

Background: Shortage of isoprenoids causes aberrant activity of prenylated small GTPases.
Results: Inactivation of RhoA due to lack of prenylation leads to Rac1 activation and subsequent priming for IL-1β secretion.
Conclusion: RhoA inactivation contributes to the pathology of isoprenoid deficiency.
Significance: Insight into the multiple networks involving RhoA will benefit new intervention strategies in prenylation-related pathologies.
Keywords: Autophagy; Interleukin; Protein Isoprenylation; Ras Homolog Gene Family, Member A (RhoA); Ras-related C3 Botulinum Toxin Substrate 1 (Rac1); Small GTPase; Autoinflammatory-disorder; Mitochondrial Elongation
Abstract
Protein prenylation is a post-translational modification whereby non-sterol isoprenoid lipid chains are added, thereby modifying the molecular partners with which proteins interact. The autoinflammatory disease mevalonate kinase deficiency (MKD) is characterized by a severe reduction in protein prenylation. A major class of proteins that are affected are small GTPases, including Rac1 and RhoA. It is not clear how protein prenylation of small GTPases relates to GTP hydrolysis activity and downstream signaling. Here, we investigated the contribution of RhoA prenylation to the biochemical pathways that underlie MKD-associated IL-1β hypersecretion using human cell cultures, Rac1 and RhoA protein variants, and pharmacological inhibitors. We found that when unprenylated, the GTP-bound levels of RhoA decrease, causing a reduction in GTPase activity and increased protein kinase B (PKB) phosphorylation. Cells expressing unprenylated RhoA produce increased levels of interleukin 1β mRNA. Of other phenotypic cellular changes seen in MKD, increased mitochondrial potential and mitochondrial elongation, only mitochondrial elongation was observed. Finally, we show that pharmacological inactivation of RhoA boosts Rac1 activity, a small GTPase whose activity was earlier implied in MKD pathogenesis. Together, our data show that RhoA plays a pivotal role in MKD pathogenesis through Rac1/PKB signaling toward interleukin 1β production and elucidate the effects of protein prenylation in monocytes.
Introduction
The functional consequence of protein prenylation to cell signaling is not completely understood. Earlier work on prenylation of small GTPases provided experimental support that prenylation can be involved in subcellular protein localization and control of GTP hydrolysis. The prenylation modification can direct small GTPases to the membrane, or cover a nuclear localization signal, as is the case in Rac1 (1, 2). Small GTPases are heavily involved in cytoskeleton regulation and vesicular trafficking, where the prenylation moiety serves to localize GTPases to the membranes and regulate their activity.
The process of protein prenylation itself concerns the C-terminal post-translational modification of a protein with a non-sterol isoprenoid. Protein targets for prenylation express a short four amino acid sequence at the C terminus, called a CAAX box, where the modification is attached at the C (or cysteine), while A stands for aliphatic, and X can be any residue. The prenylation is executed by the farnesyl- (C-15) or geranylgeranyl (C-20) transferases. The prenylation moiety can have different functions. In case of the nuclear lamins for instance, the prenylation helps the cellular localization and provides a signal for further modification and stable integration in the membrane (3). Another major function of prenylation moieties is as a specific protein interaction domain (4). Prenylation is not always permanent. Although the thioether connecting the lipid to the protein is very stable, it has been described that the C-terminal residues can be removed, including the prenyl group, yielding an unprenylated but slightly shorter version of the protein. This is, for instance, mediated by the bacterial protease YopT (from Yersenia pestis) to alter cellular shape and increase infection rate (5, 6).
The activity of small GTPases is regulated by an extensive network of guanine-nucleotide exchange factors, or GEFs,2 and GTPase-activating proteins (GAPs). When a small GTPase is bound to GDP it is generally considered inactive. GEFs can exchange the GDP for GTP, while GAPs increase the hydrolytic activity of GTPases, to catalyze the conversion of GTP back to GDP. This creates a network of molecular on-off switches (7). As an added layer of regulation, there are proteins classified as GDP dissociation inhibitors (GDI). These proteins can prevent the binding of GEFs and GAPs thereby keeping the GTPase in its current state (8). Prenylation plays an important role in this process. One of the best studied GDIs, Rho GDI, uses the prenylation moiety on GTPases as an important part of its interaction surface with its targets, not only inhibiting GEF/GAP binding but controlling localization at the same time (4).
One obvious disease where protein prenylation is affected is Mevalonate Kinase Deficiency (MKD). MKD is caused by mutations in the mevalonate kinase protein (9, 10). This protein, as a key enzyme in the pathway that synthesizes cholesterol, produces an intermediate for isoprenoid synthesis. Previous studies have shown that MKD causes depletion and shortage of both farnesylpyrophosphate and geranylgeranylpyrophosphate, both substrates used for protein prenylation (11). Patients suffer from periodic fever episodes induced by uncontrolled release of Interleukin-1β (IL-1β). Although both isoprenoid versions are depleted, the inflammation related symptoms are caused specifically by a lack of geranylgeranylpyrophosphate (12). The exogenous addition of geranylgeranylpyrophosphate to patient cells or cell cultures restores the normal regulation of IL-1β secretion. The MKD phenotype can be mimicked in vitro by exposing cells to statins, which are compounds that inhibit HMG-Coa reductase, the enzyme directly upstream of mevalonate kinase. Inhibition of the enzyme geranylgeranyltransferase leads to a similar MKD phenotype (13).
