

Background: Aβ amyloid formation is associated with Alzheimer disease.
Results: Non-chaperone proteins prevent amyloid formation and reduce the cytotoxicity of the Aβ peptide.
Conclusion: Non-chaperone proteins may affect the onset and development of Alzheimer disease by interfering with Aβ peptide aggregation.
Significance: Non-chaperone proteins can function as a chaperone protein to regulate the pathway of the Aβ fibrillation in proteostasis providing a new strategy in the treatment of Alzheimer disease.
Keywords: Alzheimer Disease, Amyloid-β (AB), Protein Aggregation, Protein-Protein Interaction, Spectroscopy, Aggregates, Fibrillation, Toxicity
Abstract
Many factors are known to influence the oligomerization, fibrillation, and amyloid formation of the Aβ peptide that is associated with Alzheimer disease. Other proteins that are present when Aβ peptides deposit in vivo are likely to have an effect on these aggregation processes. To separate specific versus broad spectrum effects of proteins on Aβ aggregation, we tested a series of proteins not reported to have chaperone activity: catalase, pyruvate kinase, albumin, lysozyme, α-lactalbumin, and β-lactoglobulin. All tested proteins suppressed the fibrillation of Alzheimer Aβ(1–40) peptide at substoichiometric ratios, albeit some more effectively than others. All proteins bound non-specifically to Aβ, stabilized its random coils, and reduced its cytotoxicity. Surprisingly, pyruvate kinase and catalase were at least as effective as known chaperones in inhibiting Aβ aggregation. We propose general mechanisms for the broad-spectrum inhibition Aβ fibrillation by proteins. The mechanisms we discuss are significant for prognostics and perhaps even for prevention and treatment of Alzheimer disease.
Introduction
The correlation between Alzheimer disease (AD)2 and the occurrence of extracellular amyloid β (Aβ) peptide plaques (oligomers/fibrils) in brain has been well established since the pioneering work of Glenner and Wong in 1984 (1). Aβ oligomers that precede the formation of fibrillar oligomers (FOs), fibrils, and amyloid have been shown to be the most toxic form of Aβ. These prefibrillar oligomers (PFOs) are now considered to be a prime link between cognitive impairment/neurodegeneration and Aβ plaques (2). Although the Aβ fibrils have been recognized as a less toxic structural variant of Aβ, these fibrils may drive the formation of oligomers by secondary nucleation (3).
Several small molecules are known to interfere with oligomerization and fibrillation of Aβ, for instance meclocycline sulfosalicylate, hemin, and hematin (4). Also, certain specifically engineered cyclic peptides have similar inhibitory effects on fibrillation (5–7). However, these organic compounds are potentially toxic. Even more effective was an engineered affibody that ceased the aggregation of Aβ (8) and stabilized the structure of the Aβ oligomers (9). Also, chaperone proteins, which are the body's own defense against protein misfolding, have been recently found to interfere with Aβ aggregation. αΒ-crystallin (10), clusterin (11), and heat shock protein (Hsp)B8 (12) have all been associated with pathological lesions of AD. They also inhibit fibrillation and reduce cytotoxicity of Aβ in vitro (13).
Amyloid aggregates can sequester other proteins, which may disrupt essential cellular functions (14). As a promiscuous binder of (disordered) proteins, Aβ in its aggregated state may, therefore, also function as a molecular hub in protein interaction networks (15). For instance, Aβ binds the enzyme catalase with high affinity and inhibits its hydrogen peroxide breakdown (16). Hence catalase is associated with senile plaques. Aβ also inhibits key enzymes of mitochondrial respiration (17). These results indicate that besides chaperone proteins, other proteins interact with Aβ and may, therefore, interfere with its fibrillation. Indeed, in vitro studies revealed that albumin, membrane-related Brichos domains, and lysozyme efficiently delay the fibrillation of Aβ (18–20). It is unknown to what extent these aggregation-inhibiting activities are specific functionalities of these proteins or whether there is a more generic, broad-spectrum mechanism at work.
Here we report that proteins not known for chaperone activity can inhibit Aβ(1–40) aggregation with surprisingly high efficiency. Catalase and pyruvate kinase completely suppressed Aβ fibrillation at a molar ratio of 1:100 (protein: Aβ). But also albumin was effective, albeit at a lower molecular ratio of 1:10 (protein:Aβ). Even the whey proteins β-lactoglobulin and α-lactalbumin inhibited Aβ aggregation (but at a 1:1 molar ratio). As these whey proteins are not present in brain, they cannot have a biological function in inhibiting Aβ aggregation. In the presence of all tested proteins, the Aβ peptides retained their secondary random coiled structure. In an earlier, preliminary study, we already reported that lysozyme inhibits fibrillation at a 1:1 molar ratio (18). The tested proteins also suppressed the toxicity of Aβ aggregates. HSQC NMR spectroscopy indicated that the interactions between the tested proteins and monomeric Aβ peptide were nonspecific. These results prompt us to propose common, broad-spectrum, protein-based inhibition mechanisms of Aβ aggregation.
EXPERIMENTAL PROCEDURES
Materials
Human lysozyme (catalog no. L1667), human serum albumin (catalog no. A3782), α-lactalbumin from bovine milk (catalog no. L6010), catalase from bovine liver (catalog no. C9327), and β-lactoglobulin from bovine milk (catalog no. L3908) were purchased from Sigma. Pyruvate kinase (catalog no. 10836821) from rabbit muscle was bought from Roche Diagnosis. The Aβ(1–40) peptides (either unlabeled or 15N-labeled) were purchased from AlexoTech AB (Umeå, Sweden) and prepared according to previously described protocols; the Aβ peptides were dissolved in 10 mm NaOH to a peptide concentration of 2 mg/ml and then sonicated for 1 min in an ice bath before dilution in the appropriate buffer. The preparations were kept on ice.
ThT Fluorescence Assay
A 10 mm ThT stock solution was prepared in distilled H2O. The proteins were added in the desired amounts to aliquots of this solution. Then freshly prepared Aβ(1–40) peptide was added, yielding final samples containing 5 μm ThT, 10 μm Aβ(1–40), 50 mm Tris buffer (pH 7.4), and protein in the desired concentrations. All buffers and samples were prepared on ice to avoid the aggregation. The samples were pipetted into a well plate with 384 wells holding 45 μl each. Fluorescence measurements were recorded with an Infinite M1000 PRO microplate reader every 15 min using excitation and emission wavelengths of 446 and 490 nm, respectively. The plate was thermostated at +37 °C, and the wells were automatically shaken 30 s before each measurement.
