. A., Vasquez D., Koo S. H., Bardeesy N., Depinho R. A., Montminy M., Cantley L. C. (2005) The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 310, 1642–1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zou M. H., Kirkpatrick S. S., Davis B. J., Nelson J. S., Wiles W. G., 4th, Schlattner U., Neumann D., Brownlee M., Freeman M. B., Goldman M. H. (2004) Activation of the AMP-activated protein kinase by the anti-diabetic drug metformin in vivo: role of mitochondrial reactive nitrogen species. J. Biol. Chem. 279, 43940–43951 [DOI] [PubMed] [Google Scholar]
- 31. Nair V., Pathi S., Jutooru I., Sreevalsan S., Basha R., Abdelrahim M., Samudio I., Safe S. (2013) Metformin inhibits pancreatic cancer cell and tumor growth and downregulates Sp transcription factors. Carcinogenesis 34, 2870–2879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Abdelrahim M., Baker C. H., Abbruzzese J. L., Sheikh-Hamad D., Liu S., Cho S. D., Yoon K., Safe S. (2007) Regulation of vascular endothelial growth factor receptor-1 expression by specificity proteins 1, 3, and 4 in pancreatic cancer cells. Cancer Res. 67, 3286–3294 [DOI] [PubMed] [Google Scholar]
- 33. Abdelrahim M., Smith R., 3rd, Burghardt R., Safe S. (2004) Role of Sp proteins in regulation of vascular endothelial growth factor expression and proliferation of pancreatic cancer cells. Cancer Res. 64, 6740–6749 [DOI] [PubMed] [Google Scholar]
- 34. Jutooru I., Chadalapaka G., Abdelrahim M., Basha M. R., Samudio I., Konopleva M., Andreeff M., Safe S. (2010) Methyl 2-cyano-3,12-dioxooleana-1,9-dien-28-oate decreases specificity protein transcription factors and inhibits pancreatic tumor growth: role of microRNA-27a. Mol. Pharmacol. 78, 226–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jutooru I., Chadalapaka G., Lei P., Safe S. (2010) Inhibition of NFκB and pancreatic cancer cell and tumor growth by curcumin is dependent on specificity protein down-regulation. J. Biol. Chem. 285, 25332–25344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sreevalsan S., Safe S. (2013) The cannabinoid WIN 55,212-2 decreases specificity protein transcription factors and the oncogenic cap protein eIF4E in colon cancer cells. Mol. Cancer Ther. 12, 2483–2493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ardito C. M., Grüner B. M., Takeuchi K. K., Lubeseder-Martellato C., Teichmann N., Mazur P. K., Delgiorno K. E., Carpenter E. S., Halbrook C. J., Hall J. C., Pal D., Briel T., Herner A., Trajkovic-Arsic M., Sipos B., Liou G. Y., Storz P., Murray N. R., Threadgill D. W., Sibilia M., Washington M. K., Wilson C. L., Schmid R. M., Raines E. W., Crawford H. C., Siveke J. T. (2012) EGF receptor is required for KRAS-induced pancreatic tumorigenesis. Cancer Cell 22, 304–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Navas C., Hernández-Porras I., Schuhmacher A. J., Sibilia M., Guerra C., Barbacid M. (2012) EGF receptor signaling is essential for K-RAS oncogene-driven pancreatic ductal adenocarcinoma. Cancer Cell 22, 318–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chadalapaka G., Jutooru I., Burghardt R., Safe S. (2010) Drugs that target specificity proteins downregulate epidermal growth factor receptor in bladder cancer cells. Mol. Cancer Res. 8, 739–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Furuta E., Pai S. K., Zhan R., Bandyopadhyay S., Watabe M., Mo Y. Y., Hirota S., Hosobe S., Tsukada T., Miura K., Kamada S., Saito K., Iiizumi M., Liu W., Ericsson J., Watabe K. (2008) Fatty acid synthase gene is up-regulated by hypoxia via activation of Akt and sterol regulatory element binding protein-1. Cancer Res. 68, 1003–1011 [DOI] [PubMed] [Google Scholar]
- 41. Owen J. L., Zhang Y., Bae S. H., Farooqi M. S., Liang G., Hammer R. E., Goldstein J. L., Brown M. S. (2012) Insulin stimulation of SREBP-1c processing in transgenic rat hepatocytes requires p70 S6-kinase. Proc. Natl. Acad. Sci. U.S.A. 109, 16184–16189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Porstmann T., Santos C. R., Griffiths B., Cully M., Wu M., Leevers S., Griffiths J. R., Chung Y. L., Schulze A. (2008) SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 8, 224–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lu S., Archer M. C. (2010) Sp1 coordinately regulates de novo lipogenesis and proliferation in cancer cells. Int. J. Cancer 126, 416–425 [DOI] [PubMed] [Google Scholar]
- 44. Bergmann U., Funatomi H., Yokoyama M., Beger H. G., Korc M. (1995) Insulin-like growth factor I overexpression in human pancreatic cancer: evidence for autocrine and paracrine roles. Cancer Res. 55, 2007–2011 [PubMed] [Google Scholar]
- 45. Beitner-Johnson D., Werner H., Roberts C. T., Jr., LeRoith D. (1995) Regulation of insulin-like growth factor I receptor gene expression by Sp1: physical and functional interactions of Sp1 at GC boxes and at a CT element. Mol. Endocrinol. 9, 1147–1156 [DOI] [PubMed] [Google Scholar]
- 46. Wang L. W., Li Z. S., Zou D. W., Jin Z. D., Gao J., Xu G. M. (2008) Metformin induces apoptosis of pancreatic cancer cells. World J. Gastroenterol. 14, 7192–7198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Johnson A. C., Ishii S., Jinno Y., Pastan I., Merlino G. T. (1988) Epidermal growth factor receptor gene promoter: deletion analysis and identification of nuclear protein binding sites. J. Biol. Chem. 263, 5693–5699 [PubMed] [Google Scholar]
- 48. American Cancer Society (2011) Cancer Facts & Figures 2011, American Cancer Society, Atlanta, GA [Google Scholar]
- 49. Jutooru I., Guthrie A. S., Chadalapaka G., Pathi S., Kim K., Burghardt R., Jin U. H., Safe S. (2014) Mechanism of action of phenethylisothiocyanate and other reactive oxygen species-inducing anticancer agents. Mol. Cell. Biol. 34, 2382–2395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jiang N. Y., Woda B. A., Banner B. F., Whalen G. F., Dresser K. A., Lu D. (2008) Sp1, a new biomarker that identifies a subset of aggressive pancreatic ductal adenocarcinoma. Cancer Epidemiol. Biomarkers Prev. 17, 1648–1652 [DOI] [PubMed] [Google Scholar]
- 51. Deniaud E., Baguet J., Mathieu A. L., Pagès G., Marvel J., Leverrier Y. (2006) Overexpression of Sp1 transcription factor induces apoptosis. Oncogene 25, 7096–7105 [DOI] [PubMed] [Google Scholar]