In the context of MKD, the small GTPase Rac1 was identified as a mediator for the IL-1β hypersecretion. Rac1, with reduced prenylation due to isoprenoid shortage, was more active in MKD cell culture models. Inhibition of Rac1 in THP-1 monocyte cultures leads to normalization of IL-1β levels (14). Yet there are a number of other biochemical hallmarks of MKD, including altered autophagy, mitochondrial potential, and morphology and redox balance that cannot be explained by aberrant activity of Rac1 alone (15). Henneman et al. (16) reported that RhoA, normally prenylated, activity was increased in MKD patient-derived fibroblasts, which however do not display the autoinflammation phenotype. Here we asked what is the contribution of unprenylated RhoA to IL-1β-mediated autoinflammation in an MKD model. We find that inhibiting prenylation in the monocytoid cell line THP-1, reduces, RhoA activity. Reduced RhoA activity does not affect mitochondrial membrane potential or mitophagy, but does affect mitochondrial morphology. In addition, inactive RhoA leads to activation of Rac1 and PKB phosphorylation, thereby contributing to IL-1β gene transcription and the pathogenesis of MKD.
EXPERIMENTAL PROCEDURES
Reagents
Simvastatin and Bafilomycin A1 were purchased from Sigma-Aldrich. Mitotracker Green, Mitotracker Deep Red and GGTI-298 were bought from Millipore. C3 Transferase (Rho inhibitor) was bought from Cytoskeleton. Simvastatin was hydrolyzed to its bioactive form as previously described (17).
Cell Cultures
THP-1 cells were cultured in RPMI 1640 supplemented with 1% glutamine, antibiotics (penicillin, streptomycin) and 10% FBS. HEK293T cells were cultured in DMEM supplemented with 1% glutamine, antibiotics (penicillin, streptomycin), and 10% FBS cells. Simvastatin treatment of cells was 48 h prior to the start of the experiment and at a concentration of 10 μm unless stated otherwise in the figure legends.
Plasmids
Plasmids containing Rac1 and RhoA with and without CAAX box were made by amplifying cDNA isolated from human fibroblasts. The primers introduced a restriction site (KpnI forward, XhoI reverse) to allow further cloning. Primers: RhoA forward 5′-CGATA GGTACC ATG GCT GCC ATC CGG AAG AAA-3′, RhoA reverse 5′-CGATA CTCGAG TCA CAA GAC AAG GCA CCC AGA TTT TTT CTT CC-3′, RhoA (-CAAX) reverse 5′-CGATA CTCGAG TCA CCC AGA TTT TTT CTT CC-3′, Rac1 forward 5′-CGATA GGTACC ATG CAG GCC ATC AAG TGT GTG-3′, Rac1 reverse 5′-CGATA CTCGAG TTA CAA CAG CAG GCA TTT TCT C-3′, Rac1 (-CAAX) reverse 5′-CGATA CTCGAG TTA TTT TCT CTT CCT CTT CTT CAC-3′. The amplicons were ligated into pGEM-T vector (Promega) and sequenced to ensure the correct sequences were amplified. The RhoA and Rac1 sequences were then removed from the pGEM-T vector with KpnI and XhoI and ligated into pcDNA3 vector (Invitrogen).
Activated RhoA and Rac1 Immunoprecipitation Assays
Activated RhoA and Rac1 assessment assays were performed as described in Henneman et al. (16). Cultured THP-1 cells were washed three times with ice-cold PBS, lysed by scraping in the culture flask using lysis buffer (50 mm Tris, pH 7.4, 200 mm NaCl, 10% glycerol, 1% tergitol-type Nonidet P-40 (Nonidet P-40), 2 mm magnesium chloride (MgCl2), 0.1 mm phenylmethylsulfonylfluoride, 10 μg/ml leupeptin, 10 μg/ml aprotinin, 1 mm benzamidine, 1 mm dithiothreitol (DTT), 1 mm vanadate). The lysates were then centrifuged (10 min, 12,000 × g), and the supernatants collected. After determining protein concentrations of the supernatants with a Bradford assay, 500 μg of total protein in 500 ml was incubated (60 min, 4 °C) with bacterially produced glutathione S-transferase Ras-binding domain (GST-RBD, Rhotekin) (Reid et al. (18) (for RhoA immunoprecipitations) or GST-p21-activated Ser/Thr kinase (PAK) (Sander et al., Ref. 19) (for Rac1 pulldowns) bound to glutathione-agarose beads (Sigma). The beads were washed three times with lysis buffer followed by centrifugation (10 s, 12,000 × g). Bound proteins, i.e. active RhoA or Rac1, were eluted by boiling in SDS-sample buffer and analyzed by immunoblot analysis.
Activated RhoA and Rac1 G-LISA Assays
Rac1 and RhoA G-LISA kits (Cytoskeleton) were used to quantify the amount of GTP-bound Rac1 and RhoA according to the manufacturers' instructions. Results were normalized by quantifying total Rac1 and RhoA by Western blot. Analysis of band intensity was done with Gel Doc easy system and software (Bio-Rad).
Transient Transfection
THP-1 cells were transfected using the Amaxa Human Monocyte Nucleofector Kit (Lonza). Cells were pelleted and resuspended in 100 μl of Human Monocyte Nucleofector Solution and transferred into a cuvette together with 1 μg of DNA. A control plasmid containing GFP was cotransfected in 1:10 ratio to determine transfection efficiency. Transfection was done using a Nucleofector II (Lonza) using program Y-001. Following transfection, cells are resuspended in 500 μl of RPMI (2 mm glutamine, antibiotics, and 20% human serum) plated. Cells were analyzed 24 h post-transfection. Hek293T cells were seeded in 6-well culture plates at 50% confluence. Cells were transfected with 6 μg of DNA using low toxicity (LTX) Lipofectamine Kit (Invitrogen) according to the manufacturer's instructions. To determine the transfection efficiency, the pCMV-dsRed plasmid expressing the red fluorescent protein was used. Cells were analyzed 48 h post-transfection.
Western Blot Analysis
Western blots were performed according to standard procedure. Equal amounts of proteins were separated by 12% SDS-polyacrylamide gels and transferred polyvinylidene difluoride (PVDF-FL) membranes (Millipore). Membranes were blocked in Tris-buffered saline containing 0,3% Tween and 5% nonfat dry milk (30 min, room temperature). Membranes were probed with the following antibodies: anti-RhoA (Santa Cruz Biotechnology sc-418), anti-Rac-1 (Santa Cruz Biotechnology sc-217), anti-phospho AKT (S473) (Cell Signaling Technology #4058S), and anti-HSP90 (Cell Signaling Technology #4875S).
