

Background: Competition between pathogens and their hosts drives the evolution of molecules that give either organism an edge.
Results: Structural and biochemical data show how the parasite pseudokinase ROP5 inhibits the murine GTPase IRGa6.
Conclusion: The surfaces of both ROP5 and IRG proteins that interact in the complex are under strong selective pressure.
Significance: This highlights an extreme case of evolutionary competition.
Keywords: Crystal Structure, GTPase, Innate Immunity, Parasitology, Protein Kinase
Abstract
The Red Queen hypothesis proposes that there is an evolutionary arms race between host and pathogen. One possible example of such a phenomenon could be the recently discovered interaction between host defense proteins known as immunity-related GTPases (IRGs) and a family of rhoptry pseudokinases (ROP5) expressed by the protozoan parasite, Toxoplasma gondii. Mouse IRGs are encoded by an extensive and rapidly evolving family of over 20 genes. Similarly, the ROP5 family is highly polymorphic and consists of 4–10 genes, depending on the strain of Toxoplasma. IRGs are known to be avidly bound and functionally inactivated by ROP5 proteins, but the molecular basis of this interaction/inactivation has not previously been known. Here we show that ROP5 uses a highly polymorphic surface to bind adjacent to the nucleotide-binding domain of an IRG and that this produces a profound allosteric change in the IRG structure. This has two dramatic effects: 1) it prevents oligomerization of the IRG, and 2) it alters the orientation of two threonine residues that are targeted by the Toxoplasma Ser/Thr kinases, ROP17 and ROP18. ROP5s are highly specific in the IRGs that they will bind, and the fact that it is the most highly polymorphic surface of ROP5 that binds the IRG strongly supports the notion that these two protein families are co-evolving in a way predicted by the Red Queen hypothesis.
Introduction
Large GTPases, such as the dynamins, dynamin-related proteins, and immunity-related GTPases (IRGs),4 are characterized by their low affinities for nucleotide and their multimerization-driven cooperativity (1–3). Thus, unlike canonical small GTPases, large GTPases do not require additional partners, such as guanine exchange factors or GTPase-activating proteins (GAPs) to efficiently hydrolyze GTP. This allows large GTPases like dynamin to act independently from other adaptor and regulatory proteins to catalyze membrane fission in vitro (4, 5). Given their similar biochemical properties, it appears likely that the dynamin-related proteins and IRGs function analogously.
The IRGs are a critical component of the cell-autonomous immune response against intracellular pathogens in vertebrates. IRGs recognize, assemble on, and lead to the clearance of foreign membranes, although the mechanism is poorly understood (6). As is typical for molecules at the interface of the host-pathogen interaction, the IRGs are fast evolving (7), probably as a result of their competition with the pathogens they must control. In fact, specific alleles of IRG have recently been shown to protect mice against otherwise virulent strains of the parasite Toxoplasma gondii (8). T. gondii, which infects an estimated one-third of the world's human population, secretes the ROP5 family of pseudokinases that competitively inhibits IRG oligomerization (9, 10) and two active kinases, ROP17 and ROP18, that phosphorylate and permanently inhibit the IRGs (11–13). The ROP5 proteins are a family of closely related, catalytically inactive kinases, or pseudokinases, that are critical for Toxoplasma virulence (14, 15). Each strain of the parasite encodes multiple, divergent copies of the ROP5 genes (14, 15), and allelic variation is concentrated in hotspots of positive selection in the ROP5 pseudokinase domains (14). These differences are the greatest identified determinant of disease outcome between strains and are responsible for a >105-fold difference in virulence in a mouse model of disease (14, 15).
Pseudokinases, such as ROP5, are proteins with a kinase fold that have substitutions at key residues that make them unable to efficiently catalyze phosphoryl transfer. Pseudokinases are emerging as key regulators of cellular signaling (16) and often serve as scaffolds to organize signaling complexes (17–20) but not to amplify a signal, as an active kinase might. Pseudokinases have evolved independently from different lineages in eukaryotes, so their functions and mechanisms of action are diverse; the known structures of pseudokinases in complex with their partner proteins (17–21) have revealed a wide variety of binding modes that are often distinct from the interactions typical of an active kinase with its substrate and regulatory proteins.
ROP5 and the active Toxoplasma kinase ROP18 exhibit epistasis in their genetic effects on virulence (14), and cellular data demonstrate that ROP5, ROP17, and ROP18 are required together to inhibit IRG activation in vivo (9, 10, 22, 13). This appears to be due, at least in part, to the inhibition of IRG oligomerization by ROP5 (9, 10). Here, we present the crystal structures of two alleles of ROP5 in complex with the murine GTPase IRGa6. The structures reveal not only that ROP5 uses an unusual, polymorphic binding surface to bind IRGa6 but also that ROP5 binding induces an allosteric rearrangement of the IRGa6 active site. This has implications for the competition of Toxoplasma with the IRGs, which are, themselves, polymorphic in the region ROP5 recognizes.
EXPERIMENTAL PROCEDURES
Mutagenesis
Mutant ROP5CI was generated using the Phusion (New England Biolabs) mutagenesis protocol with forward primer (5′-TGTACACCTCTGCCTGACTTCGTG-3′) and either reverse primer G490D (5′-TGAGTCGAATGCCAGACTGTCAGTCCC-3′) or G490F/S491P (5′-TGGGAAGAATGCCAGACTGTCAGTCCCTGG-3′).
Protein Expression and Purification
All recombinant protein was expressed in E. coli Rosetta2(DE3) (EMD Biosciences). ROP5BI and ROP5CI were affinity-purified on nickel-nitrilotriacetic acid resin (Qiagen), and the His6 tag was removed by overnight thrombin (Hematologic Technologies) cleavage at 4 °C. GST-IRGa6 was affinity-purified on glutathione-Sepharose (GE Healthcare) and eluted by on-bead thrombin cleavage overnight at 4 °C. All proteins were further purified by anion exchange and size exclusion chromatography.
Crystallization
Crystals of ROP5-IRGa6 grew at a wide range of pH (5 - 9) and PEG molecular weights. ROP5-IRGa6 complex was formed by adding equimolar amounts of individually purified ROP5 and IRGa6 in 10 mm HEPES, pH 7, 100 mm NaCl, 10 mm DTT. High quality crystals of ROP5BI-IRGa6 were grown at 18 °C by mixing equal volumes of protein solution (125 μm complex), nucleotide solution (10 mm MgCl2, 1 mm ADP, 1 mm GDP, 10 mm DTT), and a reservoir containing 20% PEG 3350, 0.1 m NH4 citrate, pH 7.0. Similarly, high quality crystals of ROP5CI-IRGa6 were grown using a reservoir containing 12% PEG 20,000, 0.1 m MES, pH 6.0. Crystals were flash-frozen in a cryoprotectant of mother liquor with 25% (ROP5BI-IRGa6) or 30% (ROP5CI-IRGa6) ethylene glycol.
