

Background: Electrostatic interactions are known accelerate binding in protein complexes with long lifetimes (minutes to hours).
Results: NMR and calorimetry were used to characterize binding kinetics in short lived (∼1 ms) protein-peptide complexes.
Conclusion: Electrostatic enhancement is much less and basal (electrostatic-free) association rates are greater than previously observed.
Significance: Electrostatics may play different roles in short lived and long lived protein complexes.
Keywords: Isothermal Titration Calorimetry (ITC), Ligand-binding Protein, Nuclear Magnetic Resonance, Protein Dynamic, Src Homology 3 Domain (SH3 Domain), Carr–Purcell–Meiboom–Gill (CPMG) Relaxation Dispersion
Abstract
Much of our knowledge of protein binding pathways is derived from extremely stable complexes that interact very tightly, with lifetimes of hours to days. Much less is known about weaker interactions and transient complexes because these are challenging to characterize experimentally. Nevertheless, these types of interactions are ubiquitous in living systems. The combination of NMR relaxation dispersion Carr–Purcell–Meiboom–Gill (CPMG) experiments and isothermal titration calorimetry allows the quantification of rapid binding kinetics for complexes with submillisecond lifetimes that are difficult to study using conventional techniques. We have used this approach to investigate the binding pathway of the Src homology 3 (SH3) domain from the Fyn tyrosine kinase, which forms complexes with peptide targets whose lifetimes are on the order of about a millisecond. Long range electrostatic interactions have been shown to play a critical role in the binding pathways of tightly binding complexes. The role of electrostatics in the binding pathways of transient complexes is less well understood. Similarly to previously studied tight complexes, we find that SH3 domain association rates are enhanced by long range electrostatics, whereas short range interactions are formed late in the docking process. However, the extent of electrostatic association rate enhancement is several orders of magnitudes less, whereas the electrostatic-free basal association rate is significantly greater. Thus, the SH3 domain is far less reliant on electrostatic enhancement to achieve rapid association kinetics than are previously studied systems. This suggests that there may be overall differences in the role played by electrostatics in the binding pathways of extremely stable versus transient complexes.
Introduction
Many biological processes rely on the rapid formation of protein-ligand complexes (1–5). Consequently, there has been great interest in elucidating the mechanisms through which binding partners are able to efficiently recognize each other in solution and adopt the correct conformations, relative positions, and orientations to form native stereo-specific complexes. This has been addressed theoretically (6, 7), by computer simulation (8, 9), and experimentally (1, 5, 10, 11). The association process is typically broken down into at least two steps. In the first step, the binding partners encounter each other through a random collision. For the small fraction of collisions in which the binding surfaces are correctly aligned (as few as 1 in 105), the molecules proceed to the second step and dock to form the native stereo-specific complex (12). If the docking step occurs much more rapidly than the time scale of productive intermolecular collisions, the reaction is said to be “diffusion-controlled,” and the association rate is viscosity-dependent (6). When binding is diffusion-controlled, the protein and ligand pass through a free energy maximum en route to the bound state, in which translational-rotational entropy is reduced with little compensation from favorable interactions (12, 13). The ensemble of conformations at this free energy maximum has been referred to as an “encounter complex” (6), “transient complex” (14), or “transition state” (15). We shall use the term “transition state” throughout. For these systems, binding is essentially two-state, such that significant populations are observed only for the free and/or bound states, and a negligible fraction of molecules populate the high-energy transition state at any one time. Note that the two-state diffusion-controlled binding mechanism is compatible with conformational changes occurring after the transition state, provided that they proceed sufficiently rapidly (16). Additionally, transient conformational reorganization can occur prior to the intermolecular collision, leading to a “gated” diffusion-controlled binding mechanism (17). In either case, the structure of the transition state is key to understanding the binding pathway because it represents a kinetic bottleneck. Interactions that stabilize the transition state accelerate molecular association rates (6, 14, 15).
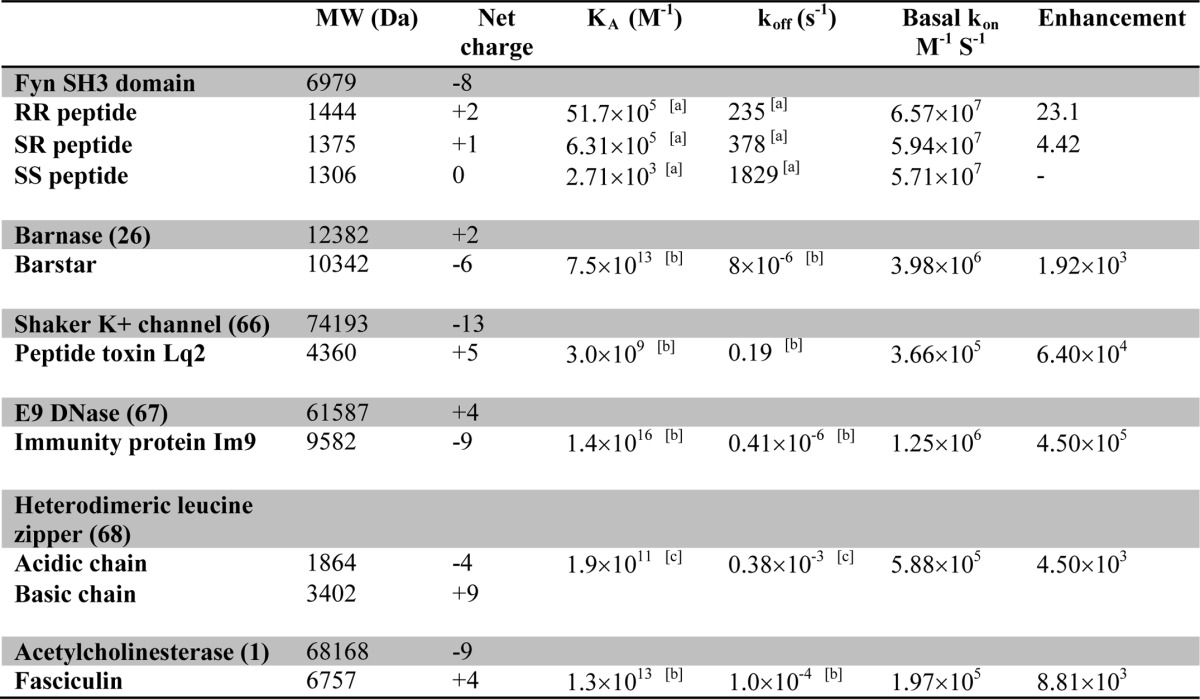
A variety of interactions have been proposed to contribute to the stability of transition states, including nonspecific hydrogen bonding, van der Waals interactions, non-polar desolvation, long range electrostatic interactions (6, 18–20), or subsets of native bound-like interactions (21). The role of electrostatics in binding pathways has been particularly well studied. For a number of systems, including barnase-barstar (5), E9-Im9 (10), IL4-IL4BP (11), and Fas-AchE (1), it has been found that electrostatic stabilization of weakly specific transition states can accelerate association by up to 4 or more orders of magnitude, compared with the basal rate (15). A combination of experimental and computational studies has led to a picture of the transition state in which the binding partners remain solvent-separated and interact almost exclusively by long range electrostatics (15, 22).
However, these investigations have focused on extremely tight protein-ligand interactions, as listed in Table 1. Equilibrium association constants have ranged from 109 to 1013 m−1 (KD nanomolar to picomolar), with dissociation rates of 10−1 to 10−6 s−1 (lifetimes of seconds to days) Much less is known about the binding pathways of weaker complexes with lifetimes on the order of milliseconds. These types of short lived protein interactions are abundant in nature, raising the important question of to what extent long lived and short lived complexes follow similar binding pathways. In what follows, we will use “long lived” to refer to complexes that bind with <nm KD and have lifetimes on the order of minutes or longer. We will use “short lived” to refer to complexes with at least micromolar KD and lifetimes on the order of milliseconds. Several studies of short lived complexes have derived structural models of transient complexes that precede formation of the fully bound state (23–25). However, these investigations did not extract kinetic parameters or identify the interactions governing association rates, which is key to understanding binding pathways.
TABLE 1.
Electrostatic enhancement of protein association rates
a Measurement obtained at I = 2 mm.
b Measurement obtained at I = 25 mm.
c Measurement obtained at I = 74 mm.
The binding kinetics of short lived complexes are challenging to measure experimentally. In typical stopped-flow binding measurements, the protein and ligand are rapidly mixed, and the extent of the reaction is monitored as a function of time using techniques including spectroscopic absorbance (21), fluorescence (10, 26, 27), enzymatic activity (1, 28, 29), and surface plasmon resonance (30, 31). These techniques are limited by the requirement that the lifetimes of the free and bound states must be severalfold longer than the time required to completely mix the protein and ligand solutions; otherwise, the reaction proceeds nearly to completion before the start of the measurement period.
The combination of nuclear magnetic resonance (NMR) spectroscopy and isothermal titration calorimetry (32) can characterize binding kinetics that are too rapid for standard stopped-flow techniques, on the order of 104 s−1 (33). In this approach, a substoichiometric amount of ligand is added to a protein sample whose signals are monitored by NMR spectroscopy. The resulting dynamical exchange between the free and bound states modulates the chemical shifts of nuclei in the protein on the millisecond to microsecond time scale, leading to signal broadening. The broadening is quantified by NMR relaxation dispersion experiments (34–36), which yield the dissociation rate constant, koff. The equilibrium association constant of the interaction, Ka, is determined by ITC or NMR titration experiments. The second-order association rate constant is then given by the equation, kon = Ka·koff. These two parameters report on the binding pathway because Ka is related to the free energy difference between the free and bound states, whereas kon is related to the free energy difference between the free state and the transition state.