Flow Cytometry
Phospho-PKB levels were detected by flow cytometry by fixing cells with Fixation buffer (BD Cytofix) for 15 min at 37 °C. Next cells were pelleted and resuspended in Perm Buffer III (BD Phosflow) and incubated for 30 min on ice. Cells were subsequently washed twice and stained with anti-phospho-AKT-PE (S473) (BD Phosflow #560378) for 30 min (room temperature, dark, shaking). Cells were washed twice and resuspended in flow cytometry buffer (PBS, 2% FCS) for acquisition.
For measurement of mitochondrial relative presence and mitochondrial membrane potential, cells were washed once in PBS, resuspended in staining mix containing RPMI without phenol red (Invitrogen), 50 nm Mito Tracker Deep Green, and 50 nm Mito Tracker Deep Red and incubated (30 min, 37 °C, dark). Cells were subsequently pelleted and resuspended in culture medium. Data were acquired with a FacsCanto II (BD Bioscience) and were analyzed with FACS Diva or Flowjo analytical software.
Confocal Microscopy
Hek293T cells were seeded in 24-well culture plates on 1.5-mm glass coverslips pre-coated with 2% poly-l-lysine solution (0.1 w/v in H20, Sigma-Aldrich) for 30 min at room temperature. Plates were washed twice in PBS, after which cells were added on plates. After 24 h of incubation at 37 °C, cells were treated with Bafilomycin A1 (10 nm, 4 h). The culture medium was removed, and coverslips were washed twice in PBS. Cells were fixed in 3.7% paraformaldehyde (MERC) for 10 min (room temperature) and washed twice with PBS. Residual PBS was carefully removed from coverslips and they were mounted on prolong containing DAPI. THP-1 cells were washed with PBS and stained in RPMI (w/o) phenol red with 100 nm Mitotracker green and 150 nm Mitotracker red for 30 min at 37 °C. Cells were washed and plated on WillCo wells coated with Cell-Tak (BD Biosciences) in RPMI (w/o) phenol red and 10% FBS. All images were obtained with 1.3× optical zoom using “Plan-Apochromat” 63 × 1.40 oil DIC M27 objective on a Zeiss LSM710, and processed using Zen 2009 software (Zeiss Enhanced Navigation). Images were analyzed with ImageJ software. Cell count was obtained by automated nuclei count in the DAPI channel. (Threshold for brightness was set, followed by automated analysis with the following settings: size (square pixels) 75-infinity and circularity 0.05–1.00). Autophagosomes were quantified is the same way using the GFP channel (settings: size (square pixels) 5–75 and circularity 0.20–1.00).
RNA Isolation and Quantification
RNA was isolated by dissolving cell pellets in TRIpure (RnD) and following manufacturer's protocols. Isolated RNA was converted to cDNA using iScript (Bio-Rad) according to the manufacturer's instructions. Detection was done with CF-96 (Bio-Rad) using SYBR green (Bio-Rad), 100 ng of cDNA was used per reaction. Primers used: GAPDH Forward 5′-GTCGGAGTCAACGGATT-3′, reverse 5′-AAGCTTCCCGTTCTCAG-3′, NLRP3 Forward 5′-CTTCCTTTCCAGTTTGCTGC-3′, reverse 5′-TCTCGCAGTCCACTTCCTTT-3′, CASP1 Forward 5′-ATAGCTGGGTTGTCCTGCAC-3′, reverse 5′-GCCAAATTTGCATCACATACA-3′, IL-1β Forward 5′-AGAAGAACCTATCTTCTTCGAC-3′, reverse 5′-ACTCTCCAGCTGTAGAGTGG-3′, IL-18 Forward 5′-TCCCCAGCTTGCTGAGCCCT-3′, reverse 5′-GTTGGCAGCCAGGAGGGCAA-3′.
Statistics
Error bars shown represent S.E. unless stated otherwise in the figure legends. Statistical test between two variables was done using the Mann-Whitney test. In figures, one asterisk (*) indicates a p value < 0.05, two (**) indicate a p value of < 0.01.
RESULTS
Prenylation-deficient RhoA Has Reduced GTP Binding Activity in THP-1 Cells
Prenylation status of small GTPases affects their activation to varying extent among small GTPase family members and the tissue or cell lines studied (20, 21). Activated Rac1 facilitated IL-1β-driven autoinflammation in a culture model of MKD (14). Rac1 and RhoA can be reciprocally regulated (22, 23), suggesting the possibility that deficiency in RhoA prenylation may cause Rac1 activation and thereby drive IL-1β-driven autoinflammation. We tested this hypothesis in a culture-based MKD model, where THP-1 monocytic cells were pretreated 24 to 48 h with 10 μm simvastatin (13, 15). Simvastatin targets HMG-CoA reductase, the rate-limiting enzyme in the cholesterol synthesis cascade. Inhibition of HMG-CoA reductase is a well-established system to inhibit protein prenylation and mimic MKD. Simvastatin-treated THP-1 cells were lysed, and GTP-bound Rac1 or RhoA was immunoprecipitated from the lysate (Fig. 1, A and B, respectively). The total amount of Rac1 and RhoA protein was also quantified and β-actin included as a loading control. We found that Rac1 activity is indeed increased upon simvastatin treatment; however RhoA activity was significantly reduced. Of note, an opposite effect was seen by Henneman et al., where treatment of fibroblasts of both healthy individuals, and MKD patients with low dose simvastatin caused increased GTP-bound RhoA (16). This apparent discord may relate to the different cell types used. In support of our data, a study by Hiraoka et al. on monocyte motility found that pitavastatin treatment of THP-1 cells reduced RhoA activity (24), corroborating our finding of reduced RhoA activity in THP-1 cells.