Data Collection, Structure Determination, and Refinement
The diffraction data were collected at beamline 11.1 of the Stanford Synchrotron Radiation Laboratory at a wavelength of 0.979 Å and a temperature of 100 K. Indexing, integration, and scaling of the diffraction data were performed using the XDS suite (23). Because both ROP5BI-IRGa6 and ROP5CI-IRGa6 crystallized in the low symmetry space group P1, data sets from multiple crystals were merged for each complex (see Table 1). Initial phases for the two complexes were obtained by molecular replacement using Phaser (24) with the structures of ROP5BI (3Q60) and IRGa6-GDP (1TPZ) used as initial search models. Probably due to the conformational changes we eventually discovered in the structure, molecular replacement was only successful by first searching for ROP5, followed by IRGa6. Manual rebuilding in Coot (25) and refinement in Phenix (26) led to a final 1.70 Å structure of ROP5BI-IRGa6 and a 1.72 Å structure of ROP5CI-IRGa6, which were deposited in the Protein Data Bank (accession numbers 4LV5 and 4LV8, respectively). Both structures show good stereochemistry (97.1 and 97.4% favored for ROP5BI-IRGa6 and ROP5CI-IRGa6, respectively) from Ramachandran plots, as validated by the program MOLPROBITY (27).
TABLE 1.
Crystallographic information
Values in parentheses are for the highest resolution shell.
| ROP5BI-IRGa6-GDPa | ROP5CI-IRGa6-GDPb | |
|---|---|---|
| Data collection | ||
| Space group | P 1 | P 1 |
| Cell dimensions | ||
| a, b, c (Å) | 52.6, 54.7, 85.6 | 53.4, 53.7, 86.8 |
| α, β, γ (degrees) | 99.0, 106.6, 106.9 | 104.7, 95.3, 111.0 |
| Resolution (Å) | 1.70 Å | 1.72 Å |
| Rmerge | 5.8 (183.1) | 9.2 (202.8) |
| I/σI | 19.19 (1.15) | 19.43 (1.52) |
| CC(1/2) (52) | 100.0 (53.7) | 100.0 (65.9) |
| Completeness (%) | 99.5 (99.6) | 99.9 (99.9) |
| Redundancy | 8.07 (7.61) | 13.8 (13.7) |
| Refinement | ||
| Resolution (Å) | 1.70 | 1.72 |
| No. of reflections | 92,476 | 90,512 |
| Rwork/Rfree | 0.1923/0.2277 | 0.1951/0.2298 |
| No. of atoms | 6647 | 6676 |
| Protein | 6081 | 6065 |
| Ligand/ion | 83 | 96 |
| Water | 483 | 515 |
| B-factors | 38.00 | 35.90 |
| Protein | 37.50 | 35.50 |
| Ligand/ion | 31.52 | 38.32 |
| Water | 44.80 | 41.10 |
| RMSD | ||
| Bond lengths (Å) | 0.008 | 0.007 |
| Bond angles (degrees) | 1.106 | 1.128 |
a Three independent data sets were merged.
b Four independent data sets were merged.
Interface Analysis and Figure Generation
The interface residues listed in Table 2 were determined using NCONT in CCP4 (28) with a 4 Å cut-off. Polymorphisms were analyzed with DnaSP version 5 (29). Figures were rendered using PyMOL (Schrödinger, LLC, New York).
TABLE 2.
Interface contacts
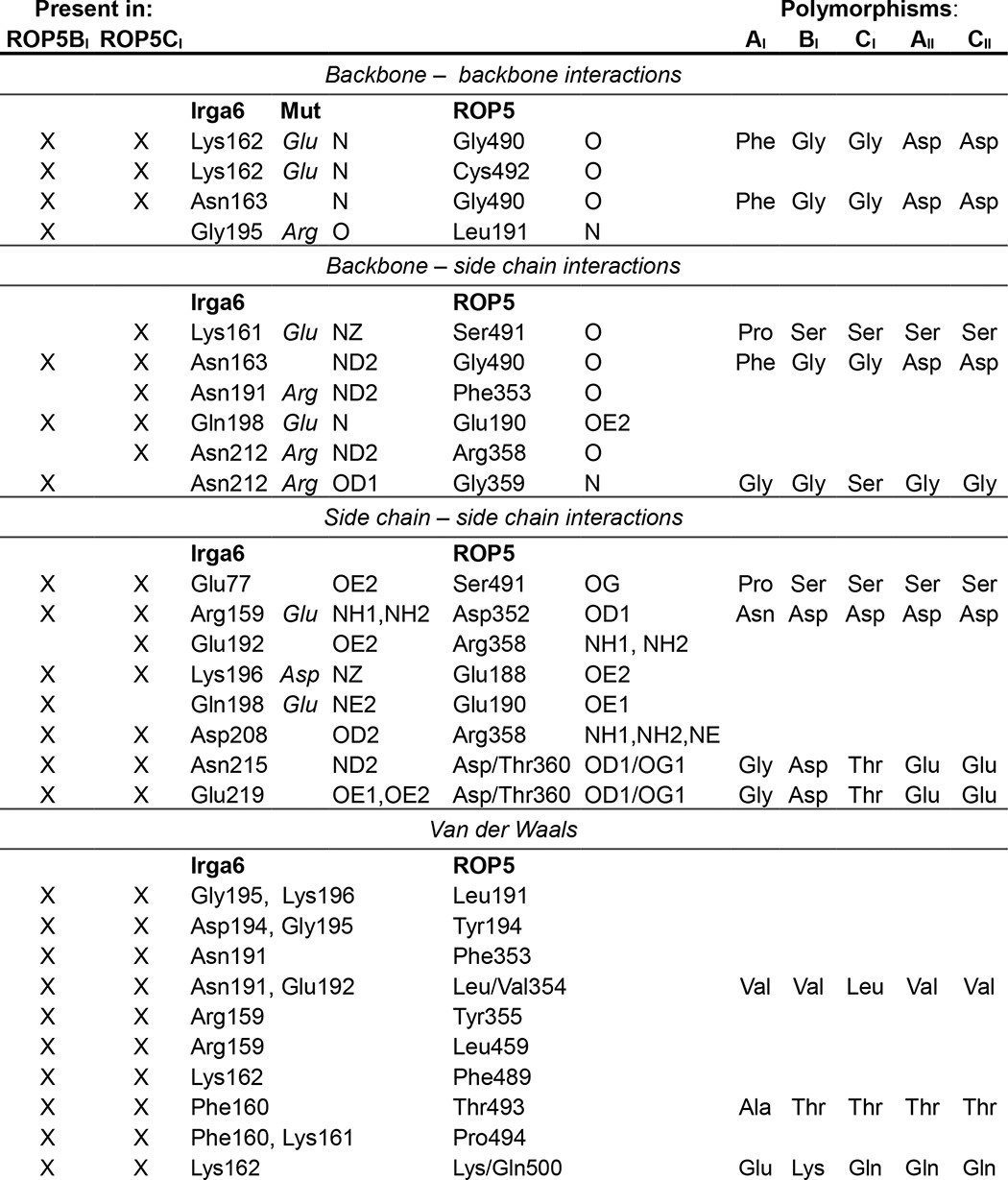
Interface residues from ROP5 and IRGa6 that directly contact the other protein (within 4 Å) are listed. An “X” indicates that the interaction is present in the structure of IRGa6 in complex with ROP5BI and/or ROP5CI. Polymorphisms are noted in the right-hand columns. Mutations in IRGa6 that were previously shown to reduce ROP5 binding (9) are indicated in the “Mut” column.