Each sample was prepared in duplicate, and average fluorescence signals were calculated after subtracting the base-line fluorescence of control samples without Aβ(1–40) peptide. The fluorescence intensity (I) data were fitted to a sigmoid curve with a sloping base line using the equation, I(t) = k1t + A/(1 + exp(−k2(t − t½), where the parameter k1 describes the sloping base line, A is the amplitude, k2 is the elongation rate constant, and t½ is the time of half-completion of the aggregation process (21).
For the disaggregation assay, a stock of Aβ(1–40) peptide aggregates was obtained by incubating 50 μm Aβ(1–40) peptide in 50 mm Tris (pH 7.4) at 200 rpm for 18 h at 30 °C. Then, 5 μm aggregates were diluted and mixed with the test proteins and ThT for the fluorescence assay. The protocol and buffer conditions were the same as described above. Fluorescence was measured after 0, 5, 10, and 15 h. For the fibrillation control assay, we prepared 10 μm Aβ and 10 μm ThT in Tris buffer (pH 7.4) in the presence and absence of non-chaperone proteins and then incubated them in Eppendorf tubes in a 37 °C shaker at 200 rpm.
CD Spectroscopy
A Chirascan CD unit from Applied Photophysics was used to monitor the kinetics of a 10 μm solution of Aβ(1–40) peptide dissolved in 5 mm sodium phosphate buffer at pH 7.3 in the presence and absence of test proteins. CD spectra at 30-min intervals were recorded between 190 and 260 nm using a step size of 2 nm and a slit size providing 1-nm resolution. The sample was thermostated at 37 °C in a 10-mm path length quartz cuvette (lot: 119.004-QS, Hellma Analytics) and mechanical rotary stirring (22 rpm) with an 0.7-cm magnet was used to speed up aggregation.
Cell Viability Assay
Neuroblastoma SH-SY5Y cells were used with a maximum passage number of 15. Cells were cultured in Dulbecco's modified Eagle's medium, a 1:1 mixture of DMEM and Ham's F-12 medium, and 10% supplemental fetal bovine serum containing 1% (v/v) penicillin/streptomycin at 37 °C, 5% CO2 in a 75-cm2 flask (Greiner Bio-one, catalog no. 658170). To avoid using trypsin, cells were detached by 5 mm EDTA, PBS for 5 min in 37 °C. Then cells were resuspended at a concentration of 200,000 cells/ml in DMEM/F-12 containing 1% (v/v) penicillin/streptomycin. The resuspended cells were plated at a volume of 50 μl and a cell density of 20,000 cells/well in a 96-well plate. The plated cells were incubated for 48 h at 37 °C at 5% CO2. Aβ40 oligomer-enriched fractions were prepared at a concentration of 100 μm in the presence of the test proteins at 25 °C for 100 min in PBS with 1 mm EDTA. The Aβ oligomer solutions were diluted to final concentrations of 30 μm in wells in the presence of the different proteins. As a control, the PBS-dissolved test proteins in 50 μl of medium were added to control wells and incubated for 48 h. After 48 h the plate was equilibrated at room temperature for ∼30 min. CellTiter-Glo® Luminescent Cell Viability Assay (Promega, catalog no. G7571) compound was added to each well, and then the contents in the plate were mixed using an orbital shaker for 2 min to induce cell lysis (22). Luminescent intensity was measured (1000-ms integration time) with the Infinite M1000 PRO 384-well microplate reader. Measurements from three independent experiments were analyzed statistically to calculate average values and S.D.
ANS Fluorescence Assay
Aβ oligomers were prepared in a similar fashion as for the cell viability assay. 100 μm Aβ(1–40) peptide was incubated with PBS at room temperature for 2 h in an Eppendorf tube. Aliquots of 20 μl oligomeric Aβ and 0.4 μl of proteins (500 μm in PBS) were pipetted into a 384-well plate (OptiPlate 384, PerkinElmer Life Sciences), and then 1 μl of 50 mm ANS was added. ANS fluorescence spectra between 400 and 600 nm were recorded with an Infinite M1000 PRO microplate reader (excitation at 350 nm). ANS fluorescence intensity of Aβ oligomers was calculated after subtracting the intensity of non-chaperone proteins or buffers, which were handled similarly.
NMR Spectroscopy
A Bruker Avance 500-MHz spectrometer was used to record 1H,15N HSQC spectra at +5 °C of 100 μm 15N-labeled Aβ(1–40) peptide in 20 mm sodium phosphate buffer at pH 7.3 (90/10 H2O/D2O) both in the absence and presence of the different proteins. The spectrometer was equipped with a triple-resonance cryogenically cooled probe head, and the spectra were referenced to the water signal. All NMR measurements were done at +5 °C to slow down the aggregation process. The assignment of the amide peaks for the Aβ(1–40) peptide is known from previous work.
Bioinformatics
The molecular weight, theoretical PI, and amyloidogenic regions of proteins were calculated using the ExPASy server and Waltz software (23), respectively. Total solvent-excluded surface area and total solvent-accessible surface area were calculated using the Chimera software suite (24).
RESULTS
All Test Proteins Suppress Aβ Peptide Fibrillation
We monitored the effect of various concentrations of catalase, pyruvate kinase, albumin, lysozyme, α-lactalbumin, and β-lactoglobulin on the fibrillation of 10 μm Aβ using a ThT assay (Fig. 1, Table 1). All test proteins inhibited Aβ fibrillation. The fluorescence increase characteristic of Aβ fibrillation significantly dropped in the presence of 25 nm pyruvate kinase and did not occur at all in 100 nm pyruvate kinase (Fig. 1a). Fibrillation of Aβ was hardly observed in 100 nm catalase (Fig. 1e).
FIGURE 1.

ThT aggregation kinetics of 10 μm Aβ peptide in the presence of the test proteins pyruvate kinase (a), α-lactalbumin (b), albumin (c), lysozyme (d), catalase (e), and β-lactoglobulin (f). For pyruvate kinase (panel a), which was most active in preventing fibrillation, we tested five different concentrations: 0 nm (black), 25 nm (olive), 100 nm (magenta), 500 nm (cyan), and 1000 nm (blue). For the other test proteins we used five slightly higher concentrations: 0 nm (black), 100 nm (olive), 500 nm (magenta), 1 μm (cyan), and 10 μm (blue). Red curves (all coinciding with the horizontal axis) are controls in which no Aβ was added to 20 μm test proteins. The curves show the average of three independent experiments.
TABLE 1.
