

Abstract
Cadmium (Cd) is a toxic, heavy industrial metal that poses serious environmental health hazards to both humans and wildlife. Lately, Cd and Cd containing compounds have been classified as known human carcinogens and epidemiological data show causal associations with prostate, breast and lung cancer. The molecular mechanisms involved in Cd-induced carcinogenesis are poorly understood and are only now beginning to be elucidated. The effects of chronic exposure to Cd have recently become of great interest due to the development of malignancies in Cd-induced tumorigenesis in animal. Briefly, various in vitro studies demonstrate that Cd can act as a mitogen, stimulate cell proliferation, inhibit apoptosis and DNA repair, and induce carcinogenesis in several mammalian tissues and organs. Thus, the various mechanisms involved in chronic Cd exposure and malignant transformations warrant further investigation. In this review, we will focus on recent evidence of various leading general and tissue specific molecular mechanisms that follow chronic exposure to Cd in prostate, breast and lung transformed malignancies. In addition, this review considers less defined mechanisms such as epigenetic modification and autophagy, which are thought to play a role in the development of Cd-induced malignant transformation.
Keywords: Cancer, Malignant, Transformation, Prostate, Breast, Lung
Introduction
Cadmium (Cd) is a heavy metal classified as a compound that is hazardous to human health by the International Agency for Research on Cancer (IARC) (1). Cd is highly toxic; Acute exposure to Cd causes inflammation followed by cough, dryness and irritation of the nose and throat, headache, dizziness, chest pain, pneumonitis, and pulmonary edema (2). An estimated 70% of Cd used in the metal mining and refining industries, construction, shipyard and Cd-containing batteries is dissipated into the environment (CCCC 8th report) (3). These are considered to be the major sources of human exposure (4, 5). To some extent, diet and tobacco use are additional sources (6). Thus, inhalation, dermal contact and ingestion through air, soil, sediment and water are the primary routes of exposure to Cd. In addition, Cd accumulates in the body over a long period of time (1-3 decades) due to its low excretion levels (7). Epidemiological surveys suggest that long-term exposure to Cd is strongly correlated with an increased risk of prostate, genitourinary, breast, lung and colon cancers as well as hepatocellular carcinoma in humans (4, 8-11). Suggesting that low level exposure to Cd can potentially become a health hazard over the course of one’s lifetime.
Although several epidemiological studies have demonstrated correlation of a strong risk of developing various cancers from Cd exposure in humans (12-18), the precise molecular mechanisms by which Cd-induced malignant transformation occurs in different tissue types are yet to be fully characterized. In this review, we will discuss the possible molecular mechanisms involved in Cd-induced carcinogenesis.
Molecular Mechanisms of Cd Induced Carcinogenesis
Long term environmental exposure to Cd acts as a carcinogen in humans. Chronic exposure of Cd to normal epithelial cells cultured in vitro transforms them to malignant cells, and this is further confirmed in animal models (19). At higher concentrations of Cd exposure, biosynthesis of DNA, RNA, and protein is inhibited. Although direct interaction of Cd with DNA is very minimal, it may act indirectly through epigenetic mechanisms by altering the signaling events upstream of DNA repair and apoptosis [see review (20). Inhibition of DNA repair by Cd is also likely to play an important role in Cd-induced carcinogenesis (20-22). Cd inhibits the binding efficiency of the tumor suppressor p53 to DNA (23) (24), which in turn inhibits base excision repair of DNA damaged by UV light exposure in HeLa cells (25). Cd also inhibits the binding of xeroderma pigmentosum group A (XPA) to DNA. This protein responsible for recognizing DNA damage (26). Human 8-oxo-dGTPase, an enzyme that protects against the incorporation of 8-oxo-dGTP into DNA, is also inhibited by Cd. Thus, Cd is capable of inhibiting DNA repair on various levels and leads to genomic instability, which is associated with tumorigenesis (21).