In Vitro GTPase Activity Assay
10–20 μm IRGa6 was incubated at 37 °C with 2 mm GTP (Sigma) in reaction buffer (10 mm HEPES, pH 7.4, 100 mm KCl, 10 mm MgCl2, 10 mm DTT) either with or without the addition of ROP5. All protein was of crystallographic purity. Individual 5-μl reactions were initiated at time points between 0 and 40 min such that all reactions could be halted simultaneously with the addition of 100 μl of BIOMOL Green (Enzo) to measure free inorganic phosphate according to the manufacturer's instructions. A standard curve was measured with each independent experiment.
IRG Oligomerization
Oligomerization of IRGa6 was measured by an increase in turbidity at A400 and was carried out essentially as described previously (30). Briefly, oligomerization of 60 μm IRGa6 alone or in the presence of 100 μm wild-type or mutant ROP5CI was initiated by the addition of 5 mm GTP. Assays were conducted at 25 °C in 20 mm HEPES, pH 7.0, 100 mm NaCl, 10 mm MgCl2, 10 mm DTT.
RESULTS
The ROP5-IRGa6 Interface Is Distal from the Protein Active Sites
Previous work demonstrated that ROP5 can physically interact with and inhibit the IRG system in vivo (9, 10, 22). Although the ROP5-IRGa6 interface was partially mapped (9), the analysis was based on limited mutagenesis and low resolution NMR data. To determine the molecular mechanism of ROP5 inhibition of the IRGs at atomic resolution, we solved the crystal structure of murine IRGa6 in complex with the pseudokinase domains of each of two allelic isoforms of ROP5, ROP5BI and ROP5CI, at 1.70 and 1.72 Å resolution, respectively (Fig. 1 and Table 1). The two alleles of ROP5 bind IRGa6 in essentially the same orientations (Fig. 1B).
FIGURE 1.
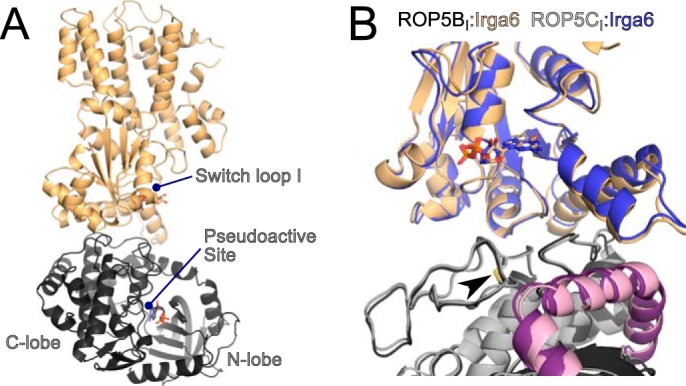
ROP5BI and ROP5CI bind IRGa6 in a similar conformation distal from the pseudokinase and GTPase active sites. A, the structure of IRGa6 (orange) bound to ROP5BI (gray) is shown with their respective bound nucleotides shown in sticks. B, an overlay of the two structures near the ROP5-IRGa6 interface is shown. Cα RMSD for the complex is 1.6 Å over 754 atom pairs, and the Cα RMSD values for the individual IRGa6 and ROP5 chains are 1.1 Å over 398 atom pairs and 0.9 Å over 356 atom pairs, respectively. The ROP5 N-terminal extension is highlighted in pink, and the conserved disulfide bridge (Cys-458/492) that stabilizes the loop in ROP5 that interacts with IRGa6 helix 3 is indicated with an arrowhead.
As suggested by previously published NMR data (9), the region of ROP5 that binds IRGa6 is on the opposite face of the pseudokinase domain from the activation loop and substrate recognition site (Fig. 1A). This is consistent with the idea that pseudokinases have evolved away from catalysis to adopt roles such as protein-protein interaction (16, 31, 32). Instead, the region of ROP5 that binds IRGa6 is the face sometimes recognized by specific kinase regulatory proteins (33). Specificity at this surface is achieved by discrimination of three-dimensional structure rather than simply primary sequence (as is typical in substrate recognition). For ROP5, the IRG-binding surface includes two structural elements that define the parasite-specific family of kinases to which ROP5 belongs (32, 34): a loop rigidified by a conserved disulfide bridge and an N-terminal helical extension nested between the N- and C-lobes of the kinase domain (Fig. 1B).
Although IRGa6 adopts slightly different conformations to interact with the two ROP5 isoforms, it retains an identical active site conformation in the two structures (Fig. 1B). The surface of IRGa6 that interacts with ROP5 is a face near but not including the GTPase active site. This surface of IRGa6 does, however, contain a number of highly conserved residues that have been implicated in IRG activation and oligomerization through mutagenesis studies (9, 30) (Fig. 2 and Table 2). Importantly, several of these IRG residues have been previously mutated and shown to disrupt ROP5 binding (Table 2). These data are consistent with the proposed model that ROP5 competitively inhibits IRG oligomerization by sterically occluding its multimerization (9).
FIGURE 2.