FIGURE 1.

Lack of prenylation changes Rac1 and RhoA activation and signaling. A, pull down of GTP-bound Rac1 and RhoA (B) in THP-1 cells treated with various concentrations of simvastatin. Reducing the prenylation of both proteins changes their activation status. The amount of GTP-bound Rac1 is increased, while GTP-bound RhoA decreases with increasing simvastatin concentration. Left panels show representative blots of one experiment; right panels show bar graph of quantified blots as average of at least three independent experiments. C, transfection of Rac1 and RhoA (D) or its prenylation-dead mutants in THP-1 cells increases PKB signaling. Representative blots are shown of three independent experiments. HSP90 was used as loading control. E, unprenylated Rac1 and RhoA both contribute to increased p-PKB. THP-1 cells transfected with Rac1 and RhoA or its prenylation dead mutants are stained for p-PKB and analyzed with flow cytometry. Simvastatin-treated cells were used as positive control.
What is the effect of the increased Rac1 or RhoA activity on downstream signaling? To this end, we measured the phosphorylation status of protein kinase B (PKB) in transiently transfected THP-1 cells with either a plasmid with the wild-type protein, or a prenylation deficient mutant that lacks the CAAX box. Indeed, Rac1 activation can increase pPKB without the need for additional stimuli (14). We found that transfection of both Rac1 and Rac1-caax led to increased Rac1 levels, but only the mutant increased phosphorylation of PKB (Fig. 1C). The effect of the RhoA plasmids on the total amount of RhoA was small, however WT RhoA suppresses PKB phosphorylation. An effect that can be abrogated by rendering RhoA prenylation deficient (Fig. 1D). As a second approach to address PKB phosphorylation, we analyzed the cells by flow cytometry (Fig. 1E). Simvastatin-treated cells were used as a positive control. While transfection with the wild-type Rac1 or RhoA proteins did not alter the phosphorylated PKB level, prenylation-deficient mutants did increase p-PKB levels although not to the same extent as simvastatin treatment. We therefore conclude that the increased phosphorylation of PKB is mediated by both Rac1 and RhoA-mediated signaling and that the latter effect is largely mediated via Rac1.
Lack of Prenylation on RhoA Has No Effect on Autophagy
In a previous study we have shown that impaired mitophagy, i.e. autophagy of damaged mitochondria, contributes to MKD-associated IL-1β hypersecretion (15). The precise mechanism by which defective isoprenylation interferes with mitophagy remains elusive. We therefore investigated if RhoA prenylation modulates autophagosome formation. We used a HEK293T cell line where LC3 is N-terminally fused with GFP to enable direct visualization of autophagosomes. LC3 (or ATG8) is an essential protein for autophagosome formation. It is recruited to autophagosomes upon their assembly and is incorporated into the autophagosomal membrane. It can therefore be used to identify autophagosomes. (25) We transfected the cells with RhoA or the prenylation-deficient RhoA mutant. Transfected cells were left untreated or treated with bafilomycin A1. Bafilomycin A1 impairs the acidification of lysosomes and indirectly inhibits autophagy (26), resulting in an accumulation of autophagosomes. We found that basal autophagy levels were unchanged in cells overexpressing either RhoA variant compared with controls (Fig. 2, A and B). Treatment with bafilomycin A1 resulted in equal accumulation of autophagosomes in all conditions (Fig. 2, A and B). We recently reported altered prenylation on autophagy by simvastatin treatment, where bafilomycin did not increase the amount of autophagosomes, suggesting a block in autophagosome development (15). Taken together, these data support that RhoA prenylation status does not alter or interfere with autophagy, surmising that simvastatin targets other molecules than RhoA alone. Of note, RhoA-caax mutant cells expressed a more stretched cell morphology than untreated, empty vector or wild-type RhoA-transfected HEK293T cells, confirming prior work on the regulatory role of RhoA on the cytoskeleton (27, 28, 38). Thus, while RhoA-caax modulates cell morphology, most likely as a consequence of deregulating the RhoA signaling, unprenylated RhoA does not affect autophagy.
FIGURE 2.

Lack of prenylation on RhoA does not affect autophagy. A, HEK293T LC3-GFP cells are transfected with RhoA or the prenylation dead mutant. Following treatment with either vehicle or bafilomycin 1A cells are fixed, and nuclei are counterstained with DAPI. During confocal microscopy the ability of the cells to form autophagosomes was assessed. Transfection both variants of RhoA does not hamper autophagosome formation, both with and without bafilomycin A1. The only notable difference is the altered cell morphology in the RhoA mutant. B, bar graph with the quantification of number of autophagosomes per cell. Each condition is based on at least 120 cells. No significant differences were observed among any of the conditions within the untreated or bafilomycin 1A groups.