In cell culture models, Cd induces many biochemical changes, including aberrant gene expression and signal transduction (Figure 1). Importantly, E-cadherin dysfunction, inhibition of DNA methylation, activation of c-fos, c-jun and c-myc and induction of general stress response genes such as metallothionein (MT) and heat-shock proteins are associated with Cd-induced transformation (26). In addition, Cd has been shown to influence transcription factor activation and total translation levels. For example, transcription factors NF-kB and AP-1 become activated by Cd in both cell culture and in animal (27-29). In contrast, inhibition of NF-kB activation has been reported in Cd-treated cells (30). Epigenetic mechanisms associated with Cd carcinogenesis and malignant transformation include aberrant DNA methylation events that emerge during malignant transformation, some of which occur in an agglomerative fashion (31). Agglomerative DNA hypermethylation was found in Cd-transformed prostate epithelial cells, linking chronic Cd exposure in vitro with hypermethylation of large chromosomal regions (31). Cd-induced aberrant DNA methylation of nucleosomes that are normally trimethylated at histone H3 lysine-27 affect gene expression in stem cells (31).
Figure 1. Expression pattern of Cd induced gene expression in prostate, breast and lung cancer.
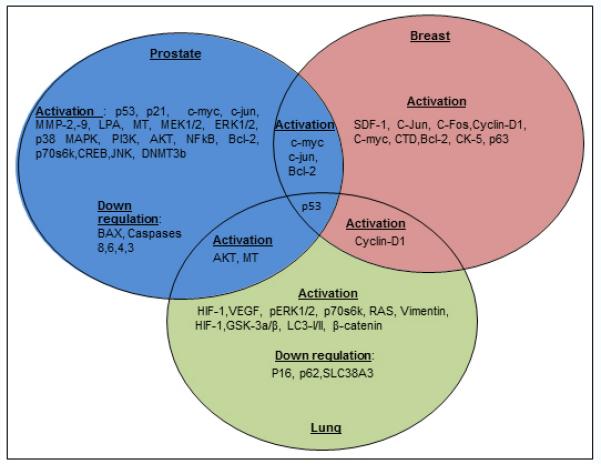
In prostate cancer Cd induced cycle regulatory proteins, pro-surival transcription factors and gene expression in normal prostate epithelial cells. Also, it simultaneously down regulated pro-apoptotic genes such as BAX and caspase 8,6,4,3 in transformed prostate cells. Interestingly, following Cd exposure, prostate and breast cancer share activation of c-myc, c-jun and bcl-2 expression. It also appears that p53 activation is a common event in all three tissue types (prostate, breast and lung) following chronic Cd treatment. Cyclin-D1 expression was found in both breast and lung cancer, however, there is no report in prostate epithelial cells. Similarly, lung and prostate cancer cells shares the common pro-survival gene AKT along with MT following Cd treatment.
Reactive oxygen species and Cd Induced Carcinogensis
Reactive oxygen species (ROS) are often implicated in Cd-induced cytotoxicity in a variety of cell culture models and lead to mitochondrial dysfunction, inhibition of respiration, induction of oxidative stress and lipid peroxidation (32, 33). Cd causes oxidative damage by inducing lipid peroxidation, DNA strand breaks, and chromosomal aberrations (34). More specifically, the depletion of glutathione and protein-bound sulfhydryl groups (35), which have been implicated in carcinogenesis, which disrupts antioxidant defense mechanisms and results in increased lipid peroxidation, oxidative DNA damage (21, 33) and alteration of the cellular redox signaling network (36, 37). Cd-induced ROS has been implicated in carcinogenesis (21), in part by disruption of antioxidant defenses, resulting in increased lipid peroxidation and oxidative DNA damage (33) (21). Depletion of cellular antioxidants has been suggested as the mechanism by which Cd facilitates exacerbation of ROS related cellular and DNA damage, thus further promoting carcinogenesis. In addition, prolonged oxidative stress causes changes in cellular redox homestasis and leads to abnormal activation of redox-sensitive signaling molecules (38). Further oxidative stress, such as observed in Cd-exposure, can cause genetic and epigenetic changes, uncontrolled cell growth, and abnormal cellular signaling, all of which are primary mechanisms involved in metal-mediated carcinogenesis (39) (40) (41).
Cadmium and Prostate Cancer
Prostate cancer is one of the most frequently occurring cancers in men. The etiology of prostate cancer development is complex and is associated with a multitude of potential contributing factors (23, 42, 43) (Table 1). Experimental evidence has shown that long-term exposure of normal human prostate epithelial cells (RWPE-1) to Cd in culture transforms them to malignant phenotypes (19, 44). However, the molecular mechanisms associated with this transformation remain elusive.
Table 1.