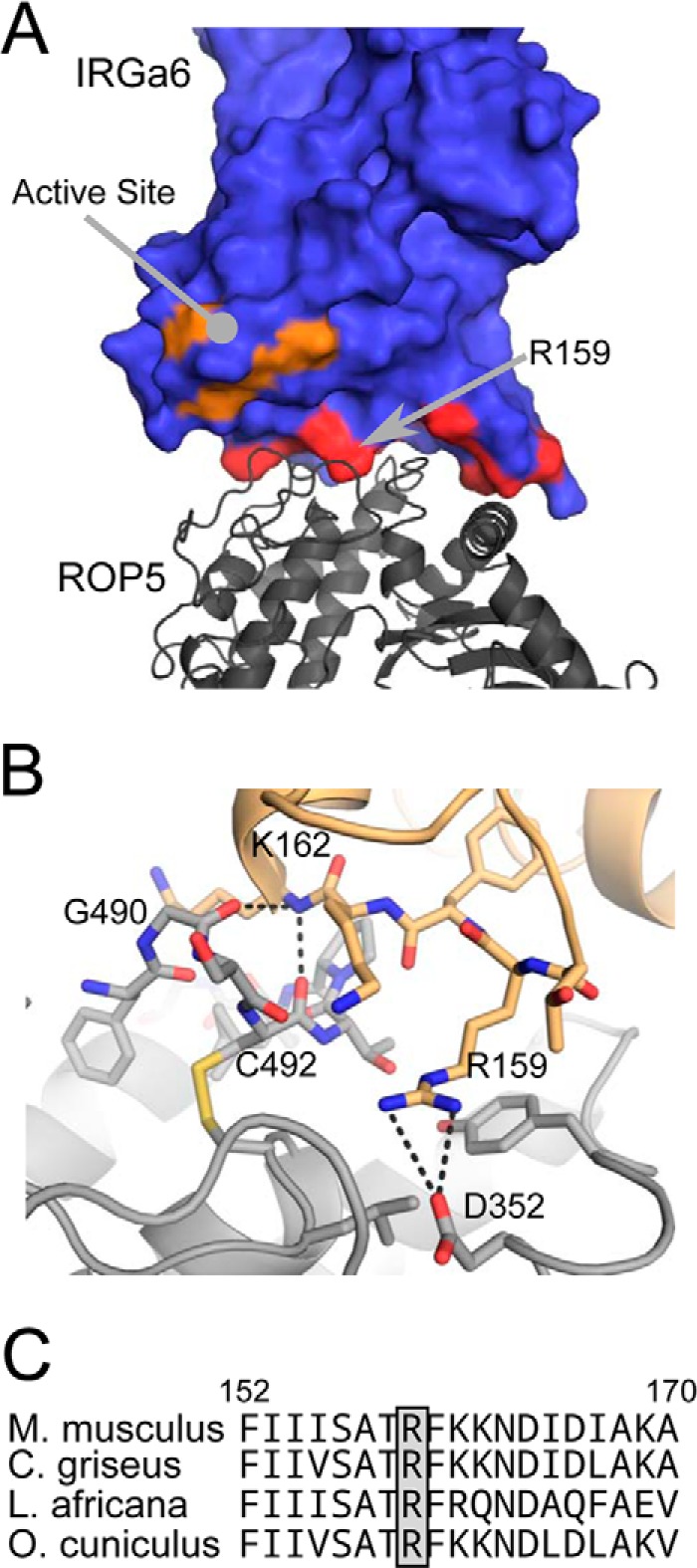
ROP5 binds a region of IRG required for oligomerization that is distinct from the GTPase active site. A, the structure of ROP5-IRGa6 is shown with IRGa6 as a surface and ROP5 as a gray schematic. IRGa6 residues whose mutation disrupts oligomerization are highlighted in red and orange (such residues in the active site are orange). B, contacts between ROP5 (gray) and IRGa6 (orange) in the region around IRGa6 helix 3 and Arg-159 are highlighted. C, an alignment of sequences surrounding the conserved Arg-159 in IRGa6 from diverse mammalian species (mouse, Chinese hamster, African elephant, and rabbit) are shown.
ROP5 Binding Alters the Conformation of the IRGa6 Active Site
NMR data previously indicated that ROP5 binding to IRGa6 alters the conformation of the GTPase active site (9), although the extent of these changes was unknown. Consistent with this observation, the IRGa6 switch loop adopts a helical conformation in ROP5-IRGa6-GDP that differs radically from the conformation in the IRGa6-GDP structure (35) (Fig. 3). Unlike other proteins that modulate GTPase active site conformation, such as RhoGAPs (36), RhoGDIs (37), or GPR/GoLoco motif proteins (38), ROP5 makes direct contacts neither with the switch loops of its target GTPase nor with the IRGa6-bound nucleotide. ROP5 binding does, however, cause significant conformational changes throughout IRGa6 (Fig. 3). A major change occurs near IRGa6 helix 3, which exhibits ∼5-Å translation of the helix base (residue 162) as well as ∼20° rotation upon ROP5 binding. This necessitates a structural rearrangement within the GTPase active site, in which catalytic Lys-82 swaps an interaction with the GDP β-phosphate for a salt bridge with Asp-106 in the switch loop (Fig. 4, A and B). The residues that coordinate Mg2+ are also rearranged upon ROP5 binding, apparently excluding a highly ordered Mg2+ from the active site and binding two water molecules in its place (Figs. 3C and 4, A and B). We cannot, however, rule out that Mg2+ is somehow involved in binding, although we see no evidence of an ordered cation in these structures.
FIGURE 3.

ROP5 binding induces conformational changes throughout the IRGa6 structure. A, the ROP5-bound structure of IRG is shown colored according to Cα displacement compared with the published ROP5-unbound IRGa6-GDP structure (Protein Data Bank code 1TPZ). B, the ROP5-bound structure of IRGa6 (orange) is overlaid on the published IRGa6-GDP structure (Protein Data Bank code 1TPZ). In both panels, helix 3 is indicated with a closed arrowhead, and switch loop I is indicated with an open arrowhead. Cα RMSD of IRGa6 between the two structures is 2.1 Å over 399 atom pairs (2.6 Å over the G-domain alone; 166 atom pairs). C, stereo image of switch loop I and the bound GDP in the ROP5-IRGa6 structure superposed with the 2Fo − Fc electron density map countered at 2.0σ.
FIGURE 4.

ROP5 inhibits IRGa6 GTPase activity. The active sites of IRGa6-GDP (A) and ROP5-IRGa6-GDP (B) are shown with residues that coordinate GDP, associated waters, and Mg2+. C, 20 μm IRGa6 was incubated with GTP in either the presence or absence of 150 μm ROP5CI, and released phosphate was measured over time. Error bars, S.D. (n = 3). D, 10 μm IRGa6 was incubated with 2 mm GTP and varying concentrations of either ROP5BI or ROP5CI for 40 min, phosphate release was measured, and activities were normalized to IRGa6 activity without ROP5. Error bars, errors in slope by GraphPad Prism (n = 3 per data point).
The ROP5 partner kinases, ROP17 and ROP18, phosphorylate Thr-102 and Thr-108 on the IRG switch loop I (11–13). Although ROP5 does not directly contact the switch loop, the closest pseudokinase residue is ∼15 Å away, suggesting that an additional conformational change may be required to allow the kinases simultaneous access. Alternatively, the kinases may be in dynamic exchange with the pseudokinase as they continuously inactivate each successive IRG protein that targets the parasitophorous vacuolar membrane. Further work is necessary to differentiate these two models.
ROP5 Binding Inhibits IRG GTPase Activity
Structural changes in these key regions motivated us to ask whether ROP5 stabilizes an active or off-pathway conformation of IRG. Previous work has led to the model that IRG proteins must multimerize to efficiently turnover GTP (3, 30), complicating the dissection of inhibition of oligomerization and activity. Because ROP5 binding blocks IRG oligomerization in vivo and in vitro (9), ROP5-IRGa6 would be expected to be catalytically inhibited. We tested IRGa6 GTPase activity in the presence and absence of ROP5. ROP5CI strongly inhibits IRGa6 catalytic activity (Fig. 4C), whereas ROP5BI is a less efficient inhibitor (Fig. 4D). It is important to note, however, that both isoforms of ROP5 are able to inhibit IRG activation in vivo (9, 10), suggesting that this difference in in vitro potency may be of limited significance in vivo.