Src homology 3 (SH3)2 domains are typical of weakly binding proteins that form short lived complexes. These ∼60-amino acid modules are found in the context of larger multidomain proteins and mediate protein-protein interactions in signal transduction pathways (37, 38). The type I ligand recognized by many SH3 domains comprises a short peptide sequence, +XΦPXΦP, where P is proline, Φ is a hydrophobic amino acid, X is any amino acid, and + is a positively charged residue, typically arginine or lysine (38). The positions in the motif are numbered −3 to 3 such that the conserved + occupies site −3. Dissociation constants for SH3 domains are usually on the order of micromolar, and the lifetimes of their complexes are on the order of milliseconds (33, 39). We previously used NMR and ITC to study the binding kinetics of the SH3 domain from the Fyn tyrosine kinase. We showed that this protein binds a type I target peptide in a two-state manner with millisecond time scale kinetics and a temperature dependence of kon that is consistent with diffusion-limited association (33). In the current study, we have focused on how positively charged residues within the SH3 binding motif and negatively charged residues in the binding site on the SH3 domain interact to influence association kinetics. Three different dodecapeptides were employed: one with a typical type I consensus motif with the conserved Arg−3 preceded by Ser−4 (SR, net charge +1), a second containing R−4R−3 (RR, net charge +2), and a third containing S−4S−3 (SS, net charge 0). We analyzed the contributions of the conserved Arg−3 and “extra” Arg−4 residues to the stabilities of the fully bound and transition states by comparing the binding affinities and association rates of the RR, SR, and SS peptides. Data were extrapolated to both zero ionic strength, where long range electrostatic interactions are maximal, and infinite ionic strength, where long range electrostatics are fully attenuated. We find that the conserved Arg−3 and the “extra” Arg−4 residues stabilize both the encounter and fully bound complexes via similar long range electrostatic interactions, which implies that the Fyn SH3 domain and peptide are fairly close in the transition state. Notably, Arg−3 additionally stabilizes the bound state via short range interactions that are not screened by adding salt. Likely candidates include a network of salt bridges and hydrogen bonds previously observed for this residue in the bound complex by NMR (40) and x-ray crystallography (Protein Data Bank entry 4EIK). These short range interactions involving Arg−3 are not present in the transition state and are only made after the molecules have crossed the free energy barrier, near the end of the binding pathway. Thus, the SH3-peptide transition state is fairly compact and stabilized primarily by long range electrostatic interactions, as seen for other protein-ligand systems (5, 14, 15, 22). However, there are surprising differences between the system studied here and the high-affinity long lived complexes investigated previously. We find that the electrostatic-free basal association rate for this system is significantly greater, whereas the electrostatic association rate enhancement is several orders of magnitude less. This result suggests that short lived complexes may rely less on electrostatic enhancement to achieve rapid binding than do high-affinity long lived complexes.
MATERIALS AND METHODS
Protein and Peptide Preparation
Wild-type chicken Fyn SH3 domain (residues 81–148) was expressed and purified as described previously (41). RR (Ac-WSLAR−4R−3P−2L−1P0P1L2P3-NH2),SR (Ac-WSLAS−4R−3P−2L−1P0P1L2P3-NH2), and SS (Ac-WSLAS−4S−3P−2L−1P0P1L2P3-NH2) peptides were purchased from the Sheldon Biotechnology Centre (McGill University) and purified to homogeneity by reverse-phase HPLC. Protein and peptide samples were dialyzed against 5 mm MES, pH 6.0, at the desired NaCl concentration. For ionic strength calculations, it was assumed that the buffer contributed 2.57 mm charged ions. Most experiments were performed at 40 °C. This temperature was chosen to optimize the NMR CPMG relaxation dispersion profiles obtained with the RR and SR peptides and lies within the physiological temperature range of 40–42 °C (42).
ITC
Titrations were performed at 40 °C using a VP-ITC instrument (MicroCal LLC, Northampton, MA). Measurements were taken in triplicate for each NaCl concentration and consisted of 56 injections of 5 μl. RR peptide data were collected at 0.00, 0.01 0.03, 0.05, 0.10, 0.20, 0.30, 0.40, 0.50, 1.00, 1.50, and 2.00 m NaCl. For the SR and the SS peptide, measurements were taken at 0.00, 0.03, 0.20, and 0.30 m NaCl. Titrations were repeated for the SS peptide at 20 °C. Data were fitted using the one-set-of-independent-sites model in the Origin Software package (OriginLab, Northampton, MA) and in-house MATLAB scripts (MATLAB R2013a, The MathWorks, Natick, MA).
NMR
1H-15N HSQC titration spectra, referenced to an internal 4,4-dimethyl-4-silapentane-1-sulfonic acid standard (43), were collected at 40 °C and 800 MHz for samples containing ∼0.3 mm protein and 16 different peptide concentrations. In the case of the SS peptide titration, chemical shift peak trajectories were fit globally according to Ref. 44,
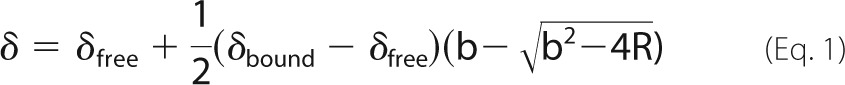 |
where δfree and δbound are the chemical shifts of the free and bound protein, R = [peptide]total/[protein]total, and b = 1 + R + (Ka × [protein]total)−1.
1H-decoupled 15N CPMG experiments (36) with power compensation schemes (33) were performed at 40 °C and 500 and 800 MHz on 1 mm samples of the Fyn SH3 domain, free in solution (0 m NaCl), in the presence of 0.1 mm SS, SR, and RR peptides (0, 0.2, 0.3 m NaCl) and with saturating amounts of the RR peptide (0 m NaCl). Measurements were repeated at 20 °C for the SH3 domain alone (0 m NaCl) and in the presence of 0.1 mm SS peptide (0, 0.2, and 0.3 m NaCl). Experiments employed a constant relaxation delay, Trelax, of 40 ms and 23 values of νCPMG = 1/(2τCP) ranging from 25 to 1000 Hz, where τCP is the delay between consecutive pulses. Transverse relaxation rates, R2, were calculated for each cross-peak signal at each νCPMG value according to the following,
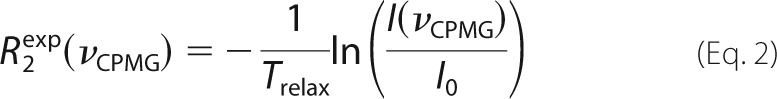 |
where I(νCPMG) is the peak intensity at a given value of νCPMG, and I0 is the signal intensity with Trelax = 0. The experimental uncertainties in peak intensities (σI) were considered to be the same for all values of νCPMG for a given residue at a given ionic strength and static magnetic field. CPMG experiments were repeated for a subset of νCPMG values (Ndup different values), with nj replicates obtained at the jth repeated νCPMG value. The nj replicates obtained at (νCPMG)j for a given residue, ionic strength, and static magnetic field yield an S.D. value, sj. The uncertainty in σI was calculated as follows,
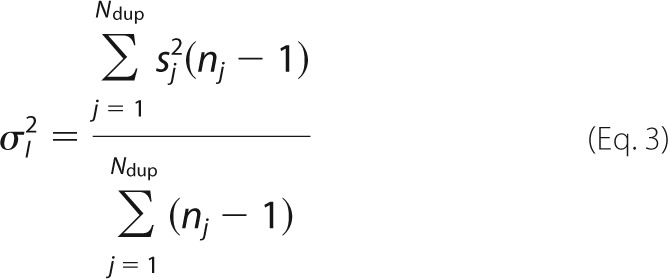 |
and the associated errors in transverse relaxation rates, σR2, are given by the following.
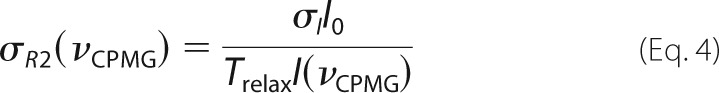 |
CPMG data were fitted using a two-state model of conformational exchange,
 |
where P is the protein, L is the ligand, PL is the complex, koff is the dissociation rate constant, kon is the association rate constant, [L] is the concentration of free peptide, and the difference between the precession frequencies of an 15N nucleus of the protein in the free and bound states is given by Δω. Dispersion profiles were fit to equations valid for the fast time scale regime (45) (Δω ≪ (koff + kon[L])) or all time scale regimes (46) based on the parameter α,
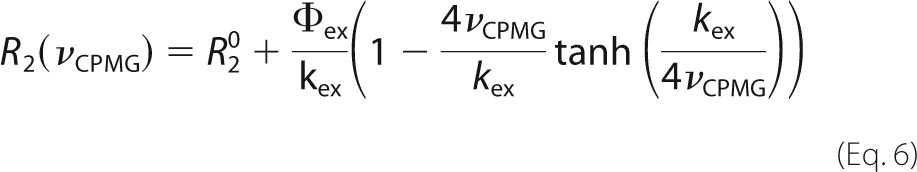 |
where Rex is the total exchange contribution to R2, and B0 is the static magnetic field (47). Profiles with α > 1.5 were considered to be in the fast exchange regime and were analyzed according to Ref. 45 as follows,
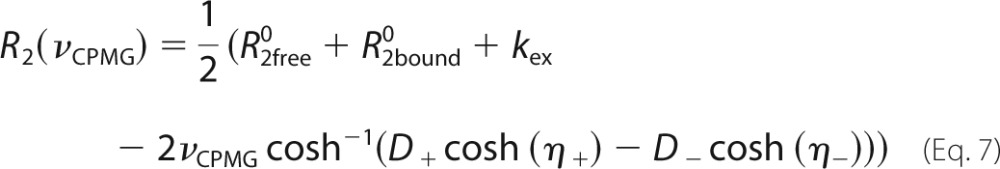 |
where pfree and pbound are the relative populations of free and bound protein (pfree + pbound = 1), kex = koff + kon[L], Φex = pfreepboundΔω2, and R20 is the population-weighted average (broadening-free) transverse relaxation rate (R20 = pfreeR2free0 + pboundR2bound0). All other dispersion profiles were analyzed according to Ref. 46 as follows.