Gene expressions of Cd-induced prostate epithelial malignant transformation.
| S.No | Reference | Cell type/model |
Cd conc. | Molecular Events |
|---|---|---|---|---|
| 1 | Achanzar et al.(2000) | RWPE-1 | 10μm | p53, c-myc, c-jun ↑ |
| 2 | Achanzar et al.(2001) | RWPE-1 | 10μm | MMP-2, MMP-9↑ |
| 3 | Achanzar et al.(2002) | CTPE/Tumor | 10μm | Bcl-2 ↑, BAX, Caspase 8,6,4,3 ↓ |
| 4 | Arriazu et al.(2003) | Rat | 60ppm | p53, LPA ↑ |
| 5 | Gaddipati et al.(2003) | RWPE-1 | .025-10μm | MT ↑ |
| 6 | Misra et al.(2003) | 1LN | .01-1μm | MEK1/2, ERK1/2, p38 MAPK, JNK1/2, AKT, PI3K, p70s6k, NFkB, CREB ↑ |
| 7 | Qu et al.(2006) | RWPE-1 | 10μm | MT, JNK ↑ |
| 8 | Qu et al.(2007) | CTPE | 10μm | Bcl-2 ↑, BAX, JNK1/2 ↓ |
| 9 | Benbrahim et.al.(2007) | RWPE-1 | 10μm | DNMT3b↑ |
| 10 | Albrecht et al.(2008) | RWPE-1 | 3-12μm | MT ↑ |
| 11 | Aimola et al.(2012) | RWPE-1, CTPE |
10μm | p53, p21 ↑ |
These transformed prostate cells give rise to aggressive tumors in nude mouse models (19, 20), which further supports the potential for Cd-induced carcinogenesis in humans. The rat is an ideal experimental model for human prostate cancer (45-47), because the morphological similarities between human prostatic intraepithelial neoplasia (PIN) and dysplastic changes in rat prostate are experimentally similar (48). Several studies have demonstrated that Cd induces preneoplastic (hyperplastic) lesions and tumors (adenocarcinomas) in rat prostate (49-52). There are also useful mouse models. Cd transformed human prostate epithelial cells (CTPE) form malignant tumors in nude mice, which morphologically and biochemically resemble human prostatic carcinoma (19). Thus, Cd transformed human prostate epithelial cells may be useful for examining the molecular events involved in Cd-induced carcinogenesis in the prostate.
Based on the current literature, Cd has multiple molecular targets and thus, Cd-induced prostate cancer is likely to develop through more than one signaling pathway. (For a summarized table of Cd-induced gene expression in prostate cells, please refer to Table 1.) Cd can affect cell proliferation and differentiation, cell cycle progression, DNA synthesis and repair, apoptosis and other cellular activities (53, 54). Lysophosphatidic acid (LPA), a phospholipid growth factor involved in a variety of cellular processes such as proliferation, differentiation, migration, inflammation, angiogenesis, wound healing, cancer invasion and survival is one of the most investigated molecules in experimental models (55-57). Cd-treated rats showed significantly higher LPA-1 expression in dysplastic lesions compared with that in normal prostate epithelium in controls (42). In addition, LPA may induce proliferation and survival of androgen-independent prostate cancer cells (55).
Cd inhibits DNA repair in various in vitro models of prostate cancer (22) Activation of p53 governs multiple cellular events including inhibition of angiogenesis, cell cycle regulation, DNA repair and genomic stability. Inhibition of p53 activation in Cd-treated cultures results in abrogation of apoptosis, suggesting that p53 activation is necessary for Cd induced apoptosis (23). Interestingly, short-term treatment of normal prostate epithelial cells with Cd induced apoptosis, however, chronic exposure to Cd allows the cells to eventually become resistant to apoptosis during malignant transformation (58). These results were confirmed in CTPE cells that were resistant to Cd-induced apoptosis even after weeks of being maintained in Cd-free medium, when compared with normal RWPE-1 control cells (59). Analysis of molecular signaling in CTPE cells revealed that the ratio between Bcl-2/Bax was 5 times higher than in the parental RWPE-1 cell line, which conferred greater resistance to apoptosis induction (59). The Bcl-2/Bax dimer abrogates the pro-apoptotic function of Bax and protects the cells from apoptotic stimuli (60). CTPE cells also display reduced expression of several other pro-apoptotic genes, including caspases −8, −4, −6, −3 and −10a at both the protein and mRNA levels when compared to RWPE-1(59). The caspases are an integral part of the apoptotic machinery. They are involved in both commitment to and execution of programmed cell death, and reduction of caspase activity has been shown to obstruct apoptosis (61). Thus, the acquisition of apoptotic resistance may be an important characteristic of Cd-induced malignant transformation. Qu et al. suggest that Bcl-2 activation inhibits the JNK-mediated pro-apoptotic signal transduction pathway by demonstrating that the BH4 domain of Bcl-2 binds to JNK, thus blocking phosphorylation of JNK in CTPE cells (62). Markers of stress and inflammatory responses such as p38, JNK, c-fos, c-myc and metallothionein (MT) are also upregulated in a Cd-dose dependent manner in normal prostate epithelial cells in vitro (62-65).