Polymorphic Residues Cluster in the ROP5-IRG Interface
To determine whether ROP5 binding to IRG may be driving the evolution of either molecule, we first used IRG sequences from diverse, non-laboratory-derived mouse strains (8) to map the sequence variation of three IRG proteins (IRGa6, IRGb6, and IRGb2) on our crystal structure of the ROP5-IRGa6 complex (Fig. 5). IRGa6 appears to be under purifying selection across the sequences examined (Tajima's π(a)/π(s) = 0.18), with few non-synonomous substitutions. Other IRG family members, including IRGb6 and IRGb2, are highly substituted both in the ROP5 binding site and in switch loop I, which is the location of the threonine residues targeted for phosphorylation by the active kinases ROP17 and ROP18 (11–13). IRGb6 is a second major target of ROP5 inhibition (9, 13). IRGb2 has been shown to act as a protector of the IRG system from Toxoplasma inhibition, possibly by sequestering ROP5 from interaction with its functional targets, such as IRGa6 and IRGb6 (8).
FIGURE 5.

A schematic representation of the IRGa6-ROP5 complex with polymorphic residues shaded according to their level of substitution in each of three IRG family members.
IRGb6 is highly polymorphic, and many of these polymorphisms occur at the ROP5 interface or border the switch loop phosphosites targeted by ROP17 and ROP18 (Fig. 5). Given the high level of polymorphism throughout IRGb6, it is not clear that the polymorphic regions near the ROP5 interface are overrepresented. As predicted by Lilue et al. (8), however, IRGb2 shows a strong enrichment for substitutions near the ROP5 interface, suggesting that there is indeed some evolutionary pressure on this IRG that may result from its interaction with ROP5.
Like the IRGs, ROP5 is encoded by an expanded locus that harbors 4–10 genes in each of the three major Toxoplasma strains (14). Mapping of the ROP5 polymorphisms on the complex structure demonstrated that ROP5 uses a highly polymorphic surface to recognize IRG; 24 of 51 residues that differ between alleles (14, 39) of ROP5 are located on this surface (Fig. 6). Whereas ROP5BI and ROP5CI inhibit murine IRG activation in vivo, ROP5AI, ROP5AII, and ROP5CII do not (9, 10, 22). Consistent with this observation, the IRG-binding interfaces of ROP5BI and ROP5CI are highly substituted when compared with alleles unable to inhibit IRG (Fig. 6, B–D). It should be noted, however, that alleles unable to inactivate the IRG system in breeds of mice tested to date still have a substantial number of substitutions in the IRG binding interface. For example, this region of ROP5AI differs at 16 residues from ROP5AII (Fig. 6, C and D), suggesting that these alleles still experienced positive selective pressure on the sequence that composes this surface. Thus, these ROP5 alleles may be important inhibitors of IRG in another genetic context or may use the same surface to bind other targets.
FIGURE 6.
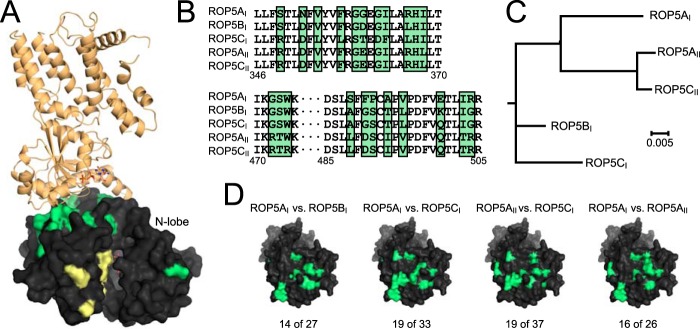
ROP5 uses a polymorphic surface to bind IRGa6. A, overview of the ROP5-IRGa6 structure. ROP5 is shown as a black surface with polymorphic residues shaded green or yellow (the latter are in the pseudoactive site). IRGa6 is shown in orange. B, an alignment of regions of ROP5 sequence that are hotspots of polymorphism (green shading) and involved in the interaction with IRGa6. C, a phylogenetic tree of five ROP5 alleles in which all residues not located at the interface with IRGa6 were held constant. D, pairwise comparison of different ROP5 alleles reveals that all alleles of ROP5 appear to have experienced diversifying selective pressure on the interface with IRGa6 (polymorphic interface residues shaded green; note that the orientation of ROP5 shown in A has been rotated to show the full binding surface in D). The numbers of polymorphic residues at the IRGa6 interface versus in the entire sequence are indicated.
These varying patterns of substitutions on the IRG proteins as compared with ROP5 highlight the crux of the host-pathogen evolutionary arms race: whereas a pathogen's effector often evolves with a singular function (to inhibit a host process), the host target protein must evolve not only to evade inhibition by the pathogen but also to maintain its critical function. This may help to explain why the IRG family has expanded to >20 members in many non-primate mammals but with apparently little functional redundancy (7).
The ROP5-IRGa6 interface is relatively small, burying only ∼960 Å2 of surface area. Consistent with this small surface area, few residues form direct side chain-side chain interactions between the two proteins (Table 2 and Fig. 2B). Although 24 of the residues in the ROP5 pseudokinase domain that differ between alleles are clustered near the surface involved in recognizing IRGa6, only 8 of these residues make direct contacts with IRGa6. This suggests that many of these polymorphisms may have provided a stabilizing or long distance tuning of the three-dimensional surface. Alternatively, these polymorphic residues may be involved in initiating the recognition of IRG, whereas the GTPase is in a conformation not captured in our structure. Those polymorphic ROP5 residues that do contact IRGa6 are largely located in and around a loop containing Cys-492 that is rigidified by a conserved disulfide bridge. Several of the ROP5 residues in this region form backbone hydrogen bonds with the loop at the base of IRGa6 helix 3 (Table 2 and Fig. 2B). Just proximal to this interaction, IRGa6 Arg-159 is buried in a pocket in ROP5 that is capped by Asp-352 (Fig. 2B). Consistent with this, a charge swap mutation of Arg-159 has previously been shown to disrupt ROP5 binding (9). Intriguingly, Arg-159 is well conserved in IRG family members and among species (Fig. 2C) and appears to be required for IRGa6 function (30).
Given the patterns of polymorphism observed, we next asked what effect the polymorphic residues in ROP5CI might have on ROP5 inhibition of IRGa6 activity. Two of the most polymorphic positions in ROP5, Gly-490 and Ser-491, contact the base of IRGa6 helix 3, and those positions in IRGa6 have been previously shown to disrupt ROP5-binding (9) (summarized in Table 2). We expressed ROP5CI protein that had been mutated to reflect the substitution from either the ROP5AI (G490F/S491P) or the type II allele (G490D) at these positions (Fig. 7). These alleles of ROP5 do not inhibit IRG in vivo (9, 10, 22). Given the nonconservative nature of these mutations, they would be predicted to clash with IRGa6 binding according to our structure (Fig. 7A). Both of the mutants expressed well and were soluble to similar concentrations as wild-type protein, suggesting that they folded correctly. Although wild-type ROP5CI was able to inhibit both oligomerization and GTPase activity of IRGa6, neither of the mutants showed any inhibition (Fig. 7, B and C). These data are consistent with a model where these residues form critical contacts with IRGa6 helix 3.
FIGURE 7.