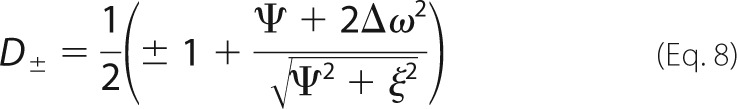 |
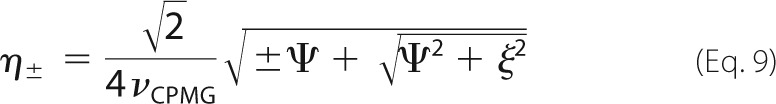 |
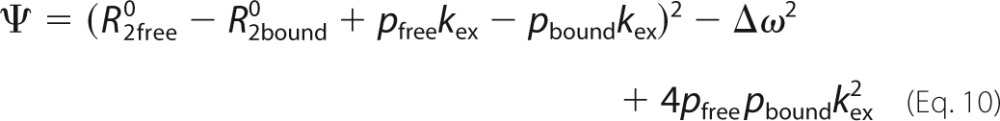 |
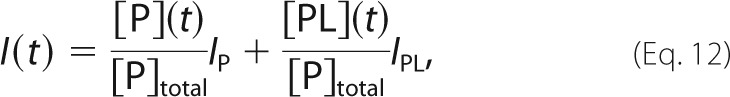 |
We performed the analysis assuming that R2free0 = R2bound0. This is typically done in analyses of CPMG data because it is not possible to reliably extract separate values for R2free0 and R2bound0 (48). In this case, the approximation is probably valid because binding of the peptide is unlikely to substantially alter 15N transverse relaxation rates in the protein.
In the case of the RR and SR peptides at 40 °C with 0, 0.2, and 0.3 m NaCl, the data sets contained mixtures of dispersion profiles in the fast and intermediate time regimes, and data for all residues of a given sample were fitted simultaneously to extract global values of pbound and kex, whereas Δω2 or Φex, R20(800 MHz), and R20(500 MHz) were obtained on a per-residue basis (49). For the SS peptide at 20 °C with 0 and 0.2 m NaCl, all but one of the dispersion profiles were in the fast time regime. In these cases, the value of pbound was calculated based on the total concentrations of protein and peptide and the ITC-derived binding constants and was held fixed in the CPMG fits. Global values of kex were extracted, whereas Δω2, R20(800 MHz), and R20(500 MHz) were obtained on a per-residue basis. Values of koff were subsequently calculated as (1 - pbound)kex. Errors in the extracted parameters were estimated using a Monte Carlo approach in which group fitting was performed on random subsets of the residues selected using a bootstrap procedure (50). Analyses were performed using scripts written in MATLAB (MALAB R2011b, The MathWorks, Natick, MA).
Electrostatic Enhancements
Log10(Ka) values for the SS peptide were fitted as a linear function of ionic strength. Interpolated values were used to calculate the affinity enhancements of the SR and RR peptides. The corresponding linear interpolation was also used to calculate kon enhancements. The ionic strength enhancements were plotted as a function of fion, Equation 22, setting R = 4 Å, which roughly corresponds to the size scale of a single Arg residue. The plot was not highly sensitive to the choice of R.
Stopped-flow Binding Kinetics
The kinetics of binding were characterized by rapidly mixing the SH3 domain and peptide and monitoring the progress of the reaction via intrinsic tryptophan fluorescence of the protein, which increases upon interacting with proline-rich peptides (51). We used a version of the RR peptide free of Trp and Tyr fluorophores (VSLARRPLPPLP); thus, the signal derives entirely from protein residues. We fit the binding data according to the following equation,
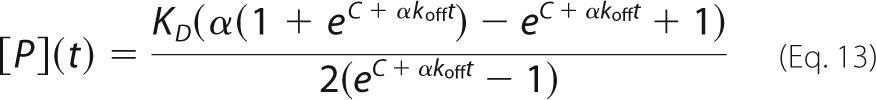 |
where I(t) is the fluorescence signal at time t, [P](t) and [PL](t) are the concentrations of free protein and bound complex at time t, [P]total = [P] + [PL] is the total concentration of protein, and IP and IPL are the fluorescence intensities of the free and bound states. The time dependence of [P](t) is given by Ref. 52 as follows,
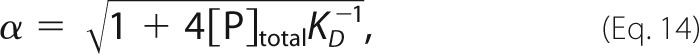 |
where
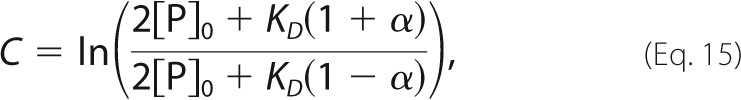 |
and
koff is the dissociation rate constant, KD is the equilibrium dissociation constant, and [P]0 is the initial concentration of free protein at t = 0, which is assumed to be equal to the initial concentration of free ligand, [L]0 = [P]0. Data were analyzed by non-linear least-squares fitting with the values of [P]total and KD held constant, while the values of IP, IPL, koff, and [P]0 were varied to minimize the function,
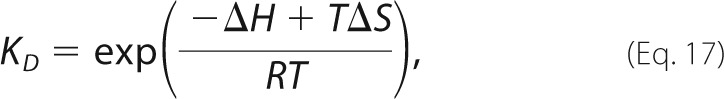 |
where I(t)calculated is given by Equation 1, and the sum runs over all data points. The value of [P]total was obtained from the absorbance of the protein sample at 280 nm (53), the concentration of the peptide was determined by amino acid analysis (54), and the value of KD was extrapolated from temperature-dependent ITC data for the Fyn SH3/RR peptide interaction (see below). Adjusting the value of [P]0 accounts for binding that occurs during the dead time of the instrument (∼4 ms). The uncertainty in koff was obtained by fitting random sets of the 100 individual stopped-flow traces selected according to the bootstrap method (50) and averaged. The error was taken as the S.D. value obtained after 100 iterations.
Experiments were performed using a Bio-Logic SFM 400 instrument coupled to a MOS-250 spectrometer using Biokine version 2.5 software for data acquisition. Intrinsic tryptophan fluorescence of the Fyn SH3 domain was measured with an excitation wavelength of 276 nm and a bandwidth of 20 nm. Emission at a wavelength of 320 nm and a bandwidth of 10 nm was recorded for 150 ms with measurements taken every 0.02 ms. The detector photomultiplier was operated at 860 V. For each trace, 201 μl of protein (1 μm) and 201 μl of RR peptide (1 μm) were mixed for a total injection volume of 402 μl at a flow rate of 13.5 ml s−1 at 5 °C.
NMR-derived Binding Kinetics
We previously characterized the binding kinetics of the RR peptide using NMR and ITC over a temperature range of 10–50 °C (33). In this prior work, we employed both CPMG and ZZ magnetization exchange NMR experiments at 20 °C, obtaining very similar values for koff: 11.2 and 12.8 s−1, respectively. At 10 °C, only ZZ exchange experiments were used, yielding koff = 4.5 s−1. CPMG experiments were not performed because spectral broadening is minimal at this temperature. We extrapolated the expected kinetic and thermodynamic behavior to 5 °C according to the following,
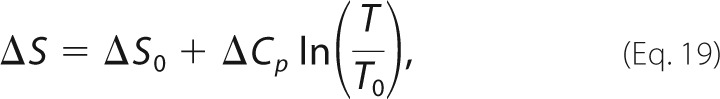 |
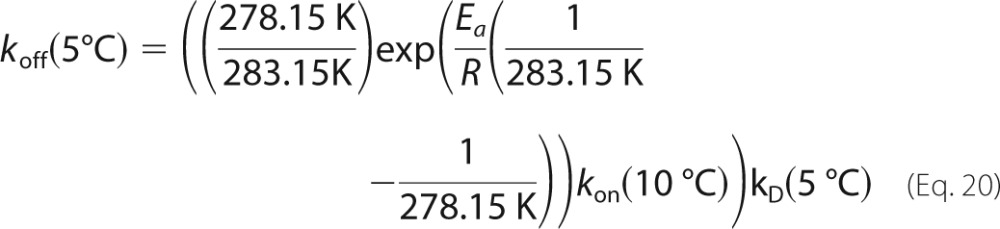 |
where T0 = 303.15 K, ΔH0 = 15.4 kcal mol−1, ΔS0 = 20 cal mol−1 K−1, ΔCp = 352 cal mol−1 K−1 (33), T = 278.15 K, R is the universal gas constant, and the equation,
 |
where kon(10 °C) = 1.03 ± 0.05 × 108 m−1 s−1 (33), and Ea = 3.4 ± 0.5 kcal mol−1. This yielded KD(5 °C) = 36.7 nm and koffNMR(5 °C) = 3.3 ± 0.2 s−1. For the sake of comparison, KD(10 °C) = 46.1 nm.
