

Abstract
The Rho family GTPase Rac1 has been implicated in the regulation of glucose uptake in myoblast cell lines. However, no evidence for the role of Rac1 has been provided by a mouse model. The purpose of this study is to test the involvement of Rac1 in insulin action in mouse skeletal muscle. Intravenous administration of insulin indeed elicited Rac1 activation in gastrocnemius muscle, suggesting the involvement of Rac1 in this signaling pathway. We then examined whether insulin-stimulated translocation of the facilitative glucose transporter GLUT4 from its storage sites to the skeletal muscle sarcolemma depends on Rac1. We show that ectopic expression of constitutively activated Rac1, as well as intravenous administration of insulin, caused translocation of GLUT4 to the gastrocnemius muscle sarcolemma, as revealed by immunofluorescent staining of a transiently expressed exofacial epitope-tagged GLUT4 reporter. Of particular note, insulin-dependent, but not constitutively activated Rac1-induced, GLUT4 translocation was markedly suppressed in skeletal muscle-specific rac1-knockout mice compared to control mice. Immunogold electron microscopic analysis of endogenous GLUT4 gave similar results. Collectively, we propose a critical role of Rac1 in insulin-dependent GLUT4 translocation to the skeletal muscle sarcolemma, which has heretofore been predicted solely by cell culture studies.
Keywords: GLUT4, Akt, AS160, electron microscopy
Insulin-stimulated transport of glucose into skeletal muscle cells occurs through redistribution of the glucose transporter GLUT4 from intracellular storage vesicles to the plasma membrane. This process is crucial for physiological glucose homeostasis because skeletal muscle is responsible for up to 75% of insulin-dependent glucose disposal in humans, and abnormalities in the regulation of skeletal muscle glucose uptake lead to insulin resistance in type 2 diabetes (1–5).
Despite the physiological importance of glucose uptake in skeletal muscle, the mechanism for GLUT4 translocation in response to insulin remains obscure at a molecular level. Poor understanding of insulin signaling in skeletal muscle may be ascribed, at least in part, to the lack of in vitro experimental systems that represent the highly organized structure and function of mature skeletal muscle. In fact, the majority of our understanding of signaling mechanisms was obtained from studies using cultured myocyte lines. Recently, a confocal imaging technique to follow trafficking of fluorescence-labeled GLUT4 in living muscle fibers was developed, elucidating different insulin action between the sarcolemma and inner transverse tubules (6–8). Moreover, transgenic mice expressing an exofacial epitope-tagged GLUT4 reporter in muscle were employed to analyze GLUT4 trafficking in response to insulin and contraction (9, 10). However, to our knowledge, these new experimental techniques have not been applied to test the involvement of a particular signal-transducing enzyme in glucose uptake, as suggested by analysis of cell lines.
In mammalian cells, the Rho family of small GTPases directs diverse cellular processes accompanied by cytoskeletal rearrangements, including cell migration and cell division (11). Its role in the regulation of insulin-dependent glucose uptake in target organs, however, remains poorly understood. In adipocytes, TC10 has been implicated in insulin-stimulated translocation of GLUT4 to the plasma membrane, whereas TC10 may not play a pivotal role in muscle cells (12, 13). Instead, a role of another Rho family GTPase Rac1 has been proposed (13–17). Particularly, it was surprising that ectopic expression of an activated Rac1 mutant by itself induced GLUT4 translocation in L6 myoblasts (17). Whereas activated Rac1 did not affect the activity of Akt, a protein kinase implicated in insulin-stimulated glucose uptake, basal Akt activity was required for Rac1-induced GLUT4 translocation (17). Furthermore, inhibition of the endogenous Rac1 protein blocked insulin-induced GLUT4 translocation (14, 15, 17). These findings were important in that they proposed a novel critical function of Rac1 but were based solely on cell culture studies. To our knowledge, there are no reports demonstrating the pivotal role of Rho family GTPases, including Rac1, in GLUT4 translocation to the skeletal muscle sarcolemma in vivo.
In this study, we show a critical role of Rac1 in insulin-stimulated translocation of GLUT4 to the skeletal muscle sarcolemma by the use of a novel method to examine insulin action in vivo. An exofacial epitope-tagged GLUT4 reporter was ectopically expressed in skeletal muscle, and its translocation to the sarcolemma in response to intravenous administration of insulin was compared between conditional rac1 knockout and control mice. Rac1 requirement for insulin-induced GLUT4 translocation to the sarcolemma is further confirmed by immunogold electron microscopic analysis.
MATERIALS AND METHODS
Materials
Commercially available antibodies against Rac1 (mouse monoclonal; BD Biosciences, San Jose, CA, USA), β-tubulin (mouse monoclonal; Sigma-Aldrich, St. Louis, MO, USA), GLUT4 (rabbit polyclonal; Millipore, Bedford, MA, USA), Akt (rabbit polyclonal; Cell Signaling, Beverly, MA, USA), phospho-Akt(T308) (rabbit polyclonal; Cell Signaling), AS160 (rabbit polyclonal; Millipore), phospho-AS160(T642) (rabbit polyclonal; Novus Biologicals, Littleton, CO, USA), and caveolin3 (mouse monoclonal; BD Biosciences) were used. Antibodies against Myc (mouse monoclonal and rabbit polyclonal) and hemagglutinin (HA; rat monoclonal) epitope tags were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA) and Roche Applied Science (Basel, Switzerland), respectively. Anti-mouse IgG, anti-rabbit IgG, and anti-rat IgG antibodies conjugated with Alexa Fluor 488/546/647 were purchased from Invitrogen (Molecular Probes, Eugene, OR, USA). Horseradish peroxidase-conjugated anti-mouse IgG and anti-rabbit IgG antibodies were purchased from Amersham (Piscataway, NJ, USA).