Polymorphisms determine ROP5 inhibition of IRGa6. A, the predicted structural consequences of mutating ROP5CI (black) at polymorphic residues that interact with IRG (orange) helix 3 are shown shaded. In particular, mutation of Gly-490 is predicted to clash with Asn-163 in IRG helix 3 and with the short helical stretch at IRG residues 129–132. B, oligomerization of IRGa6 was monitored after the addition of GTP in the presence or absence of wild-type or mutant ROP5CI. C, IRGa6 GTPase activity was measured, as in Fig. 4, in the presence 150 μm wild-type or mutant ROP5CI. Error bars, S.D.
In comparing the ROP5-bound structure of IRGa6 with previously published structures, we observed that the structure of IRGa6M173A (Protein Data Bank entry 1TQ6) was in a similar conformation as our ROP5-IRGa6 complex structures. This mutant was originally designed to destabilize an observed crystallographic dimer interface to test the interface's importance for oligomerization (35). Although the conformational differences seen in this structure were originally thought to be due to crystallographic contacts, when we examined the structure more closely, we observed helical rotations, switch loop, and active site residue conformation similar to those we describe here for wild-type IRGa6 when bound to ROP5 (Fig. 8). Met-173 sits on the terminus of helix 3, which may form part of the allosteric switch that regulates the inactivating conformational change upon ROP5 binding (Fig. 3). Consistent with this idea, IRGa6M173A was reported to have decreased catalytic activity (35). Taken together with the published structure of IRGa6M173A, our data suggest that IRGa6M173A samples the conformation that IRGa6 occupies when bound to ROP5, with a similar biochemical outcome. Furthermore, these similarities strengthen the idea that the ROP5-bound conformation of IRGa6 is an off-pathway, inhibited conformation.
FIGURE 8.

IRGa6M173A occupies a conformation similar to that of ROP5-bound wild-type IRGa6. The previously published structure (Protein Data Bank code 1TQ6; green) of the mutant M173A Irga6 bound to GMPPNP (GNP) is overlaid with the published structure of Irga6-GDP (Protein Data Bank code 1TPZ; gray) (Cα RMSD of 2.5 Å over 393 atom pairs) (A) or ROP5-bound Irga6 (orange) (Cα RMSD of 1.9 Å over 383 atoms) (B). Closed arrowhead, helix 3; open arrowhead, switch loop I. Additional helical rotations are indicated with a small closed gray arrowhead. C, the IRGa6 active site of 1TQ6 is overlaid with that of the ROP5-bound structure. Residues that coordinate the nucleotide and associated waters are shown as sticks. D, the published structure factors for 1TQ6 were used to calculate the 2.7 Å 2Fo − Fc map, which is shown contoured at 1.5σ around the bound nucleotide. Note that although the 1TQ6 coordinates include a bound Mg2+, there is no density at the ion's position, nor is there evidence for the octahedral coordination typical of a Mg2+. This suggests that the conformation of M173A IRGa6 captured in this structure, like ROP5-bound IRGa6, excludes a highly ordered Mg2+ from the active site.
DISCUSSION
We have demonstrated that ROP5 is an inhibitor of IRG that blocks its oligomerization and thus its GTPase activity, through a mechanism that is revealed by our crystal structures. ROP5 binds a conserved surface of IRGa6 that is required for oligomerization (30). ROP5 binding induces the rearrangement of the IRGa6 active site, suggesting that it allosterically stabilizes a less active conformation of IRGa6.
Whereas the IRG GTPase subdomain has a Rho family fold, ROP5 binds a surface that is not shared by canonical Rho family regulators. Whereas RhoGDIs and RhoGAPs bind directly to their target GTPase switch loops (36, 37), ROP5 binds distal from both the IRGa6 switch loops and active site. In addition, Rho-GDI binding to its GTPase buries an unstructured region of the GDI, whereas ROP5 binds IRGa6 using a well folded pseudokinase domain whose conformation is unchanged upon binding. This is a striking evolutionary adaptation in which a normally catalytic kinase fold has evolved to recognize and inhibit an enzyme through a mechanism not related to catalysis.
The existence of this inhibited conformation of IRGa6 and the allosteric switch that ROP5 exploits to trigger it suggests that native (mammalian) inhibitors also exist to regulate these proteins. Indeed, the IRGM proteins, which act as critical negative regulators of the IRG system, have been proposed to act as GDIs (40, 41), although this has not been tested experimentally.
One of the primary mechanisms by which GDIs for small G proteins (e.g. RhoGDIs) regulate their target G-proteins is by solubilizing prenyl groups on the GTPase, thus stripping the GTPase from the membrane and altering its subcellular localization (42). Many of the IRGs, including IRGa6, are myristoylated in vivo, raising the question of whether binding to ROP5 may influence the myristoyl accessibility. Because the ROP5-IRGa6 crystal structures were solved with bacterially expressed protein that is not myristoylated, the ROP5-IRGa6 complex can clearly be formed in the absence of myristoylation. It should be noted that ROP5 has been shown to interact not only with IRGa6, which is natively myristoylated, but also with the unmyristoylated IRGb6 (9); thus, any potential interaction with a myristoyl group is not likely to be the primary determinant of binding. The N terminus of IRGa6 is on the opposite side of the protein from the ROP5-binding site (ROP5 is ∼65 Å away from IRGa6 residue 14, the most N-terminal residue that is ordered in our structure), which also makes it unlikely that an N-terminal myristoyl could reach the ROP5 pseudokinase domain. A more likely scenario for modification of myristoyl accessibility due to ROP5 binding would be that ROP5 stabilizes an IRGa6 conformation that allows the burying of the myristoylated N terminus in IRGa6, itself, as is seen in Arf1 (43). Alternatively, the myristoyl group might be recognized by an additional ROP5 domain that is not present in our crystal structure. Regardless, the lack of myristoylation in these recombinant proteins prevents a deeper investigation of these hypotheses.
Large GTPases, including dynamins and the IRGs, exhibit cooperative GTPase activity that is mediated by intermolecular activation through oligomerization. As such, regulation of these proteins is thought to be based largely on regulation of oligomerization rather than direct modulation of GTPase conformation and localization by effector proteins. This paradigm is directly challenged by the report that the phox homology domain of mammalian phospholipase D is able to directly stimulate dynamin GTPase activity (44). Regulatory partners for small GTPases alter both catalytic activity and subcellular localization (42, 45). Targeting and timing of large GTPase oligomerization at the correct membrane is critical to their function. This is particularly true of the IRG system, which must discriminate between “foreign” and “self” membranes as it forms a heterogeneous assembly containing multiple families of large GTPases (46, 47) as well as recruiting autophagy (48, 49), oxidative (49), and inflammasome (50) machinery. Because targeting to this assembly requires functional GTPase domains (30, 40, 51), regulation of these immune GTPases by binding partners could tune the specific GTPase family members present and the downstream effects that they potentiate.
IRGM, a negative regulator of the system, has been suggested as a mark of “self” membranes, protecting them from destruction by the IRG system (40, 41). Intriguingly, the surface on IRGa6 that ROP5 binds is required both for IRG activation and for interaction with IRGM (30). It is therefore attractive to propose that the parasite pseudokinase ROP5 evolved to act as an allosteric molecular mimic of IRGM to inhibit the IRG system, thus disguising the Toxoplasma vacuolar membrane as self and enabling the parasite to efficiently evade clearance by this cell-autonomous mechanism.