Cadmium and Breast Cancer
Cd exposure has long been associated with the development of breast cancer (66) (67, 68). Recent clinical studies revealed that high concentrations of Cd (ranging from 3.2 to 86.9 g/g) were found in breast tissues of breast cancer patients (68), which was significantly higher than in normal breast tissue controls (0.022 μg/g) (69). Although these studies suggest a correlation between Cd exposure and incidence of breast cancer, they fail to determine whether Cd is a major source for the etiology of breast cancer. Molecularly, Cd mimics estrogen and acute exposure to Cd is known to promote estrogen receptor (ER)-mediated pro-survival signaling and cell growth (70, 71). Hence, Cd is often referred to as a metalloestrogen. However, the role of Cd acting on ER as part of a mechanism for carcinogenesis in breast remains unclear. Results in MCF-7 breast epithelial carcinoma cells suggest that prolonged exposure to Cd results in development of more aggressive cancer phenotypes including increased cell growth, migration and invasion (72). In addition, Cd has been shown to induce various pro-survival and cell cycle regulatory genes (Table 2). Chronic exposure of MCF-7 cells to Cd induces higher levels of SDF-1 expression in an estrogen receptor alpha (ERα)-dependent manner by altering the molecular dynamics between ERα and c-jun/c-fos (72) ,and inhibiting p53 activation. In the presence of Cd, the anti-cancer drug 5-fluorouracil (5-FU) fails to induce apoptosis in MCF-7 cells Furthermore, in a study using Cd concentrations as low as 1μM, significant cell proliferation in three ERα-positive breast cancer cell lines (MCF-7, T-47D and ZR-75-1 was associated with upregulation of pro-survival genes such as CycD1, c-myc and CTD. Subsequent silencing of the ERα or blocking the receptor with a competitive antagonist mitigated the stimulatory effect of Cd on ER+ breast cancer cell lines (MCF-7, T-47D and ZR-75-1), thus suggesting the involvement of ERα in mediating the cellular effects of Cd (73). However, despite our current understanding, there remains little literature addressing the mechanisms by which Cd induces transformation of normal breast epithelial cells.
Table 2.
Gene expressions of Cd-induced breast epithelial malignant transformation.
| S.No | Reference | Cell type/model |
Cd conc. | Molecular Events |
|---|---|---|---|---|
| 1 | Meplan et al, (1999) | MCF-7 | 10-30μm | p53↑ |
| 2 | Ponce et al. (2013) | MCF-7 | 10μm | SDF-1, c-jun, c-fos ↑ |
| 3 | Siewit et al. (2010) | MCF-7,T-47D, | 10−6M | CycD1, c-myc, CTD↑ |
| 4 | Asara et al. (2013) | MCF-7 | 5μm | p53, pp53, c-myc, Bcl-2↑ |
| 5 | Benbrahim et al. (2009) | MCF-10a | 2.5μm | CK5, p63↑ |
Normal human breast epithelial MCF-10A cells treated with Cd (2.5μm) for 40 weeks transformed into cells displaying a basal-like phenotype. These transformed MCF-10A cells displayed increased colony formation, invasive potential, and loss of contact inhibition (74). This suggests that ER-negative human breast epithelial cells can undergo transformation with chronic Cd exposure, and that Cd as a metalloestrogen in this case is unlikely. Benbrahim-Talla et al. report that chronic Cd treatment induced altered DNA methylation status of their MCF-10A cells, thereby altering gene expression, such as oncogenes, which may play a role in carcinogenesis. In addition, Roy et. al report that MCF-12A and MCF-12F showed significantly high induction of cholinephosphotransferase (CPT) activity, the terminal enzyme in de novo synthesis of phoshpatidylcholine, following treatment of 10 and 25 μM Cd respectively. In MCF-12F, which is the floating form of MCF-12A, a large number of nucleotide mutations were discovered following Cd treatment (75). Suggesting that even low dose and acute exposure to Cd can bring about DNA damage in normal breast cancer cells. Thus giving insight into the possible initial mechanisms of action regarding Cd-induced carcinogenesis.