pB-GLUT4myc7-green fluorescent protein (GFP) (18) was kindly provided by Jonathan Bogan (Yale University School of Medicine, New Haven, CT, USA) and Harvey Lodish (Whitehead Institute for Biomedical Research, Massachusetts Institute of Technology, Cambridge, MA, USA). The cDNA encoding GLUT4myc7-GFP was isolated from pB-GLUT4myc7-GFP and subcloned into pCAGGS (19) (kindly provided by Jun-ichi Miyazaki, Osaka University, Osaka, Japan), generating pCAGGS-GLUT4myc7-GFP. The cDNA encoding triple HA-tagged Rac1(G12V) was produced by the PCR and subcloned into pCAGGS, generating pCAGGS-HA×3-Rac1(G12V). pGEX2T-Myc-PAK(67–150) was previously described (17).
Animal procedures
All animal experiments were carried out according to institutional guidelines of Kobe University Graduate School of Medicine. C57BL/6 (wild-type) mice were purchased from CLEA Japan (Tokyo, Japan). rac1flox/flox mice (20) were mated with muscle creatine kinase (MCK)-Cre transgenic mice (21) to generate rac1flox/flox; MCK-Cre (henceforth referred to as m-rac1-KO) mice. We routinely crossbred rac1flox/flox mice with m-rac1-KO mice and compared phenotypes of sibling male rac1flox/flox and m-rac1-KO mice on a mixed 129Ola-C57BL/6 genetic background. Age-matched wild-type and rac1+/+; MCK-Cre mice on a mixed 129Ola-C57BL/6 genetic background were also used as additional controls in some experiments.
Immunoblot analysis with anti-Rac1 and anti-β-tubulin antibodies
Mouse tissues were isolated following perfusion with 3.7% paraformaldehyde in phosphate-buffered saline [PBS(−)] and homogenized by an Ultra-Turrax homogenizer (IKA, Staufen, Germany) in RIPA buffer [10 mM Tris-HCl (pH 7.4), 1% Nonidet P-40, 0.1% sodium dodecyl sulfate, 0.1% sodium deoxycholate, 0.15 M NaCl, and 1 mM ethylenediaminetetraacetic acid]. Following incubation for 30 min on ice, homogenates were centrifuged at 18,000 g for 15 min, and supernatants were subjected to immunoblot analysis, as described previously (17).
Immunoblot analysis with anti-GLUT4, anti-Akt, anti-phospho-Akt(T308), anti-AS160, and anti-phospho-AS160(T642) antibodies
After food deprivation for 16 h, mice were anesthetized with pentobarbital sodium (100 mg/kg body wt, i.p.), and insulin (5 IU/kg body wt; Eli Lilly, Indianapolis, IN, USA) was administered intravenously. Insulin injection was confirmed by measuring the blood glucose level. Following insulin injection, gastrocnemius muscle was isolated and homogenized by an Ultra-Turrax homogenizer in RIPA buffer supplemented with 100 mM NaF, 10 mM Na4P2O7, and 1 mM Na3VO4. Following incubation for 30 min on ice, homogenates were centrifuged at 18,000 g for 15 min, and supernatants were subjected to immunoblot analysis, as described previously (17). Intensities of the bands were quantified using ImageJ (U.S. National Institutes of Health Bethesda, MD, USA).
RT-PCR
Gastrocnemius muscle was isolated following perfusion with 3.7% paraformaldehyde in PBS(−). The total cellular RNA was isolated from gastrocnemius muscle using the Sepasol-RNA I super reagent (Nacalai Tesque, Kyoto, Japan), according to the manufacturer's instructions. cDNAs were synthesized using the SuperScript III first-strand synthesis system for RT-PCR (Invitrogen), and then amplified using the Expand High Fidelity PCR System (Roche Applied Science) and specific primer sets according to the manufacturer's instructions: for Rac1, 5′-TGGTATCCTGAAGTGCGACA-3′ and 5′-CTTGAGTCCTCGCTGTGTGA-3′ (30 cycles); for Rac2, 5′-GTGGCGTTCTTTCCCAGTTA-3′ and 5′-CGAGAGAGGTGTCAGGAAGG-3′ (30 cycles); for Rac3, 5′-CGATTGAACGGCTGCGGGAC-3′ and 5′-TCCAGGTACTTGACGGAACC-3′ (30 cycles); and for glyceraldehyde-3-phosphate dehydrogenase (GAPDH), 5′-TGAAGGTCGGTGTGAACGGATTTGG-3′ and 5′-ACGACATACTCAGCACCAGCATCAC-3′ (25 cycles). Amplified DNA samples were separated by agarose gel electrophoresis and visualized by ethidium bromide staining.