RhoA Controls Mitochondrial Morphology, but Not Function
Another recently observed but poorly understood phenomenon is the increase of mitochondrial potential and the change of mitochondrial morphology with deficient isoprenylation. (15) Several studies have identified a role for RhoA and RhoA signaling in mitochondrial trafficking, protection, and mitochondrial-mediated apoptosis (29–31). We therefore investigated whether RhoA prenylation affects mitochondrial trans-membrane potential. To this end, we treated transfected HEK293T cells with mitotracker green and mitotracker deep red. Mitotracker green stains all mitochondria and provides a measure of the mitochondrial mass per cell. The affinity of mitotracker deep red for mitochondria increases with potential, thereby allowing comparison of mitochondrial potential by flow cytometry. Transfection of wild-type RhoA or RhoA-caax did not modulate either mitochondrial mass or potential (Fig. 3A). When normalized for mitochondrial mass, through division of the deep red signal (potential) by the green signal (mass), the resulting calculated potentials again showed no difference (Fig. 3B). RhoA may modulate mitochondrial morphology (32), considering that GTPases can control mitochondrial fusion of small vesicles into tubular structures and therefor might be involved in the observed change in mitochondrial morphology with impaired prenylation (15). THP-1 cells were therefore stained with the same mitotrackers and subsequently taken for live cell microscopy analysis, and enumeration of the percentage of cells expressing elongated mitochondria. We attained RhoA inactivation by culture of untransfected THP-1 cells in the presence of C3 transferase from Clostridium botulinum linked to a cell penetrating moiety. To avoid electroporation or transfection-related cell damage, which might affect mitochondrial morphology unrelated to RhoA or RhoA-caax. Inactivation of RhoA led to a significant increase of cells with elongated mitochondria (Fig. 3, C and D). This induction of tubular mitochondrial morphology we had previously observed as a consequence of simvastatin treatment, however simvastatin also led to increased mitochondrial potential. Thus, RhoA inactivation may account for elongation of mitochondria, but does not increase mitochondrial potential, which may require additional signals. It should be noted that the C3 transferase also inhibits RhoB and RhoC, and we therefore cannot unequivocally state that the effect are only mediated by RhoA. However is currently no literature describing signaling for either protein leading to mitochondrial morphology, potential or mitophagy. Furthermore, most reports seem to indicate that RhoB and RhoC become active (GTP bound) when unprenylated, while the C3 transferase leaves them inactive (although in different cell types) (33, 34). Considering that the same change in mitochondrial morphology is seen with simvastatin treatment and Rho inhibition, where as far as we know the only common factor is RhoA inactivation, we propose that the mitochondrial elongation is a consequence or RhoA inactivation and RhoB and RhoC do not play an important role in the process.
FIGURE 3.

Mitochondrial morphology, but not potential, changes with reduced RhoA activity. A, THP-1 cells transfected with RhoA and RhoA-caax are stained with mitotracker green and mitotracker deep red to determine mitochondrial mass and potential. Both do not significantly change with RhoA and RhoA-caax. Bar graphs represent the average of four independent experiments for mitotracker green (left panel) end mitotracker deep red (right panel). B, mitochondrial potential is determined by the ratio of the deep red signal (potential) over the green signal (mitochondrial mass). No changes in potential are observed. Bar graph represents the average of four independent experiments. C, mitochondrial morphology is gauged by staining THP-1 cells with mitotracker green and deep red and live cell imaging. Treatment with the RhoA inhibitor leads to a significant increase of cells with elongated mitochondria. Bar graph shows average of three independent experiments, each condition contained at least 100 cells per experiment. Two representative pictures are shown (D).
RhoA Inhibition Primes Cells for IL-1β Secretion
One of the hallmarks of MKD is hypersecretion of the pro-inflammatory cytokine IL-1β. IL-1β is present in an inactive form (pro-IL-1β) in cells and requires proteolytic cleavage by caspase-1 to become bioactive. The caspase-1 required is, in immune mediated processes, part of a large protein complex called the inflammasome. Such inflammasome-mediated IL-1β secretion requires two signals, one that primes cells to form inflammasome components, and one that activates the inflammasome (35). Previous studies have reported that decreased prenylation primes THP-1 cells for IL-1β hypersecretion (13, 15). We next asked how inactivation of RhoA, as happens in MKD models through reduced prenylation, may affect priming of THP-1 cells. We decided to use the C3 transferase as in Fig. 3, C and D. Simvastatin treatment was taken for a comparison of the situation in MKD models. Inhibition of RhoA led to a strong increase of IL-1β mRNA, even more robust than achieved by simvastatin treatment (Fig. 4A). However the levels of the closely-related IL-1β counterpart, IL-18, were unaltered (Fig. 4B). Simvastatin treatment led to a slight up-regulation of the inflammasome component NLRP3, while RhoA inactivation did not raise this significantly (Fig. 4C). Messenger RNA of another inflammasome component and IL-1β maturation enzyme, caspase-1, was unaltered by both treatments (Fig. 4D). The mere inhibition of RhoA unleashed the production of increased amounts of IL-1β mRNA.
FIGURE 4.
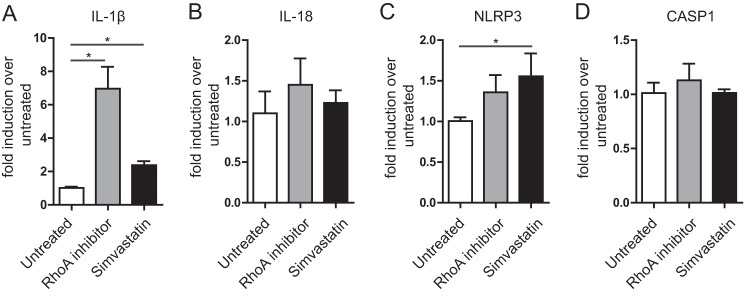
RhoA inhibition provides a priming signal for IL-1β release. Messenger RNA of THP-1 cells treated with RhoA inhibitor is isolated, and induction of four genes is determined in comparison to untreated control cells. Simvastatin treatment is taken as the broad-ranging prenylation inhibitor. Of the four genes, only IL-1β (A) is significantly up-regulated. The related cytokine IL-18 (B) and the inflammasome components NLRP3 (C) and caspase-1 (D) show no difference. All genes are normalized to GAPDH; bar graphs represent the average of three independent experiments.