RESULTS AND DISCUSSION
NMR Titration Experiments
Series of protein 1H-15N correlation HSQC NMR spectra were collected for the Fyn SH3 domain titrated with RR, SR, and SS peptides (Fig. 1) at 40 °C. Exchange between the free and bound states was intermediate on the NMR chemical time scale (47) for the RR and SR peptides and fast for the SS peptide. The signals for many of the same protein residues were displaced by the addition of each peptide (Fig. 1, a–c). This suggests that all three peptides occupy the same binding site, consistent with the idea that the ΦPXΦP motif is the minimal recognition sequence of the SH3 domain (37). The chemical shift displacements are color-coded on a structure of an SH3-peptide complex in Fig. 2. SS peptide titrations were repeated in the presence of 0, 0.2, and 0.3 m NaCl at 40 °C. Peak displacements (Fig. 1, d and e) were analyzed to extract the equilibrium association constants, Ka, yielding values in good agreement with the ITC-derived measurements (Table 2). A further SS peptide titration was performed at 20 °C in the presence of 0 m NaCl. Under these conditions, exchange was on the intermediate time scale; however, peak shifts were essentially identical to those obtained at 40 °C.
FIGURE 1.

a–c, regions of 15N/1H HSQC spectra of the Fyn SH3 domain titrated with RR (a), SR (b), and SS (c) peptides. Shown are chemical shift displacements of SH3 domain signals in the 15N (d) and 1H (e) dimensions upon titration with the SS peptide. Curves represent the global fit to a two-state binding equation.
FIGURE 2.
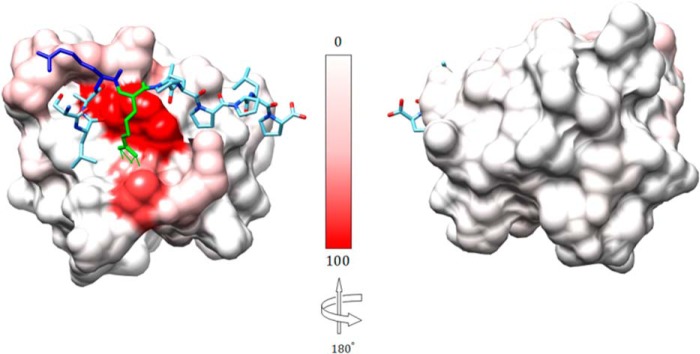
Structure of the Fyn SH3 domain in complex with a ligand identical to the RR peptide but containing Val rather than Trp at position −8 (Protein Data Bank entry 4EIK). Shown are relative 15N/1H chemical shift perturbations (Δδ2 = (10Δδ1H)2 + (Δδ15N)2), normalized to the largest perturbation (Δδ = 12.98 ppm), color-coded on the protein surface, from 0% (white) to 100% (red). Peptide residues Arg−3 (green) and Arg−4 (blue) are illustrated in ball and stick format. Hydrogen bonds between Arg−3 in the peptide and Asp17 in the Fyn SH3 domain are indicated by green lines.
TABLE 2.
Thermodynamic and kinetic binding parameters for the Fyn SH3 domain
a Population of the bound state extracted from CPMG data.
b Population of the bound state calculated based on the ITC-derived affinity and concentrations of protein and peptide used.
Isothermal Titration Calorimetry
The affinities of the Fyn SH3 domain for the RR, SR, and SS peptides were characterized by ITC at 40 °C with NaCl concentrations ranging from 0 to 2 m (Fig. 3 and Table 2), yielding equilibrium association constants (Ka) plotted in Fig. 4. The affinity of the RR peptide shows a sharp decrease with increasing salt concentration up to about 0.25 m NaCl, followed by a gradual increase. The affinity of the SR peptide also decreases sharply up to 0.25 m NaCl. This is similar to the salt dependence of protein solubility, which typically shows an initial increase or “salting in” as electrostatic interactions are screened, followed by a decrease or “salting out” as hydrophobic interactions are enhanced by increasing salt concentration (55). The hydrophobic association of pairs of methane molecules in water and the chemical potential of methane dissolved in water both increase linearly with NaCl concentration (56, 57). The affinity of the SS peptide is roughly 3 orders of magnitude weaker than those of the SR and RR peptides (at 0 m NaCl) and increases linearly with increasing salt concentration. This molecule is uncharged, and its binding to the SH3 domain is driven by non-polar interactions involving the ΦPXΦP motif that are presumably enhanced by increasing salt concentrations. Thus, the initial downward slope of the RR affinity profile is due to the screening of electrostatics, whereas the slight upward slope at high NaCl concentrations is caused by the enhancement of hydrophobic interactions involving the ΦPXΦP binding motif. As discussed below, an additional set of ITC experiments was performed for the SS peptide at 20 °C with 0, 0.2, and 0.3 m NaCl.
FIGURE 3.

Isothermal titration calorimetry raw thermograms (top) and integrated heats (bottom) for RR (a), SR (b), and SS (c) peptides binding to the Fyn SH3 domain at 40 °C and different ionic strength. Black, 0 mm NaCl added; red, 33 mm; orange, 200 mm; dark yellow, 300 mm; green, 500 mm; blue, 1000 mm; magenta, 2000 mm. Raw thermograms were obtained with 0 m added salt.
FIGURE 4.

Base 10 logarithms of affinity constants, Ka, of the Fyn SH3 domain for RR (filled circles), SR (open squares), and SS peptides (open circles) plotted as a function of ionic strength. In some cases, the error bars are within the symbols used.
NMR Kinetics Experiments
The kinetics of peptide binding were characterized using NMR CPMG relaxation dispersion experiments performed on 15N-enriched samples of the Fyn SH3 domain partially saturated with RR and SR peptides at 40 °C with NaCl concentrations of 0.00, 0.03, 0.20, and 0.30 m. Association/dissociation dynamics produce chemical shift fluctuations that contribute to 15N transverse relaxation rates, R2. CPMG experiments quench these contributions with variable trains of 180° refocusing radiofrequency pulses applied during a constant relaxation delay (34, 35). Plots of R2 versus νCPMG are shown in Fig. 5, where νCPMG = 1/(2τ), and τ is the delay between successive refocusing pulses. This spectral broadening reflects entirely exchange between the free and bound forms of the protein. Essentially flat dispersions are obtained for the protein in the absence of peptide, although the profiles of some residues in the free protein are very slightly sloped by 5 × 10−4 s−1 Hz−1. This indicates that the large dispersions that we observe are not due to internal dynamics of the protein itself because these would be evident when peptide is absent. Flat dispersions are also obtained with saturating amounts of the RR peptide. The populations of all protein and peptide species are at thermodynamic equilibrium throughout the CPMG experiment. Thus, any partly bound intermediate forms in the partially saturated sample would be present in the fully saturated sample as well. If the large dispersions were due to exchange between the bound state and an intermediate, they would also be observed for the fully saturated sample, which is not the case. Furthermore, chemical shift parameters extracted from the CPMG fits, Δω2, provide a fingerprint of the exchanging species. In this case, we obtain quantitative agreement between Δω2 and the peak displacements extracted from peptide titrations, Δδ2, as observed previously (33) (Fig. 6). If the large dispersions were due exchange between the free protein and a binding intermediate, such good agreement would not be expected, unless the intermediate had chemical shifts identical to those of the bound state, which is unlikely. Thus, the relaxation dispersion analysis reports on the kinetics of exchange between the free and fully bound states. Dissociation rate constants (koff) were extracted from the data, as described under “Materials and Methods,” and used to calculate kon, according to the equation, kon = Ka·koff.
FIGURE 5.
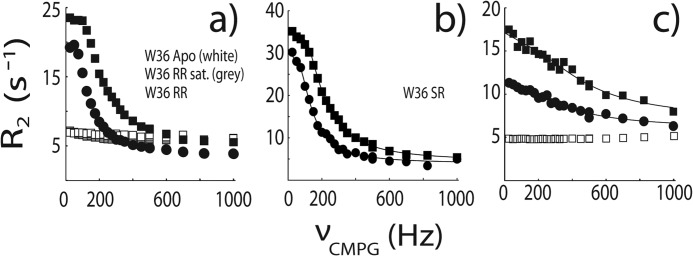
NMR 15N CPMG relaxation dispersion data for the backbone amide signal of W36 collected at 500 (circles) and 800 (squares) MHz and 0 m NaCl. In a, white, gray, and black symbols correspond to protein samples with an RR peptide/protein ratios of 0, 300, and ∼10% at 40 °C. In b, data were obtained with an SR peptide/protein ratio of ∼10% at 40 °C. In c, white and black symbols correspond to SS peptide/protein ratios of 0 and ∼10% at 20 °C. Lines indicate the global best fits as described under “Materials and Methods.”
FIGURE 6.

Comparison of Δω215N chemical shift parameters extracted from CPMG relaxation dispersion NMR experiments performed on the Fyn SH3 domain partially saturated with the RR peptide (0 m NaCl) and Δδ2 peak displacements from a comparison of the Fyn SH3 domain spectra free and saturated with the RR peptide (0 m NaCl). The line goes through the origin with a slope of 1.