Pulldown assay for activated Rac1
After food deprivation for 16 h, mice were anesthetized with pentobarbital sodium (100 mg/kg body wt, i.p.), and insulin (5 IU/kg body wt) was administered intravenously. Insulin injection was confirmed by measuring the blood glucose level. Following insulin injection, soleus and gastrocnemius muscles were isolated, frozen in liquid nitrogen, and then cut into small slices on ice. Muscle pieces were suspended in pulldown assay buffer [50 mM Tris-HCl (pH 7.4), 0.5% Nonidet P-40, 0.15 M NaCl, 20 mM MgCl2, and protease inhibitor cocktail (Nakalai Tesque, Kyoto, Japan)], and homogenized with 1 stroke (20 s) in a Potter-Elvehjem homogenizer (Iuchi, Tokushima, Japan) at 3000 rpm on ice. Homogenates were centrifuged at 18,000 g for 1 min, and supernatants were passed through cellulose acetate membrane filters (0.45-μm pore size). Supernatants were added to GST-Myc-PAK(67–150) immobilized on glutathione-Sepharose beads, and incubated at 4°C for 30 min. Glutathione-Sepharose beads were washed twice with pulldown assay buffer, and activated Rac1 bound to the beads was detected by immunoblotting using an anti-Rac1 antibody, as described previously (17).
GLUT4 reporter assay
Plasmid DNA transfer into mouse gastrocnemius muscle by electroporation was carried out essentially as described previously (22). Mice were anesthetized with pentobarbital sodium (100 mg/kg body wt, i.p.), and 100 μg of expression plasmids [pCAGGS-GLUT4myc7-GFP and pCAGGS-HA×3-Rac1(G12V) when needed] dissolved in 50 μl of TE (10 mM Tris-HCl, pH 8.0, and 1 mM ethylenediaminetetraacetic acid) was injected longitudinally into gastrocnemius muscle with a 29-gauge needle. A pair of stainless steel electrode needles fixed 5 mm apart was then inserted into the muscle belly, and square wave electrical pulses (100 V, 50 ms) were applied 3 times at 100-ms intervals, followed by 3 pulses of the opposite polarity, using a pulse generator (CUY21EDIT; Nepa Gene, Chiba, Japan). Seven days later, mice were deprived of food for 16 h and anesthetized with pentobarbital sodium (100 mg/kg body wt, i.p.). Insulin (5 IU/kg body wt) was administered intravenously. Insulin injection was confirmed by measuring the blood glucose level. Twenty minutes after injection, gastrocnemius muscle was isolated and immunostained as described below.
Isolation of gastrocnemius muscle fibers and immunofluorescent microscopy
All procedures were performed at room temperature unless otherwise specified. Gastrocnemius muscle was excised from mice euthanized by cervical dislocation, and fixed with 3.7% paraformaldehyde in PBS(−) for 30 min. Bundles of 1–3 individual fibers were teased from fixed muscle with fine forceps under stereomicroscopy. For the detection of GLUT4myc7-GFP exposed on the cell surface and HA-tagged Rac1(G12V), isolated muscle fibers were incubated in 1% BSA in PBS(−) for >30 min, treated with an anti-Myc antibody (rabbit polyclonal) for 1 h, and washed 3 times with PBS(−). Subsequently, muscle fibers were permeabilized with 0.1% Triton X-100 in PBS(−) for 5 min, washed 3 times with 0.1% Tween 20 in PBS(−), and then incubated in 0.1% Tween 20 in PBS(−) supplemented with mouse Ig blocking reagent (Vector Laboratories, Burlingame, CA, USA) overnight at 4°C. Thereafter, muscle fibers were treated with an anti-HA antibody for 30 min, and washed 3 times with PBS(−). Anti-Myc and anti-HA antibodies were detected with fluorescence-labeled anti-rabbit IgG and anti-rat IgG antibodies, respectively. For the detection of endogenous proteins (i.e., Rac1, GLUT4, and caveolin3), isolated muscle fibers were permeabilized, as described above, and treated with a specific primary antibody and a fluorescence-labeled secondary antibody.
Images were obtained using a confocal laser-scanning microscope (LSM 510 META; Carl Zeiss, New York, NY, USA). Nuclei were stained with 4′,6-diamidino-2-phenylindole and also detected under confocal microscopy. Color conversion was applied in some images to allocate a single color to each epitope by using the Zeiss LSM Image Browser version 3.5. The relative amount of GLUT4myc7-GFP exposed at the plasma membrane was estimated from measurements of Myc and GFP fluorescence signals. Intensities of Myc and GFP fluorescence signals in regions of interest as shown in Fig. 3A were quantified using ImageJ, and ratios of Myc and GFP signals (Myc/GFP) were calculated.
Figure 3.
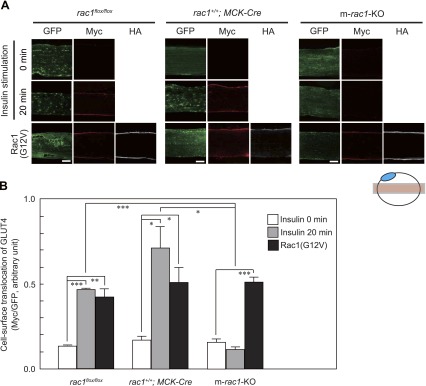
Translocation of GLUT4 to the plasma membrane in skeletal muscle fibers, as revealed by immunofluorescent staining of an exofacial epitope-tagged GLUT4 reporter. A) GLUT4myc7-GFP was expressed with or without constitutively activated HA-tagged Rac1(G12V) in gastrocnemius muscle fibers of rac1flox/flox, rac1+/+; MCK-Cre, and m-rac1-KO mice (8–10 wk old). Insulin (5 IU/kg body wt) was administered intravenously. Total GLUT4myc7-GFP was visualized by fluorescence of GFP (green). GLUT4myc7-GFP translocated to the sarcolemma and externalized to the cell surface and Rac1(G12V) were visualized by immunofluorescent staining with anti-Myc (red) and anti-HA (white) antibodies, respectively. Images were acquired from the focal plane, as depicted in schematic diagram at bottom. Blue ellipse indicates a nucleus. Scale bar = 20 μm. B) Quantification of green and red fluorescence intensity in a representative field of a gastrocnemius muscle fiber, as shown in A. Data represent mean ± se ratios of fluorescence intensity for 3 to 9 independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001.