RhoA Inactivation Increases Rac1 Activation
Having investigated some of the affected molecular pathways in MKD, we asked if RhoA and Rac1 may work in concert to cause increased IL-1β mRNA transcription, which may involve vice-versa control of each other's activity. We therefore studied the effects of RhoA inhibition on Rac1 activation. For control purposes we included the geranylgeranyl transferase inhibitor GGTI-298. We chose this inhibitor over simvastatin because simvastatin inhibits both farnesylation and geranylgeranylation, while GGTI-298 only inhibits the latter, thereby reducing off-target effects as Rac1 and RhoA are only geranylgeranylated. We chose to detect the GTP-bound fractions of both proteins using a G-LISA approach to complement our C3 transferase-based RhoA experiments. G-LISA moreover allowed us to measure both RhoA and Rac1 in the same sample. RhoA inhibition itself is enough to significantly raise the amount of GTP-bound Rac1 (Fig. 5A). The level of GTP-bound Rac1 we detected surmounted the level observed using GGTI-298, our positive control. To ascertain that RhoA was actually inactivated, we measured GTP-bound RhoA in the same samples. We found that the reduction of GTP-bound RhoA was similar when C3 transferase was included as a RhoA inhibitor as with GGTI-298; confirming the validity of using C3 transferase in these experiments. Taken together, our data support a pivotal role for RhoA in MKD-associated autoinflammation through Rac1/PKB signaling toward interleukin 1β production.
FIGURE 5.
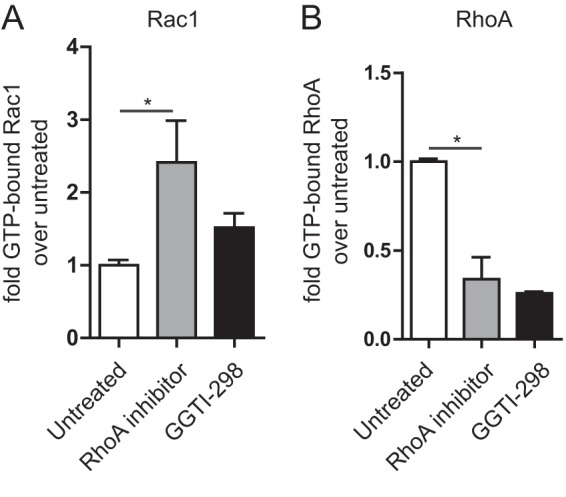
Inhibition of RhoA activity elicits Rac1 activation. A, inhibition of RhoA increases the GTP-bound portion of Rac1 in THP-1 cells. As a control, a geranylgeranyl transferse inhibitor was used. GTP-bound RhoA (B) was measured for control purposes. The G-LISA signal was normalized for total Rac1 or RhoA in the samples. The average of three independent experiments shown.
DISCUSSION
Here, we addressed the contribution of unprenylated RhoA to IL-1β hypersecretion as occurs in the autoinflammatory disease MKD. Patients suffering from MKD have severely reduced protein prenylation, but it is not fully understood how decreased prenylation is related to function of proteins that would normally be prenylated. As we and others have shown, lack of prenylation can affect activity of small GTPases (16, 24). However these effects can differ between cell types and tissues. Adding to the complexity are the large number of small GTPases, the differences in localization, and the numerous feedback and crosstalk pathways. In MKD the monocytes are the main affected cell type and responsible for the (biochemical) hallmark of the disease, IL-1β hypersecretion. Rac1 was earlier implicated in the MKD disease process. Moreover, Rac1 and RhoA have in multiple systems been shown to be reciprocally regulated (22, 23). Here we investigated the effects of loss of prenylation in a single protein, RhoA in IL-1β hypersecretion by monocytic cells, using THP-1 cells.
In a previous study we identified a defect in autophagy in MKD. Autopaghy is a complex process that requires numerous protein complexes, vesicle formation, and transport. Because of the important role of RhoA in regulation of the actin cytoskeleton, we anticipated a detrimental effect of decreased RhoA activity on autophagy. Interestingly, autophagy was not affected at all. Vesicle transport mechanisms for autophagy most likely depend more on microtubules. Many other small GTPases, in particular of the Rab family, have extensive roles in autophagy (36). It is therefore plausible that the RhoA plays a redundant role, if any, in this process.
The multiple and sometimes contradictory roles RhoA can play in different cellular systems becomes clear with the literature on the association of RhoA with mitochondria. RhoA activation has been implicated in both the protection and generation of reactive oxygen species as well as the induction and protection of apoptosis. Lack of prenylation of RhoA, without an additional signal, has in our system no effect on mitochondrial potential, nor have we seen evidence of increased cell death. We did see clear differences in the shape of mitochondria upon inactivation of RhoA. Some studies have implicated RhoA in mitochondrial trafficking, but the elongation seems a different process. Its function, if any, is unclear. We had observed this phenotype following simvastatin treatment (15). However, using simvastatin to study mitochondrial elongation would lead to numerous unprenylated proteins and affected pathways. This would make it very difficult to assess cause and consequence of mitochondrial elongation. Inhibition of RhoA could be a good approach to further elucidate the mechanism and function of mitochondrial elongation.
The inactivation of RhoA leads to increased priming of cells for IL-1β secretion. The levels of IL-1β mRNA are significantly increased. Several studies have implicated RhoA in the IL-1β secretion pathway, in particular the NF-κB signaling. It is reported that RhoA activation is necessary for the induction of G protein-coupled receptor-induced NF-κB activation (37). However, here we report an opposite effect. This again emphasizes the cell type and activation signal of specific roles of RhoA. In addition, we found it remarkable that simvastatin treatment of cells does not increase the mRNA level. This indicates that simvastatin can act on multiple pathways leading to IL-1β hypersecretion, and that some feedback mechanisms can actually reduce the amount of IL-1β mRNA or that the effect of RhoA inhibition on IL-1β mRNA is compensated in some way. Which protein or signal is responsible for the reduction of IL-1β mRNA in this system needs to be further investigated.
Finally we discovered that just by inhibiting RhoA activation, Rac1 is activated. It is already known that prenylation-deficient mutants of Rac1 are also more activated. Therefore, the lack of prenylation induced by MKD or simvastatin seems to have a synergistic role on Rac1 signaling. As Rac1 has been identified as one of the major players in IL-1β secretion, this could in part explain the hypersecretion of IL-1β in MKD.