Lag time and transition time for amyloid formation kinetics of 10 μm Aβ(1–40) peptides in the presence of 0, 0.1 μm, 0.5 μm, and 1 μm proteins, measured in 50 mm Tris buffer at pH 7.4 and +37 °C
| Control | Pyruvate kinase | α-Lactoalbumin | Albumin | β-Lactoglobulin | |
|---|---|---|---|---|---|
| tlag/h | |||||
| 0.1 μm | 3.5 | 3.7 | 2.7 | 4.0 | 3.7 |
| 0.5 μm | 4.5 | 6.0 | 4.1 | ||
| 1 μm | 5.9 | 3.7 | |||
| ttrans/h | |||||
| 0.1 μm | 2.8 | 0.9 | 3.1 | 2.9 | 1.7 |
| 0.5 μm | 2.6 | 2.4 | 2.7 | ||
| 1 μm | 2.3 | 8.0 | |||
Although α-lactalbumin, albumin, and β-lactoglobulin delayed the fibrillation of Aβ at low concentrations, their inhibition efficiencies were weaker compared with catalase and pyruvate kinase. Albumin and α-lactalbumin completely prevented Aβ fibrillation >24 h at 1 and 10 μm, respectively. And β-lactoglobulin decreased fibrillation of Aβ at concentrations >1 μm. Low concentrations of α-lactalbumin somewhat enhanced nucleation (as witnessed by a reduction in the lag time), but at higher concentrations α-lactalbumin also delayed fibrillation. We included the results we obtained earlier with lysozyme (using a different batch of Aβ) in Fig. 1d (17). As a control we ascertained that ThT fluorescence was not affected by the test proteins in the absence of Aβ (red curves in Fig. 1).
We investigated whether the test proteins could redissolve Aβ-aggregates by incubating them with preformed Aβ fibrils. Only catalase reduced the fluorescence intensity of Aβ (Fig. 2), suggesting that catalase interacts with Aβ fibrils and reverses the amyloid fibrillation. In contrast, pyruvate kinase and α-lactalbumin slightly increased the fluorescence intensity of preprepared Aβ fibrils. This might indicate that these proteins could also condense and (partially) unfold onto Aβ fibrils. Alternatively, the Aβ fibrils might nucleate the fibrillation of these test proteins. Neither lysozyme, albumin, nor β-lactoglobulin affected the fluorescence intensity of preformed Aβ fibrils. These results also indicate that the reduction in ThT signal observed when the non-chaperone proteins were incubated with soluble Aβ was not due to interference of these proteins (either through quenching or absorption) with the ThT assay. Catalase could even redissolve preformed fibrils, indicating that its protective effect cannot solely be explained by interference with nucleation events.
FIGURE 2.

a, ThT aggregation kinetics of 10 μm Aβ peptide in the presence of the test proteins pyruvate kinase, α-lactalbumin, albumin, lysozyme, catalase, and β-lactoglobulin incubated in an Eppendorf tube at 37 °C placed in a shaker at 200 rpm. The reaction vessel and the incubation conditions were different from those in Fig. 1, yet the results were very similar, suggesting that surface effects could not explain our results. b, ThT disaggregation assay of Aβ peptide in the presence of α-helical proteins. 50 μm Aβ was incubated for 18 h at 30 °C in a 200 rpm shaker (50 mm Tris buffer (pH 7.4)), and then 5 μm aggregates (after diluting the 50 μm stock in 50 mm Tris buffer (pH 7.4)) were incubated with different test proteins at 37 °C. The protocol for ThT disaggregation assay is same as for the ThT aggregation assay.
We tested if the delayed nucleation could be caused by Aβ preferentially nucleating on surfaces (air-liquid interface or the surface of the reaction vessel), the nature of which could be affected by the non-chaperone proteins, for instance by coating. To exclude such surface effects, we also incubated Aβ peptides with and without proteins in an Eppendorf tube in a 37 °C, 200 rpm shaker, which has a different geometry, air-liquid interface, and chemical composition compared with the 384-well plates. All of the proteins could also prevent or delay the fibrillation of Aβ in these different incubation conditions (Fig. 2a). The kinetics of Aβ fibrillation in Eppendorf tubes did differ slightly from fibrillation in well plates. This might be caused by differences in the composition (25) or geometry of the reaction vessels (26). However, the effects were relatively minor. The marked drop in ThT fluorescence observed after 7 h of incubation when no extra proteins were added to the Aβ peptide was caused by precipitation of the fibrils as mature amyloid.
The Test Proteins Stabilized the Random Coil Structure of the Monomeric Aβ Peptide
We investigated the effect of the test proteins on the structural transition of Aβ by CD spectroscopy. The mean residue ellipticity of Aβ was obtained after subtracting the signal of the test protein from the measured spectra (Fig. 3). As expected, Aβ in the absence of the test proteins converted its secondary structure from random coil to β-sheet in 3 h (Fig. 3). However, in the presence of substoichiometric concentrations of test protein, the Aβ peptide retained its random coil secondary structure (Fig. 3). We observed subtle differences in secondary structure changes of Aβ in the presence of the different proteins. The CD amplitudes at 200 nm increased slightly during incubation with albumin, α-albumin, β-lactoglobulin, catalase, and pyruvate kinase. In the presence of lysozyme, there was a CD up-shift at 220 nm during the incubation (Fig. 3).
FIGURE 3.

The secondary structure transition of 10 μm Aβ in the absence and presence of the test proteins: 0. 5 μm α-lactalbumin, 1.25 μm albumin, 1 μm β-lactoglobulin, 0.25 μm catalase, 1 μm lysozyme, and 0.05 μm pyruvate kinase by CD spectroscopy. In the assay, we recorded the kinetics of structural transition in the time interval of 0.5 h, black (0 h), red (0.5 h), green (1 h), blue (1.5 h), cyan (2 h), magenta (2.5 h), dark yellow (3 h), navy (3.5 h), purple (4 h), and orange (4.5 h).
The changes in lag and transition times are all correlated with the concentration of the non-chaperone proteins in a unimodal fashion (Table 1), but the correlation between the maximal fluorescence can be biphasic. See for instance Fig. 1, panel b, where the maximum fluorescence at 100 nm α-lactalbumin is lower than at 500 nm. This is consistent with our disaggregation experiment (see above and Fig. 2) and could indicate that α-lactalbumin could also condense and (partially) unfold onto Aβ fibrils.