Immunogold electron microscopy
Immunogold electron microscopic analysis was performed as described previously (23). Insulin responsiveness of subcellular GLUT4 distribution was evaluated by the χ2 test, as described previously (24). The significance of the difference of insulin responsiveness between different mouse genotypes was assessed by the Student's t test.
RESULTS
Generation of conditional rac1-knockout mice
rac1flox/flox mice (20) were mated with MCK-Cre transgenic mice (21) to generate m-rac1-KO mice, in which the rac1 gene is expected to be disrupted specifically in skeletal muscle. The MCK promoter becomes activated during late embryonic development, and the promoter activity remains at a high level thereafter (21).
m-rac1-KO mice were born without any intrauterine loss or early death as suggested by generation of rac1flox/flox and m-rac1-KO littermates at the expected Mendelian ratios (rac1flox/flox: m-rac1-KO=50.8%: 49.2%, 61 pups in total) after cross-breeding of rac1flox/flox mice with m-rac1-KO mice. In addition, m-rac1-KO mice were indistinguishable from their rac1flox/flox littermates in appearance, behavior, and growth, and anatomical and microscopic examination revealed no obvious abnormality in skeletal and heart muscles of m-rac1-KO mice. Both male and female m-rac1-KO mice were fertile.
Expression levels of Rac1 in wild-type, rac1flox/flox, and m-rac1-KO mice were examined by immunoblot analysis (Fig. 1A). As expected, Rac1 expression was largely attenuated in skeletal muscle of m-rac1-KO mice compared to wild-type and rac1flox/flox mice. Slight decrease in Rac1 expression was detected in the heart of m-rac1-KO mice, which may be ascribed to weak Cre expression in cardiac muscle (21). In contrast, the expression level remained unaffected in the liver. Loss of Rac1 expression in gastrocnemius muscle fibers of m-rac1-KO mice is also shown by immunofluorescent staining (Fig. 1B). The expression level and subcellular localization of GLUT4 remained unaffected by rac1 knockout, as shown by immunofluorescent staining (Fig. 1B). Furthermore, we examined the expression level of endogenous GLUT4 by immunoblot analysis, and found no significant difference between control and m-rac1-KO mice (data not shown). In contrast to Rac1, other Rac isoforms, Rac2 and Rac3, were not significantly expressed in gastrocnemius muscle of wild-type and m-rac1-KO mice (Fig. 1C), indicating that rac1 knockout results in a loss of total Rac activity.
Figure 1.
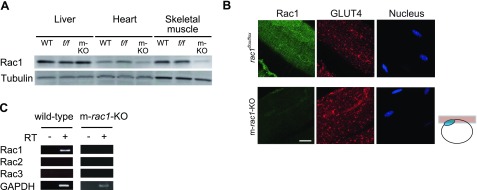
Expression of Rac1 and GLUT4 in skeletal muscle. A) Expression level of Rac1 in liver, heart, and skeletal muscle (gastrocnemius muscle) of wild-type (WT), rac1flox/flox (f/f), and m-rac1-KO (m-KO) mice (8–10 wk old) was analyzed by immunoblotting with an anti-Rac1 antibody. Expression level of tubulin was also analyzed as a loading control. Ten micrograms of protein was loaded on each lane. B) Expression level and subcellular localization of Rac1 and GLUT4 in gastrocnemius muscle fibers of rac1flox/flox and m-rac1-KO mice (8–10 wk old) were analyzed by immunofluorescent staining with specific antibodies. Nuclei were stained with 4′,6-diamidino-2-phenylindole. Images were acquired from the focal plane, as depicted in schematic diagram at right. Blue ellipse in the schematic diagram indicates a nucleus. Scale bar = 20 μm. C) Expression of 3 Rac isoforms (Rac1, Rac2, and Rac3) in gastrocnemius muscle of wild-type and m-rac1-KO mice (8–10 wk old) was analyzed by RT-PCR.
Insulin-dependent activation of Rac1 in mouse skeletal muscle
We first examined by pulldown assay whether Rac1 is activated in gastrocnemius muscle of wild-type mice following intravenous injection of insulin. Following treatment with insulin, the Rac1·GTP level in gastrocnemius muscle was indeed increased (Fig. 2). The fold increase in the Rac1·GTP level after 15-min treatment of insulin was 3.1 ± 0.75 (n=4, P<0.05). The detailed time course of Rac1 activation is shown in Supplemental Fig. 1. Rac1 activation was detected 5 min after insulin injection and continued at least for 20 min.
Figure 2.

Activation of Rac1 in insulin-stimulated mouse skeletal muscle. Insulin (5 IU/kg body wt) was administered intravenously to wild-type mice (8–10 wk old). Following stimulation with insulin for 5 or 15 min, Rac1·GTP level in soleus and gastrocnemius muscle was analyzed by pulldown assay.