Experiments with the RR peptide with 0.0 m NaCl were repeated in the presence of 15% glycerol, which translates into a 1.45-fold increase in solution viscosity, η (58). If binding rates are limited by diffusion in solution, then kon should be inversely proportional to η (6), although in practice, the slope of a kon/kon0 versus η0/η plot can diverge somewhat from the theoretical value of 1 (59). We find that the peptide binds 2.13-fold more slowly in the presence of 15% glycerol, which lies well within the range of viscosity dependences reported for diffusion-limited protein association rates (59). We also find that the association rate constant is significantly (∼4-fold) more sensitive to ionic strength than the dissociation rate constant, which is a signature of diffusion-limited binding (13). In addition, we reported previously that the apparent activation enthalpy (Ea) for kon is 3.4 kcal mol−1 (33). This matches the expected Ea = 4.5 kcal mol−1 for diffusion-limited reactions, reflecting the temperature dependence of water viscosity (16). Taken together, the NMR kinetics data strongly indicate that peptide binding to the SH3 domain is a diffusion-limited, two-state process.
In the case of the SS peptide at 40 °C, exchange kinetics are extremely rapid, leading to very small and essentially linear dispersion profiles. Due to their very small magnitudes, they are partially obscured by the slightly sloped profiles obtained for the free protein (which probably reflect some degree of internal motions unrelated to peptide binding). Therefore, in order to determine binding kinetics for the SS peptide, CPMG experiments were performed at 20 °C with 0, 0.2, and 0.3 m NaCl, conditions that produce large dispersions for the partially SS peptide-saturated protein sample, and completely horizontal dispersion profiles for the free protein, as shown in Fig. 5. Values of koff determined at 20 °C were combined with affinities (Ka), measured by ITC at 20 °C to yield association rate constants (kon). We used the previously determined value of Ea = 3.4 kcal mol−1 to extrapolate kon for the SS peptide from 20 to 40 °C according to the equation, kon(T2) ≈ (T2/T1) × kon(T1) × exp(Ea/R ×(1/T1 − 1/T2))c (60), where R is the gas constant. This leads to scaling of kon by a factor of only 1.6, which is small compared with the up to 30-fold differences in kon observed between the different peptides. Thus, the results are not sensitive to the exact choice of Ea.
Comparison of NMR and Stopped-flow Binding Kinetics
In order to cross-validate the NMR characterization of Fyn SH3 domain-peptide binding kinetics, we performed a stopped-flow spectroscopic analysis of the same system. However, the rates determined in the current study far exceed the limits of standard stopped-flow instruments (this is one of the advantages of the NMR-based approach). We therefore collected stopped-flow data at 5 °C, in order to slow the kinetics sufficiently for them to be amenable to this type of analysis. Fluorescence traces for the SH3 domain injected with buffer (green) and a peptide solution (blue) are shown in Fig. 7, with t = 0 aligned to the end of the injection. As expected, the fluorescence signal for the protein-peptide pair shows a time-dependent increase after the injection due to evolution of the binding reaction. This signal is above that of the free protein at t = 0 due to binding that occurs during the dead time of the instrument, which is about 4 ms. Analysis of the stopped-flow data yields koffSF(5 °C) = 8.9 ± 0.6 s−1, whereas the NMR kinetics method yields koffNMR(5 °C) = 3.3 ± 0.2 s−1, as described under “Materials and Methods.” These two values differ by about a factor of 3, which we believe is excellent agreement for such different experimental modalities. This gives us further confidence in the NMR-based kinetics method.
FIGURE 7.
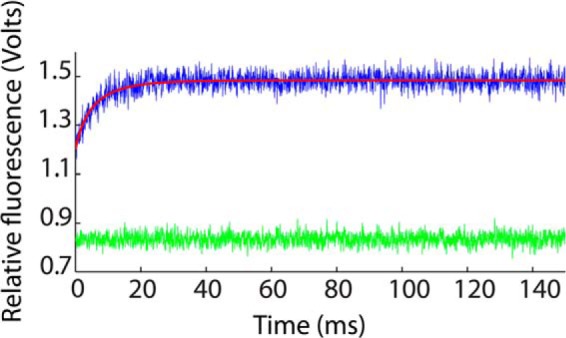
Stopped-flow fluorescence data for the Fyn SH3 domain rapidly mixed with buffer (green) and with a solution of the RR peptide (blue). Data shown are the averages of 100 individual stopped-flow traces. The best fit using Equation 12 is indicated with a red line.
Electrostatic Enhancement of Association Rates
We investigated the influence of electrostatics on SH3 domain ligand binding kinetics by varying the ionic strength of the samples, in order to attenuate long range coulombic interactions, and by comparing results for the RR, SR, and SS peptides, which carry formal charges of +2, +1, and 0. The screening of coulombic interactions by electrolyte solutions was analyzed in terms of the electrostatic attraction of two oppositely charged spheres with radii r, a center-center separation of 2r, and charges ±Z. Relative to infinite separation, the potential energy, V, is given by the expression (61, 62),
 |
 |
where e is the elemental charge, ϵ is the dielectric constant of the medium, κ is the inverse Debye length,
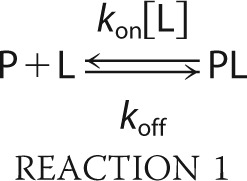 |
kB is the Boltzmann constant, and I is the ionic strength. The parameter fion describes the ionic strength dependence of the potential energy, with limiting values of 1 (full interaction strength) in the absence of salt, and 0 (zero interaction strength) at infinite salt concentration.
The logarithms of kon are plotted as a function of fion for the RR, SR, and SS peptides in Fig. 8a, together with linear regression lines. Data for the RR and SR peptides have positive slopes, as expected, because larger values of fion correspond to lower salt concentrations, greater electrostatic stabilization of the transition state, and faster association. The data for the SS peptide are close to horizontal, as expected for an uncharged ligand. The slightly negative slope may be due to weak hydrophobic stabilization of the transition state that is enhanced at high salt concentrations. Notably, the lines for the three peptides nearly intersect at fion = 0 and a kon value of about 6 × 107 m−1 s−1. This corresponds to the basal, or electrostatic-free, association rate. The close agreement of basal association rates obtained for the three different peptides gives us confidence in the reliability of the extrapolation along fion. The slopes of the lines correspond to the logarithms of the maximum electrostatic enhancement of binding with zero ionic strength, log(konenhanced/konbasal). We find that long range coulombic interactions accelerate binding of the RR and SR peptides by 23.1- and 4.42-fold, respectively, as listed in Table 1.
FIGURE 8.
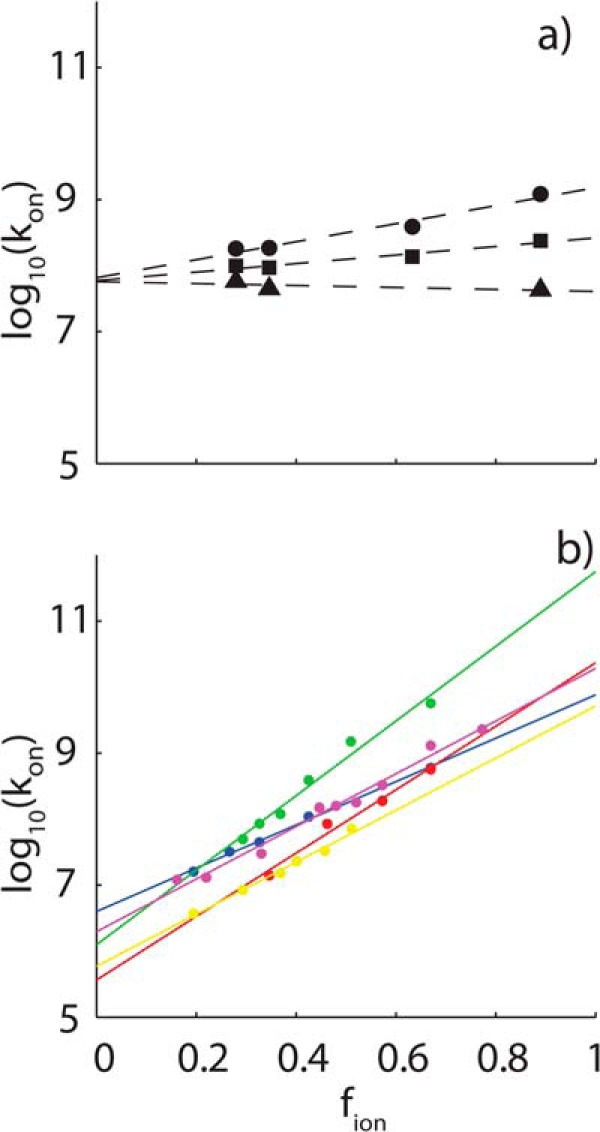
Ionic strength dependence of association rates. a, NMR and ITC-derived association rate constants (kon) for the RR (circles) and SR (squares) and SS (triangles) peptides binding to the Fyn SH3 domain, plotted as a function of fion (Equation 22), which takes values between 1 at zero ionic strength and 0 at infinite ionic strength. Dashed lines, linear regression fits of the data. b, association constants for barnase-barstar (blue), shaker K+ channel-peptide toxin Lq2 (red), E9 DNase-immunity protein Im9 (green), heterodimeric leucine zipper (yellow), and acetylcholinesterase-fasciculin 2 (magenta) plotted as a function of fion.