Taken together, we have investigated how RhoA is affected by loss of prenylation in THP-1 monocytic cells. We found that RhoA is inactivated when prenylation is lacking. Despite the numerous cellular processes that RhoA is involved in, we only found effects on mitochondrial morphology and increased levels of IL-1β mRNA. These two features correspond in part with the phenotype seen in MKD monocytes and culture-based models. Furthermore we found that inactivated RhoA leads to increased Rac1 activity. Rac1/PKB signaling was earlier implicated in IL-1β hypersecretion in MKD, in an inflammasome/caspase-1-mediated route (14). Defective mitophagy also stimulates IL-1β hypersecretion through an inflammasome-mediated route (15). We here show that suppressed prenylation of RhoA primes monocytes to autoinflammation via Rac1 activation-induced IL-1β mRNA transcription. Together these results indicate a significant role for prenylation of RhoA and Rac1in MKD and help to clarify, in part, the complex molecular processes within monocytes that underlie IL-1β-driven autoinflammatory disorders.
Footnotes
- GEF
- guanine-nucleotide exchange factor
- GAP
- GTPase-activating protein
- MKD
- mevalonate kinase deficiency
- GDI
- GDP dissociation inhibitors
- RBD
- Ras-binding domain
- PKB
- protein kinase B.
REFERENCES
- 1. Gorzalczany Y., Sigal N., Itan M., Lotan O., Pick E. (2000) Targeting of Rac1 to the phagocyte membrane is sufficient for the induction of NADPH oxidase assembly. J. Biol. Chem. 275, 40073–40081 [DOI] [PubMed] [Google Scholar]
- 2. Michaelson D., Abidi W., Guardavaccaro D., Zhou M., Ahearn I., Pagano M., Philips M. R. (2008) Rac1 accumulates in the nucleus during the G2 phase of the cell cycle and promotes cell division. J. Cell Biol. 181, 485–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lutz R. J., Trujillo M. A., Denham K. S., Wenger L., Sinensky M. (1992) Nucleoplasmic localization of prelamin A: Implications for prenylation-dependent lamin A assembly into the nuclear lamina. Biology 89, 3000–3004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hoffman G. R., Nassar N., Cerione R. A. (2000) Structure of the Rho family GTP-binding protein Cdc42 in complex with the multifunctional regulator RhoGDI. Cell 100, 345–356 [DOI] [PubMed] [Google Scholar]
- 5. Shao F., Vacratsis P. O., Bao Z., Bowers K. E., Fierke C. A, Dixon J. E. (2003) Biochemical characterization of the Yersinia YopT protease: cleavage site and recognition elements in Rho GTPases. Proc. Natl. Acad. Sci. U.S.A. 100, 904–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sorg I., Goehring U., Aktories K., Schmidt G. (2001) Recombinant Yersinia YopT Leads to Uncoupling of RhoA-Effector Interaction. Infect. Immun. 69, 7535–7543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jaffe A. B., Hall A. (2005) Rho GTPases: biochemistry and biology. Annu. Rev. Cell Dev. Biol. 21, 247–269 [DOI] [PubMed] [Google Scholar]
- 8. Dovas A., Couchman J. R. (2005) RhoGDI: multiple functions in the regulation of Rho family GTPase activities. Biochem. J. 390, 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Houten S. M., Kuis W., Duran M., de Koning T. J., van Royen-Kerkhof A., Romeijn G. J., Frenkel J., Dorland L., de Barse M. M., Huijbers W. A., Rijkers G. T., Waterham H. R., Wanders R. J., Poll-The B. T. (1999) Mutations in MVK, encoding mevalonate kinase, cause hyperimmunoglobulinaemia D and periodic fever syndrome. Nat. Genet. 22, 175–177 [DOI] [PubMed] [Google Scholar]
- 10. Drenth J. P. H., Cuisset L., Grateau G., Vasseur C., Velde-visser S. D., Van De, Jong J. G. N., Beckmann J. S., Van Der Meer J. W. M., Delpech M. (1999) Mutations in the gene encoding mevalonate kinase cause hyper-IgD and periodic fever syndrome. Nat. Genet. 22, 6–9 [DOI] [PubMed] [Google Scholar]
- 11. Lutz R. J., McLain T. M., Sinensky M. (1992) Feedback inhibition of polyisoprenyl pyrophosphate synthesis from mevalonate in vitro. Implications for protein prenylation. J. Biol. Chem. 267, 7983–7986 [PubMed] [Google Scholar]
- 12. Mandey S. H. L., Kuijk L. M., Frenkel J., Waterham H. R. (2006) A role for geranylgeranylation in interleukin-1β secretion. Arthritis Rheum. 54, 3690–3695 [DOI] [PubMed] [Google Scholar]
- 13. Kuijk L. M., Mandey S. H., Schellens I., Waterham H. R., Rijkers G. T., Coffer P. J., Frenkel J. (2008) Statin synergizes with LPS to induce IL-1β release by THP-1 cells through activation of caspase-1. Mol. Immunol. 45, 2158–2165 [DOI] [PubMed] [Google Scholar]
- 14. Kuijk L. M., Beekman J. M., Koster J., Waterham H. R., Frenkel J., Coffer P. J. (2008) HMG-CoA reductase inhibition induces IL-1β release through Rac1/PI3K/PKB-dependent caspase-1 activation. Blood 112, 3563–3573 [DOI] [PubMed] [Google Scholar]
- 15. van der Burgh R., Nijhuis L., Pervolaraki K., Compeer E. B., Jongeneel L. H., van Gijn M., Coffer P. J., Murphy M. P., Mastroberardino P. G., Frenkel J., Boes M. (2014) Defects in mitochondrial clearance predispose human monocytes to interleukin-1β hypersecretion. J. Biol. Chem. 289, 5000–5012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Henneman L., Schneiders M. S., Turkenburg M., Waterham H. R. (2010) Compromized geranylgeranylation of RhoA and Rac1 in mevalonate kinase deficiency. J. Inherit. Metab. Dis. 33, 625–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Houten S. M., Schneiders M. S., Wanders R. J., Waterham H. R. (2003) Regulation of isoprenoid/cholesterol biosynthesis in cells from mevalonate kinase-deficient patients. J. Biol. Chem. 278, 5736–5743 [DOI] [PubMed] [Google Scholar]