GLUT4 translocation as assessed by the use of an exofacial epitope-tagged GLUT4 reporter
We tried to apply the exofacial epitope-tagged GLUT4 reporter GLUT4myc7-GFP (18) to mouse skeletal muscle by expressing this reporter under the control of the CAG promoter (19). The expression plasmid pCAGGS-GLUT4myc7-GFP was introduced into gastrocnemius muscle fibers of living mice by electroporation, and GLUT4myc7-GFP expression was detected 7 d later by green fluorescence. We reproducibly observed that the subcellular localization pattern of GLUT4myc7-GFP was similar to that of endogenous GLUT4 in rac1flox/flox, rac1+/+; MCK-Cre, and m-rac1-KO mice. On intravenous injection of insulin, GLUT4myc7-GFP was translocated to the sarcolemma, and the extracellular domain containing the Myc tag was exposed to the outside in control rac1flox/flox and rac1+/+; MCK-Cre mice (Fig. 3A). The activated mutant Rac1(G12V) with an HA tag, when expressed in gastrocnemius muscle fibers with GLUT4myc7-GFP, also induced translocation of GLUT4myc7-GFP to the sarcolemma without insulin stimulation as observed in L6 myoblasts (17) (Fig. 3A). Localization of translocated GLUT4myc7-GFP and activated Rac1 in the sarcolemma was ascertained by counterstaining of the plasma membrane marker caveolin 3 (Supplemental Fig. 2). In marked contrast to control mice, insulin-dependent translocation of GLUT4myc7-GFP to the sarcolemma was totally abolished in m-rac1-KO mice (Fig. 3A). Rac1(G12V)-induced translocation, on the other hand, remained unaffected by rac1 knockout (Fig. 3A).
Insulin- or Rac1(G12V)-dependent cell surface translocation of GLUT4myc7-GFP was quantified by measuring fluorescent intensities (Fig. 3B). Fluorescent intensities of the anti-Myc antibody relative to those of GFP, indicative of functional cell-surface translocation of GLUT4 (18), were calculated (Fig. 3B). In control rac1flox/flox and rac1+/+; MCK-Cre mice, insulin and Rac1(G12V) caused statistically significant increases in the relative cell-surface GLUT4myc7-GFP level (Fig. 3B). In contrast, Rac1(G12V), but not insulin, induced significant cell surface translocation of GLUT4myc7-GFP in m-rac1-KO mice (Fig. 3B). Overall, these data strongly support the notion that Rac1 is essential for insulin-dependent translocation to the sarcolemma in mouse skeletal muscle.
GLUT4 translocation as assessed by immunogold electron microscopic analysis
Subcellular distribution of endogenous GLUT4 in unstimulated and insulin-stimulated gastrocnemius muscle was analyzed by immunogold electron microscopy (Fig. 4). GLUT4 was localized in various subcellular compartments as previously reported (24, 25). Insulin stimulation remarkably increased the number of gold particles associated with the sarcolemma in rac1flox/flox mouse muscle, whereas in m-rac1-KO mouse muscle, insulin only weakly induced preferential distribution of gold particles to the sarcolemma (Fig. 4).
Figure 4.

Subcellular distribution of GLUT4 in skeletal muscle fibers, as revealed by immunogold electron microscopy. Insulin (5 IU/kg body wt) was administered intravenously to rac1flox/flox and m-rac1-KO mice (8–10 wk old), and subcellular distribution of GLUT4 in gastrocnemius muscle fibers was analyzed by immunogold electron microscopic analysis. Arrows indicate GLUT4 labeling in sarcolemma. G, Golgi apparatus; M, mitochondrion; S, sarcolemma; SR, sarcoplasmic reticulum. Scale bar = 500 nm.
Furthermore, the distribution of gold particles in various intracellular compartments was statistically compared. Gold particles associated with different subcellular compartments, including the plasma membrane (the sarcolemma), intracellular membranes (visible membrane structures), the cytoplasm (nonvisible membrane structures), and mitochondria, were counted, and the labeling distributions in basal (unstimulated) and insulin-stimulated muscle fibers were summarized (Supplemental Table 1). First, the distributions were compared by contingency table analysis (24). Labeling distributions of basal and insulin-stimulated rac1flox/flox muscle fibers were significantly different [e.g., χ2=159, degrees of freedom=3 (2-1 columns by 4-1 rows), and P<0.001 for rac1flox/flox #1; Supplemental Table 1]. There was also significant difference between basal and insulin-stimulated m-rac1-KO mouse muscle fibers [e.g., χ2=11.2, degrees of freedom=3 (2-1 columns by 4-1 rows), and P < 0.05 for m-rac1-KO #1; Supplemental Table 1]. However, χ2 values for m-rac1-KO mice were much lower than those for rac1flox/flox mice, implying that the responsiveness to insulin was largely attenuated by conditional rac1 knockout. In fact, the difference between χ2 values obtained from 5 independent experiments for each genotype was estimated to be statistically significant by the Student's t test (Fig. 5A). Second, the insulin-dependent fold increase in the ratio of gold particles closely associated with the plasma membrane (Supplemental Table 1) was compared between rac1flox/flox and m-rac1-KO mice (Fig. 5B). The difference was again statistically significant as assessed by the Student's t test. Therefore, we conclude that insulin-dependent GLUT4 translocation was significantly impaired in m-rac1-KO mice, although a certain degree of insulin responsiveness was preserved.
Figure 5.