All Test Proteins Reduce Cytotoxicity of Aβ Aggregates
The effect of the test proteins on the cytotoxicity of the aggregates of Aβ was measured in a cell assay (Fig. 4). We first incubated 100 μm Aβ with the proteins at various concentrations for 2 h and added the aggregates to neuronal SH-SY5Y cells up to a concentration of 30 μm. After 2 days of incubation with Aβ aggregates, 20% of the cells survived, but survival significantly increased when Aβ had been allowed to aggregate in the presence of the test proteins. Cell survival was ∼100% in the aggregates of Aβ and pyruvate kinase/catalase, which was much higher than the cell survival rate after incubation with the aggregates in the presence of Aβ and α-lactalbumin/albumin/lysozyme/β-lactoglobulin. But even for the latter test proteins, a positive effect on cell survival was observed.
FIGURE 4.
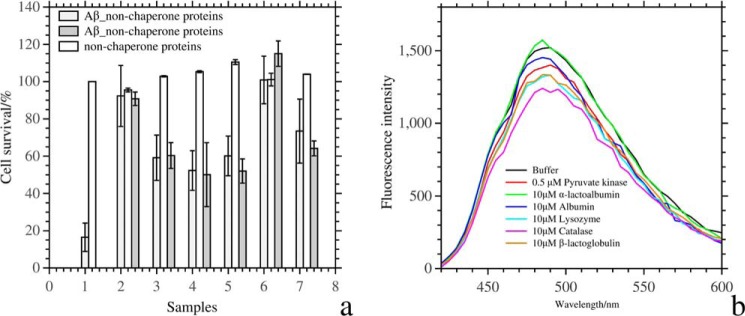
a, cell survival after incubation with Aβ aggregates that had formed in the presence and absence of the test proteins. In sample 1, the cell survival of 30 μm Aβ aggregates that had formed in the absence of test proteins is shown as light gray columns, and the cell survival of the control is in white. Samples 2, 3, 4, 5, 6, and 7 are in the presence of pyruvate kinase, α-lactalbumin, albumin, lysozyme, catalase, and β-lactoglobulin, respectively. The white column represents cell survival of 1 μm protein without Aβ aggregates in sample 2 and 2.5 μm protein in the samples 3–7. The dark gray columns are cell survival after incubation with 1 μm protein and 30 μm Aβ aggregates in sample 2 and 3 μm test protein and 30 μm Aβ aggregates in samples 3–7. The light gray columns are cell survival in the presence of 0.2 μm test protein and 30 μm Aβ aggregates in sample 2 and 0.6 μm test protein and 30 μm Aβ in samples 3–7. b, ANS fluorescence intensity of 100 μm Aβ oligomers with and without proteins. Aβ oligomers were obtained after a 2-h incubation at room temperature in an Eppendorf tube.
The Hydrophobic Nature of the Interaction between Aβ and the Test Proteins
We performed ANS fluorescence assays to examine if the hydrophobic surface of Aβ oligomers changes after adding the non-chaperone proteins. When Aβ and protein were both present, ANS fluorescence was reduced compared with identical concentrations of Aβ protein, except for α-lactalbumin (Fig. 4b). The shielding of hydrophobic surfaces that this assay indicates strongly suggests that the interaction between Aβ and the non-chaperone proteins at the very least had a hydrophobic contribution. Our hydrophobic interaction assay correlated with our toxicity assay: higher ANS fluorescence intensities went with higher cytotoxicity (27). The fluorescence assay did not allow us to ascertain whether the proteins interacted with monomer Aβ, with Aβ oligomers, or with both.
The Test Proteins Bind Monomeric Aβ in a Nonspecific Fashion
To explore the molecular interaction between monomeric Aβ and proteins, we used NMR 1H,15N HSQC spectroscopy (Fig. 5). Interactions between larger aggregates of Aβ and the test proteins would not be visible in this assay. The results from NMR spectroscopy allow us to identify the chemical shifts of each detectable residue after the addition of the test proteins. The amide chemical shifts of Aβ moved slightly after the addition of α-lactalbumin (Fig. 5a) accompanied with a decreased of the cross-peak intensity (Fig. 6a). Although no significant amide chemical shift of Aβ was observed after the addition of albumin or lysozyme (Fig. 5, b and c), the cross-peak intensities of Aβ increased significantly (Fig. 6, b and c). Upon the addition of β-lactoglobulin or catalase, there were no noticeable changes in chemical shifts (Fig. 5, d and e), and the cross-peak intensity of most Aβ residues was reduced (Fig. 6, d and e). Lysozyme and pyruvate kinase increased the cross-peak intensity of Aβ by a factor of 2 (Fig. 6, c and f), but the chemical shifts did not change (Fig. 5, c and f). These results suggest that the protein interacted with monomeric Aβ in a nonspecific fashion and that the interaction was in fast exchange, at least for those proteins where the cross-peak intensities increased (albumin, lysozyme, pyruvate kinase).
FIGURE 5.

NMR 1H,15N HSQC spectra of 90 μm15N-labeled Aβ(1–40) peptides in 20 mm sodium phosphate buffer at pH 7. 3, +5 °C, before (red) and after (blue) the addition of 18 μm α-lactalbumin (a), 5.4 μm albumin (b), 200 μm lysozyme (c), 50 μm β-lactoglobulin (d), 9 μm catalase (e), and 4.5 μm pyruvate kinase (f).
FIGURE 6.

The relative change in intensity of the HSQC-NMR cross-peaks induced by the addition of the test proteins was calculated using the spectra in Fig. 5.
DISCUSSION
Our ThT and CD data indicate that the non-chaperone proteins that we tested suppressed Aβ fibrillation by stabilizing the random coil structure of Aβ. Pyruvate kinase and catalase were the most efficient inhibitors of Aβ fibrillation, suppressing Aβ fibrillation effectively at a molar ratio of 1:100. Catalase was such an effective inhibitor that it could even reverse Aβ fibrillation. ANS fluorescence indicated that the test proteins reduced the hydrophobicity of Aβ(−oligomers), suggesting a (partially) hydrophobic interaction between Aβ and the proteins. All test proteins reduced the cytotoxicity of Aβ oligomeric aggregates. This suggests that they either prevented the formation of PFOs (the most toxic oligomeric state of Aβ) or interfered with the toxic pathways, for instance by binding and neutralizing the PFOs. Again, pyruvate kinase and catalase had the strongest effect. Pyruvate kinase increased the NMR cross-peak intensity of Aβ residues on average by a factor of about 2, but catalase reduced the cross-peak intensity.