Cadmium and Lung Cancer
Epidemiological studies suggest that occupational and environmental exposure to Cd induces lung cancer in rodents as well in humans (1, 76, 77). Chronic inhalation of Cd causes pulmonary adenocarcinomas (20, 77) in animal and in exposed human cell lines (78). Prolonged exposure of normal bronchial epithelial cells to Cd in vitro causes them to transform to a malignant state, and a similar observation was noted in a nude mouse model (79). Although the mechanisms of Cd-induced pulmonary carcinogenesis are still incompletely defined, the development of specific cell lines has greatly aided in defining the mode of Cd-mediated lung cancer development. Cd-induced, transformed human lung epithelial cells called chronic cadmium treated lung cells (CCT-LC) exhibit increased MMP-2 activity, colony formation, invasion, autonomous growth, EMT, and hyperproliferation (78). Malignant transformed lung cells as well displayed altered pro-survival, apoptotic and DNA regulatory gene expression following Cd treatment (Table 3). Chronic Cd-treatment of BEAS-2B cells for example resulted in a marked increase in cell migration and invasion compared with Cd naïve control cells. In addition, CCT-LC cells displayed a loss of p16 expression and an increase in cyclin D1 expression, both of which are commonly associated with rapidly proliferating tumor cells and lung cancers (80-82). CCT-LC cells also displayed over-expression of K-RAS, N-RAS, Vimentin and loss of SLC38A3, which are also commonly associated with a general cancer phenotype and/or specifically with lung cancers (81),(83),(84). However, the precise mechanistic details of the initiation of Cd-induced bronchial epithelial transformation have yet to be elucidated. The overexpression of major metallothionein (MT) isoforms is associated with both Cd-induced and lung cancers (85, 86). Specifically, MT-1A and MT-2A levels were shown to be significantly higher in CCT-LC as compared to control (78), suggesting that MT may increase its expression in the lung to provide protection from Cd-induced toxicity.
Table 3.
Gene expressions of Cd-induced lung epithelial malignant transformation.
| S.No | Reference | Cell type/model |
Cd conc. | Molecular Events |
|---|---|---|---|---|
| 1 | Lei et al, (2008) | 16HBE | p36↑ | |
| 2 | Jing et al. (2012) | BEAS-2B | 1μm | HIF-1,VEGF, pERK1/2, p-p70s6k, pAKT ↑ |
| 3 | Person et al. (2013) | HDL-1D | 5μm | CycD1, K-RAS, N-RAS, Vimentin, MT1/2, HIF-1 ↑, p16 and SLC38A3 ↓ |
| 4 | Park et al. (2013) | MCF-7 | 3-12μm | b-catenin, GSK-3a/β, LC3-I/II↑, p62↓ |
| 5 | Son et al. (2012) | BEAS-2B | 2μm | AKT, GSK-3β, β-catenin↑ |
| 6 | Lag et al. (2002) | Prim. Lung Epi | 1-10μm | BAX, p53↑ |
Once CCT-LC cells have acquired multiple tumor characteristics, it activates multiple signaling networks sequentially. Oxidative stress conditions are induced by antioxidative proteins Heme Oxygenase-1 (HO-1) and hypoxia inducible factor-1A (HIF-1α) (78). HIF-1 expression also occurs through Cd-induced reactive oxygen species formation associated with transformation of bronchial epithelial cells (87). Also, Cd activates AKT, GSK-3β, and β-catenin signaling in BEAS-2B human bronchial epithelial cells in a ROS-dependent manner (88). A marked induction of AKT, GSK-3β and β-catenin was maintained in tumor tissues obtained from mice that had been injected with Cd-stimulated BEAS-2B cells (88), suggesting direct involvement of ROS in Cd-induced carcinogenesis. These results also implicate a role for the AKT, GSK-3β, and β-catenin signaling pathways, as well as for c-myc and COX-2, in contributing significantly to the development of Cd-induced tumors (88). These results further support the potential of Cd to induce metastasis. Increased expression of the proliferation marker gene PCNA and the cell cycle gene CyclinD1 in Cd treated cells suggest that Cd has mitogenic potential. Coincidentally, expression of the anti-apoptosis marker Bal-2 decreased significantly as the number of 16HBE cell culture passages increased during continuous Cd treatment (89). These results suggest that apoptotic resistance could be a viable mechanism for Cd-induced malignant transformation in human bronchial epithelial cells.