Inhibition of insulin-stimulated redistribution of GLUT4 in skeletal muscle fibers by conditional rac1 knockout. The χ2 value (A) and fold increase in ratio of GLUT4 closely associated with the plasma membrane (B) in rac1flox/flox (f/f) and m-rac1-KO (m-KO) mice (8–10 wk old) (see Supplemental Table 1) are shown as means ± se for 5 independent experiments. ***P < 0.001.
Effect of rac1 knockout on the Akt signaling pathway
The effect of rac1 knockout on the Akt signaling pathway was examined in mouse skeletal muscle. Intravenous injection of insulin indeed caused phosphorylation of Akt and its substrate AS160 in wild-type mouse skeletal muscle, as observed in cultured cell lines (Fig. 6). In m-rac1-KO mice, insulin-dependent phosphorylation of Akt and AS160 remained totally unaffected, suggesting that Rac1 may not be involved in the insulin-induced activation of Akt and AS160 (Fig. 6). Mean values for relative intensities of the bands (m-rac1-KO/wild-type) were 0.98 (n=2) for phospho-Akt(T308) and 0.94 (n=2) for phospho-AS160(T642). These results also highlight the notion that the activation of the Akt pathway alone may not be sufficient for inducing GLUT4 translocation, and Rac1 presumably has a pivotal role downstream of the insulin receptor.
Figure 6.

Effect of conditional rac1 knockout on insulin-stimulated phosphorylation of Akt and AS160. Insulin (5 IU/kg body wt) was administered intravenously to wild-type and m-rac1-KO mice (8–10 wk old). Following stimulation with insulin for 5 min, phosphorylation level of Akt and AS160 in gastrocnemius muscle was analyzed by immunoblotting with phospho-specific antibodies. Relative intensities of bands are shown below the image. Sixty micrograms of protein was loaded on each lane.
DISCUSSION
Here, we demonstrate, by using m-rac1-KO mice, that Rac1 plays a crucial role in insulin-dependent GLUT4 translocation in skeletal muscle. To our knowledge, this is the first in vivo evidence for the involvement of Rho family GTPases in insulin signaling in skeletal muscle. During the preparation of this manuscript, imaging analysis of GLUT4 translocation in cardiomyocytes isolated from transgenic mice carrying an exofacial epitope-tagged GLUT4 reporter was reported (9). Our confocal microscopy-based method to visualize in vivo GLUT4 translocation to the skeletal muscle sarcolemma is similar to this study in that both utilized exofacial epitope-tagged GLUT4 as a reporter in muscle. However, the purpose of the experiment was different from each other. We intravenously injected insulin into living mice to detect GLUT4 redistribution following in vivo stimulation of skeletal muscle in contrast to the approach by Fazakerley et al. (9), where isolated cardiomyocytes were stimulated in vitro. Moreover, our results are novel and unique with respect to the demonstration of a role of a particular signaling molecule by applying the above technique to conditional knockout mice.
Recently, Schertzer et al. (10) also reported a mouse model expressing exofacially tagged GLUT4 driven by a muscle-specific promoter. In this study, an increase in the level of cell surface GLUT4 detected by an antibody against the exofacial Myc tag in response to insulin stimulation or muscle contraction was indeed detected. Thus, it is possible to test the involvement of signaling molecules, such as Rac1, in GLUT4 translocation in a way similar to our present study by crossbreeding these transgenic mice with knockout mice. In contrast to our construct, the GFP tag is not attached to the GLUT4 reporter expressed in these transgenic mice, which has an advantage in identifying GLUT4-binding proteins through immunoprecipitation (10).
Previous studies have revealed that GLUT4 was translocated to transverse tubules, as well as the sarcolemma (6, 25, 26). Time-lapse imaging demonstrated a delay in GLUT4 translocation to transverse tubules, which reflects insulin diffusion to receptors in transverse tubules (6). It remains unclear whether Rac1 is also involved in GLUT4 translocation to transverse tubules, because we concentrated on GLUT4 translocation to the sarcolemma by using a whole molecule of the anti-Myc antibody for the detection of the GLUT4 reporter in this study. We will detect translocation of GLUT4 to transverse tubules with the Fab fragment, and illustrate subcellular compartment-specific regulation of GLUT4 translocation, particularly the role of Rac1, in future studies.
Signaling cascades regulated downstream of Rac1 remain incompletely understood. Rac1-dependent actin cytoskeletal remodeling and membrane ruffle formation have been considered to be important for GLUT4 translocation, particularly GLUT4 vesicle retention beneath the plasma membrane, on the basis of cell culture studies (16, 27). Cortical actin remodeling was also critical in insulin-induced glucose uptake in rat skeletal muscle (28). Although the structure of muscle fibers is largely different from that of myoblast and in vitro differentiated myotube cultures, the regulation of cortical actin remodeling may be a crucial role of Rac1 in skeletal muscle as well, considering that Rac1 is required and even sufficient for insulin-dependent GLUT4 translocation in mature skeletal muscle as shown in this study. However, it remains possible that Rac1 may also be responsible for still uncharacterized functions in addition to the regulation of the actin cytoskeleton. Further studies of signaling pathways will reveal the detailed roles of Rac1.