A Model for Broad Spectrum Inhibition of Aβ Toxicity and Fibrillation
It is intriguing that through apparently a-specific interactions with proteins present in substoichiometric amounts, the formation of amyloid can be inhibited. We can envisage several mechanisms (which do not exclude each other): (i) monomeric peptides that spontaneously convert to a β-sheet conformation are scavenged by the globular, non-chaperone proteins and are released if they convert back to random coil, thus reducing the number of nucleation sites and reducing the number of PFOs, (ii) PFOs or other types of non-fibrillar oligomers are stabilized by the non-chaperone proteins, thus slowing down the conversion from PFO to FO that precedes fibrillation (28), (iii) the non-chaperone proteins bind to (the ends of) less toxic fibrillar oligomers, somehow preventing their growth into fibrils. If mechanism (iii) were solely responsible for the observed inhibition, we would not have observed the reduction of cytotoxicity, as the PFOs are the most toxic species. Therefore, we will focus on mechanisms (i) and (ii) (Fig. 7).
FIGURE 7.

Potential inhibition mechanism of intracellular chaperones and extracellular non-chaperone proteins. The hydrophobic and hydrophilic surface regions of the proteins are represented in orange and gray, respectively. Aβ is produced from APP by γ-secretase. Toxic oligomers of Aβ can be scavenged and potentially redissolved by intracellular chaperones and extracellular non-chaperone proteins. a–d, non-chaperone proteins can bind to Aβ oligomers directly and then prevent the nucleation of fibrils (a). Alternatively, Aβ oligomers can be converted into monomer after binding to non-chaperone proteins (b and c), or Aβ is stabilized in its monomeric state due to the interactions with non-chaperone proteins. HSP, heat shock protein
Mechanism (i) has parallels with the mode of action of chaperone proteins like Hsp90 and trigger factor. Such proteins function as holdases and unfoldases by binding to hydrophobic, aggregation-prone sequences of unfolded proteins, thus protecting them from aggregation (29, 30). Our NMR data indicated that the relative intensity of Aβ HSQC cross-peaks increased in the presence of albumin, lysozyme, or pyruvate kinase. This suggests that these proteins solubilized Aβ in its unfolded state. Furthermore, our ANS data indicated that the total hydrophobic surface area decreased upon co-incubating Aβ with these proteins. A likely explanation of these observations is that albumin, lysozyme, and pyruvate kinase weakly bind Aβ molecules with exposed hydrophobic surfaces, release them when they convert back to a less hydrophobic conformation, and thereby reduce the number of aggregation-prone hydrophobic monomers in the solution.
However, α-lactoalbumin, β-lactoglobulin, and catalase reduced the intensity of Aβ HSQC cross-peaks, indicating that a substantial amount of Aβ was no longer present in its monomeric state. This suggests that mechanism (ii) is also employed. The CD measurements show that the Aβ molecules had not adopted a β-sheet conformation in the aggregates. Perhaps these three proteins induced a dead-end aggregate of Aβ, which was neither toxic nor able to convert into toxic PFOs or fibril inducing FOs. Alternatively, α-lactoalbumin, β-lactoglobulin, and catalase had an enhanced affinity for toxic PFOs and scavenged these from solution.
It is not clear what the structural details are of mechanism (ii) in vivo. In any case we have to consider the possibility that PFO and FO formation is reduced indirectly in which the non-chaperone proteins could inhibit PFO and FO formation for instance by binding copper, a metal known to stimulate Aβ fibrillation. Catalase, α-lactoalbumin (31), and β-lactoglobulin (32) are all known to bind copper, so this indirect mechanism has to be considered. Thus, some of non-chaperone proteins may act as mediators to inhibit the formation of copper and the Aβ complex and reduce the production of reactive oxygen species in vivo.
Here we discuss a possible structural interaction-underlying mechanism (ii). It is based on the hypothesis that the structure of PFOs is similar to that of cylindrins (33). One of the hypotheses is that once such an oligomer contacts the cell membrane, its hydrophilic regions may interact with the head group of lipids, and then hydrophobic residues disrupt the tail groups, inducing weakening of the membrane, potential leaking of the cell, and eventually apoptosis. We investigated the hydrophobic and hydrophilic surface of the test proteins to test this cylindrin model (Fig. 7). Based on the surface charge distribution, we propose that once the proteins interact with Aβ oligomers, the hydrophobic and hydrophilic groups in the protein may act as the tail and head groups of cellular lipids, respectively. However, unlike the lipidic cell membrane or the amyloid oligomers, the proteins are relatively stable. The proteins may, therefore, retain their structure, but the toxic amyloid oligomer may be scavenged or even be disrupted. If the PFOs form the prime source of FOs or if FOs are captured by the proteins in a similar fashion as PFOs, such interactions could effectively reduce the number of FO nucleation seeds required for fast fibrillation. Thus, toxicity and amyloid formation would be reduced.
The Correlation between AD and the Test Proteins
Catalase, pyruvate kinase, and albumin can be found in the brain. To some extent they are related to the progression of AD. As a peroxidase enzyme, catalase may mediate the cellular levels of reactive oxygen species, which is relevant for the hypothesis of oxidative stress in AD. The activity of pyruvate kinase is significantly increased in frontal and temporal cortex of AD brains (34), but direct interaction in vivo between pyruvate kinase and Aβ remains to be investigated. The level of Aβ-albumin complex in serum is decreased in the AD brain (35). There is no direct relationship between lysozyme and AD, but lysozyme increases upon inflammation reactions, which are also triggered in the Alzheimer brain.
A Comparison with Chaperones against the Aβ Fibrillation
Extracellular chaperones and chaperone-like proteins have been reported to act as disposal mediators in the proteostasis of Aβ aggregation (36, 37). For instance, clusterin, a potent sHsp-like chaperone, sequesters prefibrillar oligomers and influences Aβ fibrillation; at a molecular ratio of 1:10 (clusterin:Aβ), suppression of Aβ fibrillation was reported (11). However, clusterin in vitro does not affect the cytotoxicity of Aβ aggregates at a molar ratio of 1:500 and increases the cytotoxicity of Aβ aggregates at a molar ratio of 1:10 (38). Like clusterin, haptoglobin and α2-macroglobulin (found in extracellular fluid), completely suppressed Aβ fibrillation at a molecular ratio of 1:10 by interacting with Aβ PFOs (39). In addition to extracellular proteins, intracellular chaperones, like heat shock proteins hsp70 and hsp90, inhibit phenotypes related to Aβ aggregation in a model of Alzheimer disease (40). Hsp70 and hsp90 interact with oligomeric aggregates and suppress Aβ fibrillation effectively at substoichiometric molecular ratio (∼1:50) (13). Similarly, hsp104 targets oligomeric intermediates and inhibits the elongation of Aβ fibrils (41).