The basal association rate for the SH3 domain is significantly greater, and the extent of electrostatic enhancement is much less, than observed previously for long lived complexes. Association rates for a data set of five different systems (13) are plotted as a function of fion in Fig. 6b, and the basal association rates and electrostatic enhancements extracted from the linear fits are listed in Table 1. These proteins bind roughly 103 to 1010-fold more tightly and dissociate 103 to 109-fold more slowly than the SH3 domain studied here. Notably, all of these basal, electrostatic association rates are 10–100-fold slower than those obtained for the SH3 domain, whereas the electrostatic rate enhancements are 102 to 104-fold greater.
It is likely that these differences are due to differences in the nature of the binding surfaces for the SH3 domain compared with the proteins in the long lived complex database. The barnase-barstar interface is fairly typical of those in the database in terms of size and complexity. It buries 1590 Å2 of accessible surface area and involves the formation of 14 specific hydrogen bonds (63). In this case, only about 1 molecular collision in 1700 aligns the binding surfaces sufficiently accurately to proceed to the fully bound state in the absence of electrostatic enhancement, assuming no charged interactions and a molecular collision frequency of 6.6 × 109 m−1 s−1 (12). In contrast, the complex formed by the SH3 domain and ΦPXΦP motif buries only 600 Å2 and is stabilized entirely by hydrophobic interactions (40). In this case, about 1 in every 160 SH3-peptide collisions is sufficiently well aligned to progress to the bound state. Docking of the SS peptide onto the SH3 binding surface involves a significantly smaller contact area with fewer specific pairwise interactions. This probably results in less stringent geometric requirements for molecular collisions to lead to successful docking of the SS peptide and a more rapid basal association rate.
A less geometrically restricted transition state could explain our observation of lower electrostatic enhancement of SH3-peptide association rates compared with that of barnase-barstar and other long lived complexes. For barnase-barstar, it was found that the transition state is stabilized by specific charged residues that are in close proximity in the stereospecific complex. This implies that these residues are correctly oriented with respect to one another at the rate-limiting step of binding (5). The SH3 domain exhibits a greater basal association rate, suggesting that the transition state is less geometrically constrained. Thus, fewer specific interactions may be formed between charged residues in this state, leading to a smaller degree of electrostatic stabilization.
Thus, the SH3 domain and target peptide do not rely as heavily upon electrostatic enhancement to achieve rapid association kinetics as do many previously studied long lived complexes. It has been noted previously that the interfaces of high-affinity complexes tend to contain more charged residues than those of low-affinity ones, and that these coulombic interactions are sometimes, but not always, thermodynamically stabilizing (64). This is consistent with higher affinity complexes relying more heavily on electrostatic enhancement to achieve rapid binding kinetics than weaker complexes; however, the question is far from resolved.
Arginine Interactions in the Transition State and Bound State
In order to clarify the role played by the peptide arginine residues in the SH3 domain binding pathway, we compared Ka and kon values for the RR and SR peptides with those of the SS peptide. The SS peptide carries no formal charges and binds in the same location as the SR and RR peptides, as evinced by similar patterns of peak movements in protein NMR titration experiments. The ratios of Ka and kon obtained for the RR and SR peptides relative to those of the SS peptide describe the contributions of the conserved (Arg−3) and “extra” (Arg−4) Arg residues to the stabilities of the fully bound and transition states. The logarithms of the RR/SS and SR/SS ratios are plotted as a function of fion in Fig. 9 and exhibit excellent linear correlations over their full ranges of roughly 2 orders of magnitude. When extrapolated to infinite salt concentration at the y intercept, the affinity of the SR peptide is still enhanced by a factor of 26, relative to the SS peptide, which translates into a stabilization of the bound state by 2.0 kcal mol−1. This enhancement is due to interactions formed by Arg−3 that are not screened by salt and therefore almost certainly involve short range forces. The slope for the RR peptide is greater than that of the SR peptide by about a factor of 2 in Fig. 9, consistent with the 2-fold difference in formal charge. However, when extrapolated to infinite salt concentration, the affinities of the two peptides are virtually identical, as evinced by the intersection of the two lines at the y axis (Fig. 7, solid lines). Thus, the “extra” Arg−4 forms long range electrostatic interactions in the bound state but no energetically significant short range (i.e. unscreened) interactions.
FIGURE 9.
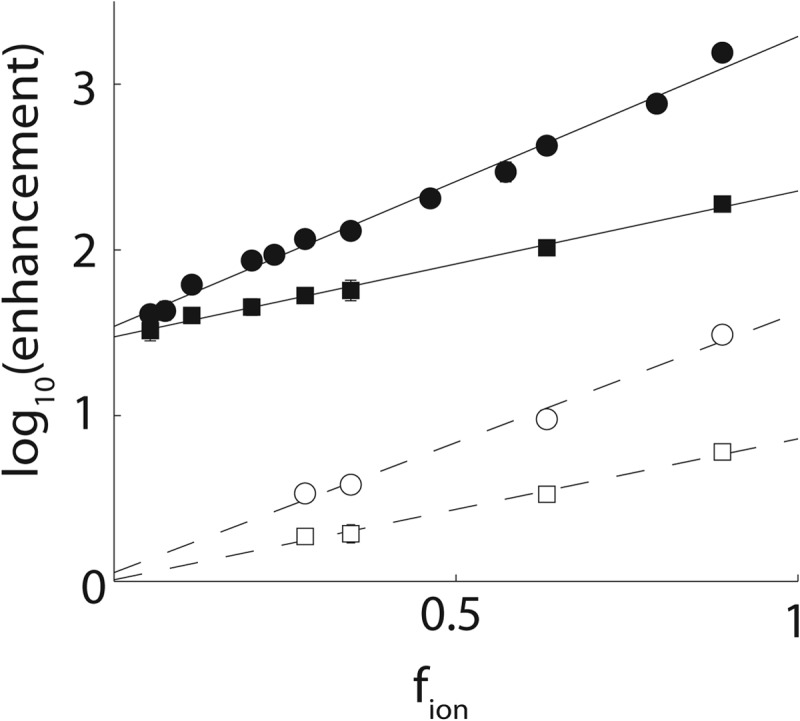
Enhancements in affinities (KaSR,RR/KaSS; solid lines, filled symbols) and association rates (konSR,RR/konSS; dashed lines, open symbols) for the RR (circles) and SR (squares) peptides relative to the SS peptide, plotted as a function of fion.
This is consistent with the x-ray crystal structure of the Fyn SH3 domain bound to a ligand very similar to the RR peptide used in this study (Trp−8 → Val−8), shown in Fig. 2 (40). The conserved Arg−3 forms a salt bridge (i.e. hydrogen-bonds) with Asp17, whereas the aliphatic portion of the residue packs against Trp36. In contrast, the “extra” Arg, Arg−4, points away from Asp17 and makes no additional hydrogen bonds. However, negatively charged Glu33 and Glu34 are within about 6.5 Å, well positioned to interact with Arg−4 electrostatically. Guanidinium ions, equivalent to the guanidino moiety of the Arg side chain, interact with the Fyn SH3 domain specifically at the peptide binding site and compete with the RR peptide (65). Together, these data strongly suggest that the guanidino moiety of Arg−3 interacts with Fyn SH3 domain via a network of hydrogen bonds, whereas Arg−4 forms looser, long range electrostatic interactions in the fully bound complex.
The pattern of enhancements seen for kon is similar but differs in an important respect. At 0 m NaCl, the RR peptide binds 5-fold more rapidly than the SR peptide, which binds 6-fold more rapidly than the SS peptide. This is consistent with both Arg−3 and Arg−4 forming long range electrostatic interactions in the transition state. However, in the extrapolation to infinite salt concentration, both RR and SR peptides bind with rates that are nearly identical to the value determined for the SS peptide. All of the interactions formed by the peptide Arg residues in the transition state are thus electrostatic and long range (i.e. are screened by salt). The short range interactions involving Arg−3 that are present in the fully bound complex are only formed after the protein-peptide pair have passed through the high-energy transition state (i.e. near the end of the binding process). The transition states of far more tightly binding partners are believed to be similarly dominated by long range electrostatic interactions and lacking in short range contacts (5, 14, 15, 22).
Ionic Strength Dependence of Dissociation Rates
For diffusion-limited binding reactions, values of kon are typically far more sensitive to ionic strength than are values of koff (1, 10, 26). In the case of the RR peptide, kon is about 4 times more sensitive to ionic strength than koff. This has been explained in terms of the structure of the transition state (13). Long range electrostatic interactions are fairly insensitive to small changes in charge separation. Thus, if the two binding partners remain close in the transition state, then long range electrostatics contribute similarly to the stabilities of the fully bound and transition states. Increasing the ionic strength screens the long range charge interactions present in both states to equal extents. Thus, the barrier to dissociation is not greatly affected, and koff is essentially unchanged. In contrast, destabilization of the transition state by added salt increases the barrier to association and slows binding rates.
However, this explanation appears inconsistent with mutational analyses of charge interactions in the fully bound and transition states. In the case of barnase-barstar, mutagenesis-derived coupling energies between charged residues in the fully bound state range from 1 to 7 kcal mol−1 (40), whereas coupling energies in the transition state are less than 0.5 kcal mol−1 (5). Similarly, the SS peptide binds 250-fold more weakly than the SR peptide but only 6-fold more slowly. The SS peptide binds 1900-fold more weakly and only 29-fold more slowly than the RR peptide in the absence of salt. These results indicate that the interactions of charged residues are much weaker in the transition state than in the fully bound state. Adding salt should significantly destabilize the bound state without significantly affecting the transition state and thus should increase dissociation rates. Our results provide a simple explanation for this apparent contradiction. We directly measured electrostatic-free basal affinities and association rates for the SS peptide, which allowed us to separate the interaction energies of Arg−3 and Arg−4 into long range “screenable” electrostatic and short range “unscreenable” components. Notably, the long range components are comparable for the fully bound and transition states. For the SR compared with the SS peptide, long range interactions enhance the affinity and association rate by factors of 9 and 6, respectively, at 0 m NaCl, compared with 250- and 6-fold total enhancements. For the RR peptide, these long range enhancement factors are 73 and 29, compared with 1900- and 29-fold total enhancements. Thus, for the Fyn SH3 domain, long range electrostatic interactions are similar in the fully bound and transition states, leading to koff values that are relatively insensitive to ionic strength. This has the implication that the peptide and SH3 domain are separated by a relatively short distance in the transition state, as seen previously for long lived protein complexes (13).