In L6 myoblasts, basal (unstimulated) Akt activity was required for constitutively activated Rac1-induced GLUT4 translocation, whereas the expression of a constitutively activated Rac1 mutant by itself did not induce the activation of Akt (17). Phosphorylation of Akt and its substrate AS160 in response to insulin administration was indeed observed in mouse skeletal muscle as shown in this study (Fig. 6). In addition, insulin signaling to Akt was totally unaffected by rac1 knockout (Fig. 6). Overall, Rac1 and Akt/AS160 pathways may function independently and collaborate with each other to induce GLUT4 translocation. Detailed mechanisms for the crosstalk between these two signaling pathways are currently being investigated in our laboratory.
In addition to glucose uptake, insulin exerts diverse effects on glucose and lipid metabolism in skeletal muscle (1–3). It remains unclear whether the role of Rac1 is restricted to GLUT4 translocation or Rac1 also regulates various enzymes implicated in glucose and lipid metabolism. If Rac1 has some cooperative role in Akt signaling, as described above, it is possible that rac1 knockout may also affect cellular responses to insulin other than GLUT4 translocation because Akt acts as a key molecule in many facets of insulin action downstream of the insulin receptor (4).
Muscle-specific knockout of GLUT4 caused insulin resistance and glucose intolerance (29). Considering that insulin responsiveness of GLUT4 was severely impaired in m-rac1-KO mice, as described in this study, m-rac1-KO mice may exhibit abnormalities in glucose homeostasis. Alternatively, overall glucose homeostasis may not be severely impaired by muscle-specific rac1 knockout, as observed in muscle-specific insulin receptor-knockout mice, which also exhibited insulin resistance in skeletal muscle (21). If Rac1 is involved not only in insulin induction of glucose uptake, but also in compensatory mechanisms for muscle insulin resistance that function only when insulin receptor-triggered signals are disrupted, m-rac1-KO mice may not develop glucose intolerance like muscle-specific insulin receptor-knockout mice. Further physiological studies employing m-rac1-KO mice will be needed to address these issues.
Supplementary Material
Acknowledgments
The authors are grateful to Jonathan Bogan (Yale University School of Medicine, New Haven, CT, USA), Harvey Lodish (Whitehead Institute for Biomedical Research, Massachusetts Institute of Technology, Cambridge, MA, USA), and Jun-ichi Miyazaki (Osaka University, Osaka, Japan) for plasmid DNAs. The authors also thank Masato Kasuga (International Medical Center of Japan) for critical reading of the manuscript and valuable suggestions and Nobuyuki Takenaka (Kobe University) for technical assistance. This investigation was supported by Grants-in-Aid for Scientific Research on Priority Areas (Systems Genomics and Life Surveyor), Scientific Research (B), the 21st Century COE Program (Center of Excellence for Signal Transduction Disease: Diabetes Mellitus as Model), and the Global COE Program (Global Center for Education and Research in Integrative Membrane Biology) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan, and by a grant from the Takeda Science Foundation.
REFERENCES
- 1. Klip A., Pâquet M. R. (1990) Glucose transport and glucose transporters in muscle and their metabolic regulation. Diabetes Care 13, 228–243 [DOI] [PubMed] [Google Scholar]
- 2. Saltiel A. R., Kahn C. R. (2001) Insulin signalling and the regulation of glucose and lipid metabolism. Nature 414, 799–806 [DOI] [PubMed] [Google Scholar]
- 3. Biddinger S. B., Kahn C. R. (2006) From mice to men: insights into the insulin resistance syndromes. Annu. Rev. Physiol. 68, 123–158 [DOI] [PubMed] [Google Scholar]
- 4. Taniguchi C. M., Emanuelli B., Kahn C. R. (2006) Critical nodes in signalling pathways: insights into insulin action. Nat. Rev. Mol. Cell. Biol. 7, 85–96 [DOI] [PubMed] [Google Scholar]
- 5. Huang S., Czech M. P. (2007) The GLUT4 glucose transporter. Cell Metab. 5, 237–252 [DOI] [PubMed] [Google Scholar]
- 6. Lauritzen H. P., Ploug T., Prats C., Tavaré J. M., Galbo H. (2006) Imaging of insulin signaling in skeletal muscle of living mice shows major role of T-tubules. Diabetes 55, 1300–1306 [DOI] [PubMed] [Google Scholar]
- 7. Lauritzen H. P., Galbo H., Brandauer J., Goodyear L. J., Ploug T. (2008) Large GLUT4 vesicles are stationary while locally and reversibly depleted during transient insulin stimulation of skeletal muscle of living mice: imaging analysis of GLUT4-enhanced green fluorescent protein vesicle dynamics. Diabetes 57, 315–324 [DOI] [PubMed] [Google Scholar]
- 8. Lauritzen H. P., Ploug T., Ai H., Donsmark M., Prats C., Galbo H. (2008) Denervation and high-fat diet reduce insulin signaling in T-tubules in skeletal muscle of living mice. Diabetes 57, 13–23 [DOI] [PubMed] [Google Scholar]