Besides extracellular and intracellular chaperones, lysozyme and albumin have been shown to bind to Aβ peptide and suppress Aβ fibrillation. Human serum albumin binds to Aβ oligomers/protofibrils in prevailing stoichiometry and affinity (1–100 nm range) and inhibits Aβ(1–42) fibril growth at substoichiometric concentrations (molecular ratio 1:18) (42, 43). Human lysozyme inhibits Aβ fibrillation at a molecular ratio of 1:1. Furthermore, the BRICHOS domain, associated with familial British dementia, interferes with Aβ aggregation before the formation of fibrils at a molecular ratio of ∼1:10 (BRICHOS domain:Aβ) (19).
Size and Surface-dependent Interaction between Aβ and Folded Proteins
To identify which aspects of these proteins and the test protein on which we report here might contribute to their apparent biological activity, we summarize some of their important biophysical characteristics in Table 2. Table 2 does suggest a positive correlation between molecular weight and inhibition efficiency. For instance, catalase, pyruvate kinase, and Hsps with molecular masses >50 kDa inhibit the fibrillation of Aβ at a molecular ratio of 1:50. We also investigated the effect of the hydrophobicity/hydrophilicity of the accessible surface area and found that the most effective inhibition of the Aβ fibrillation can be observed for proteins with a ratio of solvent-excluded surface area:solvent-accessible surface area >0.9. However, there is not a very clear biophysical, molecular property that the most effective inhibitors of fibrillation and toxicity have in common.
TABLE 2.
The comparison of properties of non-chaperone and chaperone proteins on the inhibition of the Aβ fibrillation
| Protein | Mr | State | PI | Molar effective inhibitiona | Weight effective inhibitionb | Amylogenic regionsc | SESA/SASAd |
|---|---|---|---|---|---|---|---|
| Catalase | 60 | Hexamer | 6.90 | ∼1:100 | 1:7.1 | 53–58; 81–86; 132–137; 194–202; 265–270; 277–295; 321–329; 459–464; 469–474 | 1.25 |
| Pyruvate kinase | 237 | Monomer | 7.96 | ∼1:100 | 1:1.8 | 144–148; 391–395; 506–514 | 1.00 |
| Lysozyme | 14 | Monomer | 9.38 | ∼1:1 | 1:0.3 | 73–79 | 0.89 |
| α-Lactoalbumin | 14 | Dimer | 4.92 | ∼1:1 | 1:0.3 | 70–78; 90–95; 119–125 | 0.84 |
| β-Lactoglublin | 20 | Monomer | 4.93 | ∼2:1 | 1:0.1 | 14–21; 96–101; 117–122 | 0.91 |
| Human serum albumin | 67 | Monomer | 5.85 | ∼1:10 | 1:0.6 | 9–18; 45–57; 177–182; 352–357; 574–578 | 0.99 |
| Pro-SP-C BRICHOS domain | 19 | Monomer | 6.19 | ∼1:30 | 1:6.7 | 42–51; 100–114; 119–127 | 1.00 |
| Haptoglobin 2–1 | 200 | Monomer | ∼1:10 | 1:0.2 | 13–19; 188–209; 220–225 | ||
| α2-Macro-globulin | 73 | Dimer | 6.24 | ∼1:10 | 1:0.59 | ||
| Clusterin | 70 | Dimer | 5.88 | ∼1:10 | 1:0.61 | 4–16; 364–370 | 0.93 |
| HSP 70 | 70 | Monomer or Dimer | 5.47 | ∼1:50 | 1:3.1 | 25–30; 40–46; 103–108; 141–153; 169–174; 179–184; 193–199; 205–210; 367–372; 375–381; 424–432; 439–445 | 0.98 |
| HSP 90 | 90 | Dimer | 4.94 | ∼1:50 | 1:2.4 | 30–53; 135–144; 338–342; 360–365; 490–496; 516–523; 663–674 | 0.90 |
| HSP 104 | 100 | Hexamer | 5.31 | ∼1:100 | 1:4.3 | 107–133; 278–284; 347–352; 420–429; 608–615; 628–633; 730–737; 768–775 | |
| Affibody ZAβ3 | 10 | Monomer | 6.01 | 1:100 | 1:43 | ||
| α-Synuclein | 14 | Disordered | 4.67 | Promotes Aβ fibrillation | 35–40 |
a Molecular ratio of the effective inhibition of the Aβ fibrillation in 24 h (protein:Aβ).
b Weight ratio (mg/mg) of the effective inhibition of the Aβ fibrillation in 24 h (protein:Aβ).
c Amylogenic regions were predicted by the Waltz program (24).
d Total solvent-excluded surface area/total solvent-accessible surface area.
Potential Amyloid Cross-interactions between Aβ and (Non-)folded Proteins
Amyloid proteins also affect Aβ fibrillation and toxicity. For instance, α-synuclein oligomers interact with Aβ monomers and/or oligomers and affect Aβ fibrillation (44). Such (amyloid protein:Aβ) cross-interactions may promote the progression of AD (45). This interactions may result from conformational transition of Aβ and α-synuclein (46) where the flexible amylogenic regions with more hydrophobic residues interact with each other, and electrostatic interactions form in the flexible regions. To investigate if such interactions between Aβ and the test proteins could occur, we searched for their potential amylogenic regions using the Waltz algorithm (Table 2). Despite a cluster of amylogenic regions predicted on the surface of proteins, the folded proteins, lacking a flexible hydrophobic surface, interacted with the Aβ monomer only weakly as we observed by NMR (which is in agreement with a previous report (19)). So potential amyloid cross-interactions cannot explain the effects on Aβ fibrillation that we report.
In summary, we discovered that a series of non-chaperone proteins hinder the Aβ fibrillation and reduce the toxicity of Aβ aggregates. We propose that the proteins interfere with the stability of the Aβ oligomer by hydrophobic and hydrophilic interactions of their surface. There is a positive correlation between protein surface size and Aβ-neutralizing activity, but this does not fully explain why some proteins are more active in inhibiting Aβ than others. Our results would for instance predict that high levels of catalase in the brain would not only reduce damage by neutralizing reactive oxygen species but also by reducing the toxicity of Aβ oligomers. There may be other proteins in extracellular cerebral fluids that similarly reduce Aβ toxicity to a significant extent, and monitoring or even influencing their levels in AD-prone individuals may be relevant for preventive strategies.
Footnotes
- AD
- Alzheimer disease
- Aβ
- amyloid β
- FO
- fibrillar oligomer
- PFO
- prefibrillar oligomers
- HSQC
- heteronuclear single quantum correlation
- ThT
- thioflavin T
- ANS
- 8-anilino-1-naphthalenesulfonic acid.