Acknowledgments
We are grateful to Jonathan Howard and Elliott Ross for invaluable discussions and to Felice Kelly and Eva LaDow for critical review of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant AI73756.
The atomic coordinates and structure factors (codes 4LV5 and 4LV8) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- IRG
- immunity-related GTPase
- GAP
- GTPase-activating protein
- GDI
- GDP dissociation inhibitor
- RMSD
- root mean square deviation
- GMPPNP
- 5′-guanylyl imidodiphosphate.
REFERENCES
- 1. Chappie J. S., Acharya S., Leonard M., Schmid S. L., Dyda F. (2010) G domain dimerization controls dynamin's assembly-stimulated GTPase activity. Nature 465, 435–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gasper R., Meyer S., Gotthardt K., Sirajuddin M., Wittinghofer A. (2009) It takes two to tango: regulation of G proteins by dimerization. Nat. Rev. Mol. Cell Biol. 10, 423–429 [DOI] [PubMed] [Google Scholar]
- 3. Uthaiah R. C., Praefcke G. J. K., Howard J. C., Herrmann C. (2003) IIGP1, an interferon-γ-inducible 47-kDa GTPase of the mouse, showing cooperative enzymatic activity and GTP-dependent multimerization. J. Biol. Chem. 278, 29336–29343 [DOI] [PubMed] [Google Scholar]
- 4. Bashkirov P. V., Akimov S. A., Evseev A. I., Schmid S. L., Zimmerberg J., Frolov V. A. (2008) GTPase cycle of dynamin is coupled to membrane squeeze and release, leading to spontaneous fission. Cell 135, 1276–1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pucadyil T. J., Schmid S. L. (2008) Real-time visualization of dynamin-catalyzed membrane fission and vesicle release. Cell 135, 1263–1275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kim B.-H., Shenoy A. R., Kumar P., Bradfield C. J., MacMicking J. D. (2012) IFN-inducible GTPases in host cell defense. Cell Host Microbe 12, 432–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hunn J. P., Feng C. G., Sher A., Howard J. C. (2011) The immunity-related GTPases in mammals: a fast-evolving cell-autonomous resistance system against intracellular pathogens. Mamm. Genome 22, 43–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lilue J., Müller U. B., Steinfeldt T., Howard J. C. (2013) Reciprocal virulence and resistance polymorphism in the relationship between Toxoplasma gondii and the house mouse. eLife 2, e01298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fleckenstein M. C., Reese M. L., Könen-Waisman S., Boothroyd J. C., Howard J. C., Steinfeldt T. (2012) A Toxoplasma gondii pseudokinase inhibits host IRG resistance proteins. PLoS Biol. 10, e1001358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Niedelman W., Gold D. A., Rosowski E. E., Sprokholt J. K., Lim D., Farid Arenas A., Melo M. B., Spooner E., Yaffe M. B., Saeij J. P. J. (2012) The rhoptry proteins ROP18 and ROP5 mediate Toxoplasma gondii evasion of the murine, but not the human, interferon-γ response. PLoS Pathog. 8, e1002784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Steinfeldt T., Könen-Waisman S., Tong L., Pawlowski N., Lamkemeyer T., Sibley L. D., Hunn J. P., Howard J. C. (2010) Phosphorylation of mouse immunity-related GTPase (IRG) resistance proteins is an evasion strategy for virulent Toxoplasma gondii. PLoS Biol. 8, e1000576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fentress S. J., Behnke M. S., Dunay I. R., Mashayekhi M., Rommereim L. M., Fox B. A., Bzik D. J., Taylor G. A., Turk B. E., Lichti C. F., Townsend R. R., Qiu W., Hui R., Beatty W. L., Sibley L. D. (2010) Phosphorylation of immunity-related GTPases by a Toxoplasma gondii-secreted kinase promotes macrophage survival and virulence. Cell Host Microbe 8, 484–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Etheridge R. D., Alaganan A., Tang K., Lou H. J., Turk B. E., Sibley L. D. (2014) The Toxoplasma pseudokinase ROP5 forms complexes with ROP18 and ROP17 kinases that synergize to control acute virulence in mice. Cell Host Microbe 15, 537–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Reese M. L., Zeiner G. M., Saeij J. P. J., Boothroyd J. C., Boyle J. P. (2011) Polymorphic family of injected pseudokinases is paramount in Toxoplasma virulence. Proc. Natl. Acad. Sci. U.S.A. 108, 9625–9630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Behnke M. S., Khan A., Wootton J. C., Dubey J. P., Tang K., Sibley L. D. (2011) Virulence differences in Toxoplasma mediated by amplification of a family of polymorphic pseudokinases. Proc. Natl. Acad. Sci. U.S.A. 108, 9631–9636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Boudeau J., Miranda-Saavedra D., Barton G. J., Alessi D. R. (2006) Emerging roles of pseudokinases. Trends Cell Biol. 16, 443–452 [DOI] [PubMed] [Google Scholar]
- 17. Brennan D. F., Dar A. C., Hertz N. T., Chao W. C. H., Burlingame A. L., Shokat K. M., Barford D. (2011) A Raf-induced allosteric transition of KSR stimulates phosphorylation of MEK. Nature 472, 366–369 [DOI] [PubMed] [Google Scholar]
- 18. Zeqiraj E., Filippi B. M., Deak M., Alessi D. R., van Aalten D. M. F. (2009) Structure of the LKB1-STRAD-MO25 complex reveals an allosteric mechanism of kinase activation. Science 326, 1707–1711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fukuda K., Gupta S., Chen K., Wu C., Qin J. (2009) The pseudoactive site of ILK is essential for its binding to α-Parvin and localization to focal adhesions. Mol. Cell 36, 819–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gee C. L., Papavinasasundaram K. G., Blair S. R., Baer C. E., Falick A. M., King D. S., Griffin J. E., Venghatakrishnan H., Zukauskas A., Wei J.-R., Dhiman R. K., Crick D. C., Rubin E. J., Sassetti C. M., Alber T. (2012) A phosphorylated pseudokinase complex controls cell wall synthesis in mycobacteria. Sci. Signal. 5, ra7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Christie M., Boland A., Huntzinger E., Weichenrieder O., Izaurralde E. (2013) Structure of the PAN3 pseudokinase reveals the basis for interactions with the PAN2 deadenylase and the GW182 proteins. Mol. Cell 51, 360–373 [DOI] [PubMed] [Google Scholar]
- 22. Behnke M. S., Fentress S. J., Mashayekhi M., Li L. X., Taylor G. A., Sibley L. D. (2012) The polymorphic pseudokinase ROP5 controls virulence in Toxoplasma gondii by regulating the active kinase ROP18. PLoS Pathog. 8, e1002992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zwart P. H., Afonine P. V., Grosse-Kunstleve R. W., Hung L.-W., Ioerger T. R., McCoy A. J., McKee E., Moriarty N. W., Read R. J., Sacchettini J. C., Sauter N. K., Storoni L. C., Terwilliger T. C., Adams P. D. (2008) Automated structure solution with the PHENIX suite. Methods Mol. Biol. 426, 419–435 [DOI] [PubMed] [Google Scholar]