- 18. Reid T., Furuyashiki T., Ishizaki T., Watanabe G., Watanabe N., Fujisawa K., Morii N., Madaule P., Narumiya S. (1996) Rhotekin, a new putative target for Rho bearing homology to a serine/threonine kinase, PKN, and rhophilin in the rho-binding domain. J. Biol. Chem. 271, 13556–13560 [DOI] [PubMed] [Google Scholar]
- 19. Sander E. E., van Delft S., ten Klooster J. P., Reid T., van der Kammen R. A., Michiels F., Collard J. G. (1998) Matrix-dependent Tiam1/Rac signaling in epithelial cells promotes either cell-cell adhesion or cell migration and is regulated by phosphatidylinositol 3-kinase. J. Cell Biol. 143, 1385–1398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nakagami H., Jensen K. S., Liao J. K. (2003) A novel pleiotropic effect of statins: prevention of cardiac hypertrophy by cholesterol-independent mechanisms. Ann. Med. 35, 398–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang J., Yang Z., Xie L., Xu L., Xu D., Liu X. (2013) Statins, autophagy and cancer metastasis. Int. J. Biochem. Cell Biol. 45, 745–752 [DOI] [PubMed] [Google Scholar]
- 22. Iden S., Collard J. G. (2008) Crosstalk between small GTPases and polarity proteins in cell polarization. Nat. Rev. Mol. Cell Biol. 9, 846–859 [DOI] [PubMed] [Google Scholar]
- 23. Sanders L. C. (1999) Inhibition of Myosin Light Chain Kinase by p21-Activated Kinase. Science 283, 2083–2085 [DOI] [PubMed] [Google Scholar]
- 24. Hiraoka M., Nitta N., Nagai M., Shimokado K., Yoshida M. (2004) MCP-1-induced enhancement of THP-1 adhesion to vascular endothelium was modulated by HMG-CoA reductase inhibitor through RhoA GTPase-, but not ERK1/2-dependent pathway. Life Sci. 75, 1333–1341 [DOI] [PubMed] [Google Scholar]
- 25. Chan E. Y. W., Kir S., Tooze S. A. (2007) siRNA screening of the kinome identifies ULK1 as a multidomain modulator of autophagy. J. Biol. Chem. 282, 25464–25474 [DOI] [PubMed] [Google Scholar]
- 26. Klionsky D. J., Elazar Z., Seglen P. O., Rubinsztein D. C. (2008) Does bafilomycin A1 block the fusion of autophagosomes with lysosomes? Autophagy 4, 849–850 [DOI] [PubMed] [Google Scholar]
- 27. Kaibuchi K., Kuroda S., Amano M. (1999) Regulation of the Cytoskeleton and Cell Adhesion by the Rho Family GTPases in Mammalian Cells. Annu. Rev. Biochem. 68, 459–486 [DOI] [PubMed] [Google Scholar]
- 28. Evers E. E., Zondag G. C., Malliri a, Price L. S., ten Klooster J. P., van der Kammen R. a, Collard J. G. (2000) Rho family proteins in cell adhesion and cell migration. Eur. J. Cancer 36, 1269–1274 [DOI] [PubMed] [Google Scholar]
- 29. Li D., Sewer M. B. (2010) RhoA and DIAPH1 mediate adrenocorticotropin-stimulated cortisol biosynthesis by regulating mitochondrial trafficking. Endocrinology 151, 4313–4323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Del Re D. P., Miyamoto S., Brown J. H. (2007) RhoA/Rho kinase up-regulate Bax to activate a mitochondrial death pathway and induce cardiomyocyte apoptosis. J. Biol. Chem. 282, 8069–8078 [DOI] [PubMed] [Google Scholar]
- 31. Xiang S. Y., Ouyang K., Yung B. S., Miyamoto S., Smrcka A. V., Chen J., Heller Brown J. (2013) PLCϵ, PKD1, and SSH1L transduce RhoA signaling to protect mitochondria from oxidative stress in the heart. Sci. Signal. 6, ra108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chan D. C. (2006) Mitochondrial fusion and fission in mammals. Annu. Rev. Cell Dev. Biol. 22, 79–99 [DOI] [PubMed] [Google Scholar]
- 33. Turner S. J., Zhuang S., Zhang T., Boss G. R., Pilz R. B. (2008) Effects of lovastatin on Rho isoform expression, activity, and association with guanine nucleotide dissociation inhibitors. Biochem. Pharmacol. 75, 405–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chatterjee M., van Golen K. L. (2011) Farnesyl transferase inhibitor treatment of breast cancer cells leads to altered RhoA and RhoC GTPase activity and induces a dormant phenotype. Int. J. Cancer 129, 61–69 [DOI] [PubMed] [Google Scholar]
- 35. Gross O., Thomas C. J., Guarda G., Tschopp J. (2011) The inflammasome: an integrated view. Immunol. Rev. 243, 136–151 [DOI] [PubMed] [Google Scholar]
- 36. Chua C. E. L., Gan B. Q., Tang B. L. (2011) Involvement of members of the Rab family and related small GTPases in autophagosome formation and maturation. Cell Mol. Life Sci. 68, 3349–3358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Huang S., Chen L. Y., Zuraw B. L., Ye R. D., Pan Z. K. (2001) Chemoattractant-stimulated NF-κB activation is dependent on the low molecular weight GTPase RhoA. J. Biol. Chem. 276, 40977–40981 [DOI] [PubMed] [Google Scholar]
- 38. Sander E. E., ten Klooster J.-P., van Delft S., van der Kammen R. A., Collard J. G. (1999) Rac downregulates Rho activity: reciprocal balance between both GTPases determines cellular morphology and migratory behavior. J. Cell Biol. 147, 1009–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]