REFERENCES
- 1. Glenner G. G., Wong C. W. (1984) Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 120, 885–890 [DOI] [PubMed] [Google Scholar]
- 2. Benilova I., Karran E., De Strooper B. (2012) The toxic Aβ oligomer and Alzheimer's disease: an emperor in need of clothes. Nat. Neurosci. 15, 349–357 [DOI] [PubMed] [Google Scholar]
- 3. Cohen S. I., Linse S., Luheshi L. M., Hellstrand E., White D. A., Rajah L., Otzen D. E., Vendruscolo M., Dobson C. M., Knowles T. P. (2013) Proliferation of amyloid-β42 aggregates occurs through a secondary nucleation mechanism. Proc. Natl. Acad. Sci. U.S.A. 110, 9758–9763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Necula M., Kayed R., Milton S., Glabe C. G. (2007) Small molecule inhibitors of aggregation indicate that amyloid β oligomerization and fibrillization pathways are independent and distinct. J. Biol. Chem. 282, 10311–10324 [DOI] [PubMed] [Google Scholar]
- 5. Sievers S. A., Karanicolas J., Chang H. W., Zhao A., Jiang L., Zirafi O., Stevens J. T., Münch J., Baker D., Eisenberg D. (2011) Structure-based design of non-natural amino acid inhibitors of amyloid fibril formation. Nature 475, 96–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Luo J., Otero J. M., Yu C.-H., Wärmländer S. K., Gräslund A., Overhand M., Abrahams J. P. (2013) Inhibiting and reversing amyloid-β peptide (1–40) fibril formation with gramicidin S and engineered analogues. Chemistry 19, 17338–17348 [DOI] [PubMed] [Google Scholar]
- 7. Luo J., Abrahams J. P. (2014) Cyclic peptides as inhibitors of amyloid fibrillation. Chemistry 20, 2410–2419 [DOI] [PubMed] [Google Scholar]
- 8. Hoyer W., Grönwall C., Jonsson A., Ståhl S., Härd T. (2008) Stabilization of a β-hairpin in monomeric Alzheimer's amyloid-β peptide inhibits amyloid formation. Proc. Natl. Acad. Sci. U.S.A. 105, 5099–5104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sandberg A., Luheshi L. M., Söllvander S., Pereira de Barros T., Macao B., Knowles T. P., Biverstål H., Lendel C., Ekholm-Petterson F., Dubnovitsky A., Lannfelt L., Dobson C. M., Härd T. (2010) Stabilization of neurotoxic Alzheimer amyloid-β oligomers by protein engineering. Proc. Natl. Acad. Sci. U.S.A. 107, 15595–155600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Santhoshkumar P., Sharma K. K. (2004) Inhibition of amyloid fibrillogenesis and toxicity by a peptide chaperone. Mol. Cell Biochem. 267, 147–155 [DOI] [PubMed] [Google Scholar]
- 11. Narayan P., Orte A., Clarke R. W., Bolognesi B., Hook S., Ganzinger K. A., Meehan S., Wilson M. R., Dobson C. M., Klenerman D. (2012) The extracellular chaperone clusterin sequesters oligomeric forms of the amyloid-β(1–40) peptide. Nat. Struct. Mol. Biol. 19, 79–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wilhelmus M. M., Boelens W. C., Otte-Höller I., Kamps B., Kusters B., Maat-Schieman M. L., de Waal R. M., Verbeek M. M. (2006) Small heat shock protein HspB8: its distribution in Alzheimer's disease brains and its inhibition of amyloid-β protein aggregation and cerebrovascular amyloid-β toxicity. Acta Neuropathol. 111, 139–149 [DOI] [PubMed] [Google Scholar]
- 13. Evans C. G., Wisén S., Gestwicki J. E. (2006) Heat shock proteins 70 and 90 inhibit early stages of amyloid β-(1–42) aggregation in vitro. J. Biol. Chem. 281, 33182–33191 [DOI] [PubMed] [Google Scholar]
- 14. Olzscha H., Schermann S. M., Woerner A. C., Pinkert S., Hecht M. H., Tartaglia G. G., Vendruscolo M., Hayer-Hartl M., Hartl F. U., Vabulas R. M. (2011) Amyloid-like aggregates sequester numerous metastable proteins with essential cellular functions. Cell 144, 67–78 [DOI] [PubMed] [Google Scholar]
- 15. Ferreon A. C., Ferreon J. C., Wright P. E., Deniz A. A. (2013) Modulation of allostery by protein intrinsic disorder. Nature 498, 390–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Milton N. G. (1999) Amyloid-β binds catalase with high affinity and inhibits hydrogen peroxide breakdown. Biochem. J. 344, 293–296 [PMC free article] [PubMed] [Google Scholar]
- 17. Casley C. S., Canevari L., Land J. M., Clark J. B., Sharpe M. A. (2002) β-Amyloid inhibits integrated mitochondrial respiration and key enzyme activities. J. Neurochem. 80, 91–100 [DOI] [PubMed] [Google Scholar]
- 18. Luo J., Wärmländer S. K. T. S., Gräslund A., Abrahams J. P. (2013) Human lysozyme inhibits the in vitro aggregation of Aβ peptides, which in vivo are associated with Alzheimer's disease. Chem. Commun. (Camb.) 49, 6507–6509 [DOI] [PubMed] [Google Scholar]
- 19. Willander H., Presto J., Askarieh G., Biverstål H., Frohm B., Knight S. D., Johansson J., Linse S. (2012) BRICHOS domains efficiently delay fibrillation of amyloid β-peptide. J. Biol. Chem. 287, 31608–31617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Stanyon H. F., Viles J. H. (2012) Human serum albumin can regulate amyloid-β peptide fiber growth in the brain interstitium: implications for Alzheimer disease. J. Biol. Chem. 287, 28163–28168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Luo J., Yu C.-H., Yu H., Borstnar R., Kamerlin S. C., Gräslund A., Abrahams J. P., Wärmländer S. K. (2013) Cellular polyamines promote amyloid-β (aβ) peptide fibrillation and modulate the aggregation pathways. ACS Chem. Neurosci. 4, 454–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Luo J., Mohammed I., Wärmländer S. K., Hiruma Y., Gräslund A., Abrahams J. P. (2014) Endogenous polyamines reduce the toxicity of soluble A β peptide aggregates associated with Alzheimer's disease. Biomacromolecules 15, 1985–1991 [DOI] [PubMed] [Google Scholar]