Conclusions
Most of our knowledge of protein binding kinetics derives from studies of extremely stable long lived complexes. The binding kinetics of short lived protein complexes are less well understood because these systems are challenging to characterize experimentally. We have applied a recently developed combination of NMR and ITC to study complexes of the Fyn SH3 domain and several peptides whose bound lifetimes are as short as 0.3 ms. This is beyond the range of standard stopped-flow techniques used to measure binding kinetics. We find that although the binding pathways of these transient SH3 complexes are similar to those of high-affinity long lived complexes, they differ in some important respects.
For the SH3 domain, as well as for high-affinity binding partners, the transition state is stabilized by long range electrostatic interactions, whereas short range stereo-specific contacts are formed late in the binding pathway. We find that the contributions of long range electrostatic interactions to both the fully bound and transition states are quite similar, implying that the separation between the SH3 domain and peptide is not large in the transition state. A similar situation is believed to hold true for the transition states of high-affinity systems. Thus, structural compactness and the dominance of long range electrostatic interactions appear to be universal features of diffusion-limited binding pathways involving affinities and lifetimes spanning up to 7 orders of magnitude.
However, the electrostatic acceleration of SH3 domain association rates is several orders of magnitude less than has been previously observed for stable long lived protein complexes. In contrast, the electrostatic-free basal association rate is much greater, possibly because of looser geometrical restrictions for molecular collisions to proceed to the bound state. This raises the interesting question of whether or not this is a general phenomenon. Do weakly bound, transient complexes intrinsically have more rapid basal association rates and less reliance on electrostatic enhancement than do tightly bound, long lived complexes? If so, how does this relate to the compositions and geometries of the binding interfaces? The combined NMR and calorimetric approach used here is uniquely suited for studying transient complexes to gain a better understanding of the relationships among binding affinities, basal association kinetics, and electrostatic rate enhancements and to shed light on the fundamental principles of molecular recognition.
Acknowledgment
NMR experiments were recorded at the Québec/Eastern Canada High Field NMR Facility, supported by McGill University and Groupe de Recherche Axé sur la Structure des Protéines (GRASP).
This work was supported by a grant from the Natural Sciences and Engineering Research Council (NSERC) of Canada.
- SH3
- Src homology 3
- ITC
- isothermal titration calorimetry
- CPMG
- Carr–Purcell–Meiboom–Gill
- HSQC
- heteronuclear single quantum coherence spectroscopy.
REFERENCES
- 1. Radić Z., Kirchhoff P. D., Quinn D. M., McCammon J. A., Taylor P. (1997) Electrostatic influence on the kinetics of ligand binding to acetylcholinesterase. Distinctions between active center ligands and fasciculin. J. Biol. Chem. 272, 23265–23277 [DOI] [PubMed] [Google Scholar]
- 2. Foote J., Milstein C. (1991) Kinetic maturation of an immune response. Nature 352, 530–532 [DOI] [PubMed] [Google Scholar]
- 3. Wang Y., Shen B. J., Sebald W. (1997) A mixed-charge pair in human interleukin 4 dominates high-affinity interaction with the receptor α chain. Proc. Natl. Acad. Sci. U.S.A. 94, 1657–1662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jucovic M., Hartley R. W. (1996) Protein-protein interaction: a genetic selection for compensating mutations at the barnase-barstar interface. Proc. Natl. Acad. Sci. U.S.A. 93, 2343–2347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schreiber G., Fersht A. R. (1996) Rapid, electrostatically assisted association of proteins. Nat. Struct. Biol. 3, 427–431 [DOI] [PubMed] [Google Scholar]
- 6. Berg O. G., von Hippel P. H. (1985) Diffusion-controlled macromolecular interactions. Annu. Rev. Biophys. Biophys. Chem. 14, 131–160 [DOI] [PubMed] [Google Scholar]
- 7. Zhou H. X. (1997) Enhancement of protein-protein association rate by interaction potential: accuracy of prediction based on local Boltzmann factor. Biophys. J. 73, 2441–2445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Northrup S. H., Erickson H. P. (1992) Kinetics of protein-protein association explained by Brownian dynamics computer simulation. Proc. Natl. Acad. Sci. U.S.A. 89, 3338–3342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gabdoulline R. R., Wade R. C. (2002) Biomolecular diffusional association. Curr. Opin. Struct. Biol. 12, 204–213 [DOI] [PubMed] [Google Scholar]
- 10. Wallis R., Moore G. R., James R., Kleanthous C. (1995) Protein-Protein Interactions in Colicin E9 Dnase-Immunity Protein Complexes. 1. Diffusion-Controlled Association and Femtomolar Binding for the Cognate Complex. Biochemistry 34, 13743–13750 [DOI] [PubMed] [Google Scholar]
- 11. Shen B. J., Hage T., Sebald W. (1996) Global and local determinants for the kinetics of interleukin-4/interleukin-4 receptor α chain interaction: a biosensor study employing recombinant interleukin-4-binding protein. Eur. J. Biochem. 240, 252–261 [DOI] [PubMed] [Google Scholar]
- 12. Janin J. (1997) The kinetics of protein-protein recognition. Proteins Struct. Funct. Genet. 28, 153–161 [DOI] [PubMed] [Google Scholar]
- 13. Zhou H. X. (2001) Disparate ionic-strength dependencies of on and off rates in protein-protein association. Biopolymers 59, 427–433 [DOI] [PubMed] [Google Scholar]
- 14. Alsallaq R., Zhou H. X. (2008) Electrostatic rate enhancement and transient complex of protein-protein association. Proteins 71, 320–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Alsallaq R., Zhou H.-X. (2007) Prediction of protein-protein association rates from a transition-state theory. Structure 15, 215–224 [DOI] [PubMed] [Google Scholar]
- 16. Pilling M., Seakins P. (1995) Reaction Kinetics, Chapter 6, p. 146, Oxford University Press, Oxford [Google Scholar]
- 17. Szabo A., Shoup D., Northrup S., McCammon J. A. (1982) Stochastically gated diffusion-influenced reactions. J. Chem. Phys. 77, 4484–4493 [Google Scholar]
- 18. Sommer J., Jonah C., Fukuda R., Bersohn R. (1982) Production and subsequent 2nd-order decomposition of protein disulfide anions: lengthy collisions between proteins. J. Mol. Biol. 159, 721–744 [DOI] [PubMed] [Google Scholar]
- 19. Camacho C. J., Kimura S. R., DeLisi C., Vajda S. (2000) Kinetics of desolvation-mediated protein-protein binding. Biophys. J. 78, 1094–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Camacho C. J., Weng Z., Vajda S., DeLisi C. (1999) Free energy landscapes of encounter complexes in protein-protein association. Biophys. J. 76, 1166–1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Northrup S. H., Thomasson K. A., Miller C. M., Barker P. D., Eltis L. D., Guillemette J. G., Inglis S. C., Mauk A. G. (1993) Effects of charged amino acid mutations on the bimolecular kinetics of reduction of yeast iso-1-ferricytochrome c by bovine ferrocytochrome b5. Biochemistry 32, 6613–6623 [DOI] [PubMed] [Google Scholar]
- 22. Vijayakumar M., Wong K. Y., Schreiber G., Fersht A. R., Szabo A., Zhou H. X. (1998) Electrostatic enhancement of diffusion-controlled protein-protein association: comparison of theory and experiment on barnase and barstar. J. Mol. Biol. 278, 1015–1024 [DOI] [PubMed] [Google Scholar]
- 23. Volkov A. N., Ubbink M., van Nuland N. A. J. (2010) Mapping the encounter state of a transient protein complex by PRE NMR spectroscopy. J. Biomol. NMR 48, 225–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tang C., Iwahara J., Clore G. M. (2006) Visualization of transient encounter complexes in protein-protein association. Nature 444, 383–386 [DOI] [PubMed] [Google Scholar]
- 25. Sugase K., Dyson H. J., Wright P. E. (2007) Mechanism of coupled folding and binding of an intrinsically disordered protein. Nature 447, 1021–1025 [DOI] [PubMed] [Google Scholar]
- 26. Schreiber G., Fersht A. R. (1993) Interaction of barnase with its polypeptide inhibitor barstar studied by protein engineering. Biochemistry 32, 5145–5150 [DOI] [PubMed] [Google Scholar]
- 27. Sydor J. R., Engelhard M., Wittinghofer A., Goody R. S., Herrmann C. (1998) Transient kinetic studies on the interaction of ras and the Ras-binding domain of c-Raf-1 reveal rapid equilibration of the complex. Biochemistry 37, 14292–14299 [DOI] [PubMed] [Google Scholar]