- 9. Fazakerley D. J., Lawrence S. P., Lizunov V. A., Cushman S. W., Holman G. D. (2009) A common trafficking route for GLUT4 in cardiomyocytes in response to insulin, contraction and energy-status signalling. J. Cell Sci. 122, 727–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schertzer J. D., Antonescu C. N., Bilan P. J., Jain S., Huang X., Liu Z., Bonen A., Klip A. (2009) A transgenic mouse model to study glucose transporter 4myc regulation in skeletal muscle. Endocrinology 150, 1935–1940 [DOI] [PubMed] [Google Scholar]
- 11. Bishop A. L., Hall A. (2000) Rho GTPases and their effector proteins. Biochem. J. 348, 241–255 [PMC free article] [PubMed] [Google Scholar]
- 12. Chiang S. H., Baumann C. A., Kanzaki M., Thurmond D. C., Watson R. T., Neudauer C. L., Macara I. G., Pessin J. E., Saltiel A. R. (2001) Insulin-stimulated GLUT4 translocation requires the CAP-dependent activation of TC10. Nature 410, 944–948 [DOI] [PubMed] [Google Scholar]
- 13. JeBailey L., Rudich A., Huang X., Di Ciano-Oliveira C., Kapus A., Klip A. (2004) Skeletal muscle cells and adipocytes differ in their reliance on TC10 and Rac for insulin-induced actin remodeling. Mol. Endocrinol. 18, 359–372 [DOI] [PubMed] [Google Scholar]
- 14. Khayat Z. A., Tong P., Yaworsky K., Bloch R. J., Klip A. (2000) Insulin-induced actin filament remodeling colocalizes actin with phosphatidylinositol 3-kinase and GLUT4 in L6 myotubes. J. Cell Sci. 113, 279–290 [DOI] [PubMed] [Google Scholar]
- 15. JeBailey L., Wanono O., Niu W., Roessler J., Rudich A., Klip A. (2007) Ceramide- and oxidant-induced insulin resistance involve loss of insulin-dependent Rac-activation and actin remodeling in muscle cells. Diabetes 56, 394–403 [DOI] [PubMed] [Google Scholar]
- 16. Ishikura S., Koshkina A., Klip A. (2008) Small G proteins in insulin action: Rab and Rho families at the crossroads of signal transduction and GLUT4 vesicle traffic. Acta Physiol. (Oxf.) 192, 61–74 [DOI] [PubMed] [Google Scholar]
- 17. Ueda S., Kataoka T., Satoh T. (2008) Activation of the small GTPase Rac1 by a specific guanine-nucleotide-exchange factor suffices to induce glucose uptake into skeletal-muscle cells. Biol. Cell 100, 645–657 [DOI] [PubMed] [Google Scholar]
- 18. Bogan J. S., McKee A. E., Lodish H. F. (2001) Insulin-responsive compartments containing GLUT4 in 3T3-L1 and CHO cells: regulation by amino acid concentrations. Mol. Cell. Biol. 21, 4785–4806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Niwa H., Yamamura K., Miyazaki J. (1991) Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108, 193–199 [DOI] [PubMed] [Google Scholar]
- 20. Kassai H., Terashima T., Fukaya M., Nakao K., Sakahara M., Watanabe M., Aiba A. (2008) Rac1 in cortical projection neurons is selectively required for midline crossing of commissural axonal formation. Eur. J. Neurosci. 28, 257–267 [DOI] [PubMed] [Google Scholar]
- 21. Brüning J. C., Michael M. D., Winnay J. N., Hayashi T., Hörsch D., Accili D., Goodyear L. J., Kahn C. R. (1998) A muscle-specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Mol. Cell 2, 559–569 [DOI] [PubMed] [Google Scholar]
- 22. Aihara H., Miyazaki J. (1998) Gene transfer into muscle by electroporation in vivo. Nat. Biotechnol. 16, 867–870 [DOI] [PubMed] [Google Scholar]
- 23. Kitazawa S., Kitazawa R. (2006) In situ detection of specific gene expression during and immediately after transcription at electron microscopic level. J. Struct. Biol. 153, 64–72 [DOI] [PubMed] [Google Scholar]
- 24. Mayhew T. M., Griffiths G., Lucocq J. M. (2004) Applications of an efficient method for comparing immunogold labelling patterns in the same sets of compartments in different groups of cells. Histochem. Cell Biol. 122, 171–177 [DOI] [PubMed] [Google Scholar]
- 25. Ploug T., van Deurs B., Ai H., Cushman S. W., Ralston E. (1998) Analysis of GLUT4 distribution in whole skeletal muscle fibers: identification of distinct storage compartments that are recruited by insulin and muscle contractions. J. Cell Biol. 142, 1429–1446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Marette A., Burdett E., Douen A., Vranic M., Klip A. (1992) Insulin induces the translocation of GLUT4 from a unique intracellular organelle to transverse tubules in rat skeletal muscle. Diabetes 41, 1562–1569 [DOI] [PubMed] [Google Scholar]
- 27. Randhawa V. K., Ishikura S., Talior-Volodarsky I., Cheng A. W., Patel N., Hartwig J. H., Klip A. (2008) GLUT4 vesicle recruitment and fusion are differentially regulated by Rac, AS160, and Rab8A in muscle cells. J. Biol. Chem. 283, 27208–27219 [DOI] [PubMed] [Google Scholar]
- 28. Brozinick J. T., Jr., Hawkins E. D., Strawbridge A. B., Elmendorf J. S. (2004) Disruption of cortical actin in skeletal muscle demonstrates an essential role of the cytoskeleton in glucose transporter 4 translocation in insulin-sensitive tissues. J. Biol. Chem. 279, 40699–40706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zisman A., Peroni O. D., Abel E. D., Michael M. D., Mauvais-Jarvis F., Lowell B. B., Wojtaszewski J. F., Hirshman M. F., Virkamaki A., Goodyear L. J., Kahn C. R., Kahn B. B. (2000) Targeted disruption of the glucose transporter 4 selectively in muscle causes insulin resistance and glucose intolerance. Nat. Med. 6, 924–928 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.