- 23. Maurer-Stroh S., Debulpaep M., Kuemmerer N., Lopez de la Paz M., Martins I. C., Reumers J., Morris K. L., Copland A., Serpell L., Serrano L., Schymkowitz J. W., Rousseau F. (2010) Exploring the sequence determinants of amyloid structure using position-specific scoring matrices. Nat. Methods 7, 237–242 [DOI] [PubMed] [Google Scholar]
- 24. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. (2004) UCSF Chimera. A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 25. Sluzky V., Tamada J. A., Klibanov A. M., Langer R. (1991) Kinetics of insulin aggregation in aqueous solutions upon agitation in the presence of hydrophobic surfaces. Proc. Natl. Acad. Sci. U.S.A. 88, 9377–9381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Campioni S., Carret G., Jordens S., Nicoud L., Mezzenga R., Riek R. (2014) The presence of an air-water interface affects formation and elongation of α-synuclein fibrils. J. Am. Chem. Soc. 136, 2866–2875 [DOI] [PubMed] [Google Scholar]
- 27. Bolognesi B., Kumita J. R., Barros T. P., Esbjorner E. K., Luheshi L. M., Crowther D. C., Wilson M. R., Dobson C. M., Favrin G., Yerbury J. J. (2010) ANS binding reveals common features of cytotoxic amyloid species. ACS Chem. Biol. 5, 735–740 [DOI] [PubMed] [Google Scholar]
- 28. Glabe C. G. (2008) Structural classification of toxic amyloid oligomers. J. Biol. Chem. 283, 29639–29643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Saio T., Guan X., Rossi P., Economou A., Kalodimos C. G. (2014) Structural basis for protein antiaggregation activity of the trigger factor chaperone. Science 344, 1250494–1250511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Karagöz G. E., Duarte A. M., Akoury E., Ippel H., Biernat J., Morán Luengo T., Radli M., Didenko T., Nordhues B. A., Veprintsev D. B., Dickey C. A., Mandelkow E., Zweckstetter M., Boelens R., Madl T., Rüdiger S. G. (2014) Hsp90-Tau complex reveals molecular basis for specificity in chaperone action. Cell 156, 963–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Van Dael H., Tieghem E., Haezebrouck P., Van Cauwelaert F. (1992) Conformational aspects of the Cu2+ binding to α-lactalbumin: characterization and stability of the Cu-bound state. Biophys. Chem. 42, 235–242 [DOI] [PubMed] [Google Scholar]
- 32. Divsalar A., Damavandi S. E., Saboury A. A., Seyedarabi A., Moosavi-Movahedi A. A. (2012) Calorimetric and spectroscopic investigations of β-lactoglobulin upon interaction with copper ion. J. Dairy Res. 79, 209–215 [DOI] [PubMed] [Google Scholar]
- 33. Laganowsky A., Liu C., Sawaya M. R., Whitelegge J. P., Park J., Zhao M., Pensalfini A., Soriaga A. B., Landau M., Teng P. K., Cascio D., Glabe C., Eisenberg D. (2012) Atomic view of a toxic amyloid small oligomer. Science 335, 1228–1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bigl M., Brückner M. K., Arendt T., Bigl V., Eschrich K. (1999) Activities of key glycolytic enzymes in the brains of patients with Alzheimer's disease. J. Neural Transm. 106, 499–511 [DOI] [PubMed] [Google Scholar]
- 35. Yamamoto K., Shimada H., Koh H., Ataka S., Miki T. (2014) Serum levels of albumin-amyloid β complexes are decreased in Alzheimer's disease. Geriatr. Gerontol. Int. 14, 716–723 [DOI] [PubMed] [Google Scholar]
- 36. Kim Y. E., Hipp M. S., Bracher A., Hayer-Hartl M., Hartl F. U. (2013) Molecular chaperone functions in protein folding and proteostasis. Annu. Rev. Biochem. 82, 323–355 [DOI] [PubMed] [Google Scholar]
- 37. Wyatt A. R., Yerbury J. J., Ecroyd H., Wilson M. R. (2013) Extracellular chaperones and proteostasis. Annu. Rev. Biochem. 82, 295–322 [DOI] [PubMed] [Google Scholar]
- 38. Yerbury J. J., Poon S., Meehan S., Thompson B., Kumita J. R., Dobson C. M., Wilson M. R. (2007) The extracellular chaperone clusterin influences amyloid formation and toxicity by interacting with prefibrillar structures. FASEB J. 21, 2312–2322 [DOI] [PubMed] [Google Scholar]
- 39. Yerbury J. J., Kumita J. R., Meehan S., Dobson C. M., Wilson M. R. (2009) α2-Macroglobulin and haptoglobin suppress amyloid formation by interacting with prefibrillar protein species. J. Biol. Chem. 284, 4246–4254 [DOI] [PubMed] [Google Scholar]
- 40. Fonte V., Kapulkin W. J., Kapulkin V., Taft A., Fluet A., Friedman D., Link C. D. (2002) Interaction of intracellular β amyloid peptide with chaperone proteins. Proc. Natl. Acad. Sci. 99, 9439–9444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Arimon M., Grimminger V., Sanz F., Lashuel H. A. (2008) Hsp104 targets multiple intermediates on the amyloid pathway and suppresses the seeding capacity of Aβ fibrils and protofibrils. J. Mol. Biol. 384, 1157–1173 [DOI] [PubMed] [Google Scholar]
- 42. Milojevic J., Melacini G. (2011) Stoichiometry and affinity of the human serum albumin-Alzheimer's Aβ peptide interactions. Biophys. J. 100, 183–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Milojevic J., Raditsis A., Melacini G. (2009) Human serum albumin inhibits Aβ fibrillization through a “monomer-competitor” mechanism. Biophys. J. 97, 2585–2594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ono K., Takahashi R., Ikeda T., Yamada M. (2012) Cross-seeding effects of amyloid β-protein and α-synuclein. J. Neurochem. 122, 883–890 [DOI] [PubMed] [Google Scholar]
- 45. Clinton L. K., Blurton-Jones M., Myczek K., Trojanowski J. Q., LaFerla F. M. (2010) Synergistic Interactions between Aβ, Tau, and α-synuclein: acceleration of neuropathology and cognitive decline. J. Neurosci. 30, 7281–7289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mandal P. K., Pettegrew J. W., Masliah E., Hamilton R. L., Mandal R. (2006) Interaction between Aβ peptide and α synuclein: molecular mechanisms in overlapping pathology of Alzheimer's and Parkinson's in dementia with Lewy body disease. Neurochem. Res. 31, 1153–1162 [DOI] [PubMed] [Google Scholar]