- 28. Stone S. R., Dennis S., Hofsteenge J. (1989) Quantitative evaluation of the contribution of ionic interactions to the formation of the thrombin-hirudin complex. Biochemistry 28, 6857–6863 [DOI] [PubMed] [Google Scholar]
- 29. Taylor M. G., Rajpal A., Kirsch J. F. (1998) Kinetic epitope mapping of the chicken lysozyme·HyHEL-10 Fab complex: delineation of docking trajectories. Protein Sci. 7, 1857–1867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Altschuh D., Dubs M. C., Weiss E., Zeder-Lutz G., Van Regenmortel M. H. (1992) Determination of kinetic constants for the interaction between a monoclonal antibody and peptides using surface plasmon resonance. Biochemistry 31, 6298–6304 [DOI] [PubMed] [Google Scholar]
- 31. England P., Brégégère F., Bedouelle H. (1997) Energetic and kinetic contributions of contact residues of antibody D1.3 in the interaction with lysozyme. Biochemistry 36, 164–172 [DOI] [PubMed] [Google Scholar]
- 32. Vallurupalli P., Hansen D. F., Stollar E., Meirovitch E., Kay L. E. (2007) Measurement of bond vector orientations in invisible excited states of proteins. Proc. Natl. Acad. Sci. U.S.A. 104, 18473–18477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Demers J. P., Mittermaier A. (2009) Binding mechanism of an SH3 domain studied by NMR and ITC. J. Am. Chem. Soc. 131, 4355–4367 [DOI] [PubMed] [Google Scholar]
- 34. Mittermaier A. K., Kay L. E. (2009) Observing biological dynamics at atomic resolution using NMR. Trends Biochem. Sci. 34, 601–611 [DOI] [PubMed] [Google Scholar]
- 35. Palmer A. G., 3rd, Kroenke C. D., Loria J. P. (2001) NMR methods for quantifying microsecond-to-millisecond motions in biological macromolecules. Methods Enzymol. 339, 204–238 [DOI] [PubMed] [Google Scholar]
- 36. Hansen D. F., Vallurupalli P., Kay L. E. (2008) An improved 15N relaxation dispersion experiment for the measurement of millisecond time-scale dynamics in proteins. J. Phys. Chem. B 112, 5898–5904 [DOI] [PubMed] [Google Scholar]
- 37. Yu H., Chen J. K., Feng S., Dalgarno D. C., Brauer A. W., Schreiber S. L. (1994) Structural basis for the binding of proline-rich peptides to SH3 domains. Cell 76, 933–945 [DOI] [PubMed] [Google Scholar]
- 38. Mayer B. J. (2001) SH3 domains: complexity in moderation. J. Cell Sci. 114, 1253–1263 [DOI] [PubMed] [Google Scholar]
- 39. Hansen D. F., Vallurupalli P., Lundström P., Neudecker P., Kay L. E. (2008) Probing chemical shifts of invisible states of proteins with relaxation dispersion NMR spectroscopy: how well can we do? J. Am. Chem. Soc. 130, 2667–2675 [DOI] [PubMed] [Google Scholar]
- 40. Feng S. B., Kasahara C., Rickles R. J., Schreiber S. L. (1995) Specific interactions outside the proline-rich core of two classes of Src homology 3 ligands. Proc. Natl. Acad. Sci. U.S.A. 92, 12408–12415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mittermaier A., Kay L. E. (2004) The response of internal dynamics to hydrophobic core mutations in the SH3 domain from the Fyn tyrosine kinase. Protein Sci. 13, 1088–1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bolzani R., Ruggeri F., Olivo O. M. (1979) Average normal temperature of the chicken in the morning and after 1–2 days of fasting. Boll. Soc. Ital. Biol. Sper. 55, 1618–1622 [PubMed] [Google Scholar]
- 43. Wishart D. S., Bigam C. G., Yao J., Abildgaard F., Dyson H. J., Oldfield E., Markley J. L., Sykes B. D. (1995) 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J. Biomol. NMR 6, 135–140 [DOI] [PubMed] [Google Scholar]
- 44. Macomber R. S. (1992) An introduction to NMR titration for studying rapid reversible complexation. J. Chem. Ed. 69, 375–378 [Google Scholar]
- 45. Luz Z., Meiboom S. (1963) Nuclear magnetic resonance study of the protolysis of trimethylammonium ion in aqueous solution: order of the reaction with respect to solvent. J. Chem. Phys. 39, 366 [Google Scholar]
- 46. Carver J. P., Richards R. E. (1972) A general two-site solution for the chemical exchange produced dependence of T2 upon the Carr-Purcell pulse separation. J. Magn. Reson. 6, 89–105 [Google Scholar]
- 47. Millet O., Loria J. P., Kroenke C. D., Pons M., Palmer A. G. (2000) The static magnetic field dependence of chemical exchange linebroadening defines the NMR chemical shift time scale. J. Am. Chem. Soc. 122, 2867–2877 [Google Scholar]
- 48. Ishima R., Torchia D. (2005) Error estimation and global fitting in transverse-relaxation dispersion experiments to determine chemical-exchange parameters. J. Biomol. NMR 32, 41–54 [DOI] [PubMed] [Google Scholar]
- 49. Mulder F. A. A., Mittermaier A., Hon B., Dahlquist F. W., Kay L. E. (2001) Studying excited states of proteins by NMR spectroscopy. Nat. Struct. Biol. 8, 932–935 [DOI] [PubMed] [Google Scholar]
- 50. Efron B., Tibshirani R. (1986) Bootstrap methods for standard errors, confidence intervals and other measures of statistical accuracy. Stat. Sci. 1, 54–75 [Google Scholar]
- 51. Di Nardo A. A., Larson S. M., Davidson A. R. (2003) The relationship between conservation, thermodynamic stability, and function in the SH3 domain hydrophobic core. J. Mol. Biol. 333, 641–655 [DOI] [PubMed] [Google Scholar]
- 52. Cantor C., Schimmel P. (1980) Biophysical Chemistry Part III: The Behaviour of Biological Macromolecules, Chapter 16, p. 892, W.H. Freeman and Co., New York [Google Scholar]
- 53. Pace C. N., Vajdos F., Fee L., Grimsley G., Gray T. (1995) How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 4, 2411–2423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Smith A. J. (1997) Amino Acid Analysis. Methods Enzymol. 289, 419–426 [DOI] [PubMed] [Google Scholar]
- 55. Melander W., Horváth C. (1977) Salt effects on hydrophobic interactions in precipitation and chromatography of proteins: interpretation of lyotropic series. Arch. Biochem. Biophys. 183, 200–215 [DOI] [PubMed] [Google Scholar]
- 56. Bennaim A., Yaacobi M. (1974) Effects of solutes on strength of hydrophobic interaction and its temperature-dependence. J. Phys. Chem. 78, 170–175 [Google Scholar]
- 57. Docherty H., Galindo A., Sanz E., Vega C. (2007) Investigation of the salting out of methane from aqueous electrolyte solutions using computer simulations. J. Phys. Chem. B 111, 8993–9000 [DOI] [PubMed] [Google Scholar]
- 58. Segur J. B., Oberstar H. E. (1951) Viscosity of glycerol and its aqueous solutions. Ind. Eng. Chem. 43, 2117–2120 [Google Scholar]
- 59. Schreiber G. (2002) Kinetic studies of protein-protein interactions. Curr. Opin. Struct. Biol. 12, 41–47 [DOI] [PubMed] [Google Scholar]
- 60. Eyring H. (1935) The Activated Complex in Chemical Reactions. J. Chem. Phys. 3, 107–115 [Google Scholar]
- 61. Wherland S., Gray H. B. (1976) Metalloprotein electron-transfer reactions: analysis of reactivity of horse heart cytochrome-C with inorganic complexes. Proc. Natl. Acad. Sci. U.S.A. 73, 2950–2954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Alberty R. A., Hammes G. G. (1958) Application of the theory of diffusion-controlled reactions to enzyme kinetics. J. Phys. Chem 62, 154–159 [Google Scholar]
- 63. Buckle A. M., Schreiber G., Fersht A. R. (1994) Protein-protein recognition: crystal structural-analysis of a barnase barstar complex at 2.0-angstrom resolution. Biochemistry 33, 8878–8889 [DOI] [PubMed] [Google Scholar]
- 64. Xu D., Lin S. L., Nussinov R. (1997) Protein binding versus protein folding: the role of hydrophilic bridges in protein associations. J. Mol. Biol. 265, 68–84 [DOI] [PubMed] [Google Scholar]
- 65. Zarrine-Afsar A., Mittermaier A., Kay L. E., Davidson A. R. (2006) Protein stabilization by specific binding of guanidinium to a functional arginine-binding surface on an SH3 domain. Protein Sci. 15, 162–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Escobar L., Root M. J., MacKinnon R. (1993) Influence of protein surface charge on the bimolecular kinetics of a potassium channel peptide inhibitor. Biochemistry 32, 6982–6987 [DOI] [PubMed] [Google Scholar]
- 67. Wallis R., Leung K. Y., Pommer A. J., Videler H., Moore G. R., James R., Kleanthous C. (1995) Protein-protein interactions in colicin E9 DNase-immunity protein complexes. 2. Cognate and noncognate interactions that span the millimolar to femtomolar affinity range. Biochemistry 34, 13751–13759 [DOI] [PubMed] [Google Scholar]
- 68. Wendt H., Leder L., Härmä H., Jelesarov I., Baici A., Bosshard H. R. (1997) Very rapid, ionic strength-dependent association and folding of a heterodimeric leucine zipper. Biochemistry 36, 204–213 [DOI] [PubMed] [Google Scholar]