- 27. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G. W., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Librado P., Rozas J. (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452 [DOI] [PubMed] [Google Scholar]
- 30. Pawlowski N., Khaminets A., Hunn J. P., Papic N., Schmidt A., Uthaiah R. C., Lange R., Vopper G., Martens S., Wolf E., Howard J. C. (2011) The activation mechanism of Irga6, an interferon-inducible GTPase contributing to mouse resistance against Toxoplasma gondii. BMC Biol. 9, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Reese M. L., Boyle J. P. (2012) Virulence without catalysis: how can a pseudokinase affect host cell signaling? Trends Parasitol. 28, 53–57 [DOI] [PubMed] [Google Scholar]
- 32. Talevich E., Kannan N. (2013) Structural and evolutionary adaptation of rhoptry kinases and pseudokinases, a family of coccidian virulence factors. BMC Evol. Biol. 13, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li S., Depetris R. S., Barford D., Chernoff J., Hubbard S. R. (2005) Crystal structure of a complex between protein tyrosine phosphatase 1B and the insulin receptor tyrosine kinase. Structure 13, 1643–1651 [DOI] [PubMed] [Google Scholar]
- 34. Labesse G., Gelin M., Bessin Y., Lebrun M., Papoin J., Cerdan R., Arold S. T., Dubremetz J.-F. (2009) ROP2 from Toxoplasma gondii: a virulence factor with a protein-kinase fold and no enzymatic activity. Structure 17, 139–146 [DOI] [PubMed] [Google Scholar]
- 35. Ghosh A., Uthaiah R., Howard J., Herrmann C., Wolf E. (2004) Crystal structure of IIGP1: a paradigm for interferon-inducible p47 resistance GTPases. Mol. Cell 15, 727–739 [DOI] [PubMed] [Google Scholar]
- 36. Rittinger K., Walker P. A., Eccleston J. F., Smerdon S. J., Gamblin S. J. (1997) Structure at 1.65 Å of RhoA and its GTPase-activating protein in complex with a transition-state analogue. Nature 389, 758–762 [DOI] [PubMed] [Google Scholar]
- 37. Scheffzek K., Stephan I., Jensen O. N., Illenberger D., Gierschik P. (2000) The Rac-RhoGDI complex and the structural basis for the regulation of Rho proteins by RhoGDI. Nat. Struct. Biol. 7, 122–126 [DOI] [PubMed] [Google Scholar]
- 38. Kimple R. J., Kimple M. E., Betts L., Sondek J., Siderovski D. P. (2002) Structural determinants for GoLoco-induced inhibition of nucleotide release by Gα subunits. Nature 416, 878–881 [DOI] [PubMed] [Google Scholar]
- 39. Reese M. L., Boothroyd J. C. (2011) A conserved non-canonical motif in the pseudoactive site of the ROP5 pseudokinase domain mediates its effect on toxoplasma virulence. J. Biol. Chem. 286, 29366–29375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hunn J. P., Koenen-Waisman S., Papic N., Schroeder N., Pawlowski N., Lange R., Kaiser F., Zerrahn J., Martens S., Howard J. C. (2008) Regulatory interactions between IRG resistance GTPases in the cellular response to Toxoplasma gondii. EMBO J. 27, 2495–2509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Haldar A. K., Saka H. A., Piro A. S., Dunn J. D., Henry S. C., Taylor G. A., Frickel E. M., Valdivia R. H., Coers J. (2013) IRG and GBP host resistance factors target aberrant, “non-self” vacuoles characterized by the missing of “self” IRGM proteins. PLoS Pathog. 9, e1003414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Garcia-Mata R., Boulter E., Burridge K. (2011) The invisible hand: regulation of RHO GTPases by RHOGDIs. Nat. Rev. Mol. Cell Biol. 12, 493–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Goldberg J. (1998) Structural basis for activation of ARF GTPase: mechanisms of guanine nucleotide exchange and GTP-myristoyl switching. Cell 95, 237–248 [DOI] [PubMed] [Google Scholar]
- 44. Lee C. S., Kim I. S., Park J. B., Lee M. N., Lee H. Y., Suh P.-G., Ryu S. H. (2006) The phox homology domain of phospholipase D activates dynamin GTPase activity and accelerates EGFR endocytosis. Nat. Cell Biol. 8, 477–484 [DOI] [PubMed] [Google Scholar]
- 45. Pfeffer S. R. (2012) Rab GTPase localization and Rab cascades in Golgi transport. Biochem. Soc. Trans. 40, 1373–1377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yamamoto M., Okuyama M., Ma J. S., Kimura T., Kamiyama N., Saiga H., Ohshima J., Sasai M., Kayama H., Okamoto T., Huang D. C. S., Soldati-Favre D., Horie K., Takeda J., Takeda K. (2012) A cluster of interferon-γ-inducible p65 GTPases plays a critical role in host defense against Toxoplasma gondii. Immunity 37, 302–313 [DOI] [PubMed] [Google Scholar]
- 47. Degrandi D., Kravets E., Konermann C., Beuter-Gunia C., Klümpers V., Lahme S., Wischmann E., Mausberg A. K., Beer-Hammer S., Pfeffer K. (2013) Murine guanylate binding protein 2 (mGBP2) controls Toxoplasma gondii replication. Proc. Natl. Acad. Sci. U.S.A. 110, 294–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Al-Zeer M. A., Al-Younes H. M., Lauster D., Abu Lubad M., Meyer T. F. (2013) Autophagy restricts Chlamydia trachomatis growth in human macrophages via IFNG-inducible guanylate binding proteins. Autophagy 9, 50–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kim B.-H., Shenoy A. R., Kumar P., Das R., Tiwari S., MacMicking J. D. (2011) A family of IFN-γ-inducible 65-kD GTPases protects against bacterial infection. Science 332, 717–721 [DOI] [PubMed] [Google Scholar]
- 50. Shenoy A. R., Wellington D. A., Kumar P., Kassa H., Booth C. J., Cresswell P., MacMicking J. D. (2012) GBP5 promotes NLRP3 inflammasome assembly and immunity in mammals. Science 336, 481–485 [DOI] [PubMed] [Google Scholar]
- 51. Kravets E., Degrandi D., Weidtkamp-Peters S., Ries B., Konermann C., Felekyan S., Dargazanli J. M., Praefcke G. J. K., Seidel C. A. M., Schmitt L., Smits S. H. J., Pfeffer K. (2012) The GTPase activity of murine guanylate-binding protein 2 (mGBP2) controls the intracellular localization and recruitment to the parasitophorous vacuole of Toxoplasma gondii. J. Biol. Chem. 287, 27452–27466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Karplus P. A., Diederichs K. (2012) Linking crystallographic model and data quality. Science 336, 1030–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]