Although Cd does not directly induce mutagenesis, chronic Cd treatment in human bronchial epithelial cells may impart DNA damage and decrease DNA repair capacity and genomic instability in an indirect manner. Among the over 100 DNA repair genes, hMSH2, hMLH1, ERCC1, ERCC2, XRCC1, hOGG1, and MGMT are of particular interest in this process. In 16HBE cells, Cd progressively reduced the mRNA and protein expression of these DNA repair genes (89), suggesting that DNA repair systems are diminished and DNA repair capacity is lowered during Cd-induced transformation. DNA sequence analysis in these cells revealed frame shift mutations in exons of the hMSH2, ERCC1, XRCC1, and hOGG1 genes (89), suggesting that Cd may facilitate promutagenic DNA damage and genomic instability at submicromolar concentrations. In addition, expression levels of p53 were significantly up regulated following treatment with 10μm Cd in primary epithelial lung cells (90). Thus, suggesting the involvement of Cd in DNA damage events in lung. However, no study has demonstrated weather these promutagenic effects are a direct result of Cd/DNA interactions. Induction of autophagy may play an important role in the development of Cd-induced Cd resistance and carcinogenesis. Autophagy is a process of lysosome-dependent degradation of proteins and organelles whereby portions of the cytoplasm are engulfed by double-membrane enclosed vesicles called autophagosomes, which then fuse with lysosomes. The contents of the autophagosomes are degraded within the lysosomes and recycled, thus indicating that autophagy protects cells. Recently, GSK-3β has been shown to mediate autophagy during Cd toxicity (91). Furthermore, acquisition of Cd resistance in human H460 cells, which are non-small lung carcinoma cells, has been linked directly to interruption of GSK-3α/β phosphorylation, and the resulting change in intracellular localization of GSK-3α/β regulates Cd-induced autophagy and apoptosis (92). However, the role of autophagy and the underlying mechanisms involved in Cd-induced carcinogenesis need to be further elucidated.
Conclusion and Future Directions
The initiation and progression of Cd-induced carcinogenesis are complex processes. the factors that contribute to cancer development can vary significantly in their source, duration, concentrations and route of exposure. Cd exposure may accelerate the initiation or progression of the disease when combined with other factors. While the direct Cd-related risks for cancer development is less defined, the increasing body of epidemiological evidence seems to establish chronic Cd-exposure as a viable candidate.
Overall, environmental contaminants such as heavy metals have emerged as possible high risk factors for cancer due to their increased usage in various industrial processes and widespread proliferation in food sources. Cd in particular acts as a double edge sword, by causing cell death as well as cell-survival. Induction of cell survival and proliferation, which leads to transformation, suggests that Cd is a potent carcinogen. On the other hand, it may enhance cell death by a variety of mechanisms, thus leading to tissue pathology and organ damage. In either case, the outcome of exposure to Cd on the cellular level is dependent not only upon the duration and level of exposure, but also on intrinsic tissue specificity and current metabolic state. In in vitro models of all three organs reviewed, chronic exposure to Cd has been shown to transform cells into apoptosis-resistant malignant cells. In addition, the concentration dependent effects of Cd have also been highlighted. Generally, sub-micromolar concentrations lead to proliferation or delayed apoptosis, intermediate concentrations (10 μM) can cause various types of cell death, while very high concentrations (>50 μM) can cause necrosis.
A common set of mechanisms by which most tissue types respond to chronic Cd exposure has been defined. Induction of ROS and ROS related DNA damage appears to be part of the initial events in Cd-induced toxicity in humans. However, the molecular mechanisms accounting for Cd-induced oxidative stress in mammalian organs remains incompletely defined, although the involvement of cellular mitochondria is highly plausible, given that these organelles are central to the formation of excess ROS and are known key intracellular targets for Cd. Cd-induced ROS-related genomic instability seems to be a central player in Cd-induced carcinogenesis, however other mechanisms are common in several tissue types. For example, ROS-related mitotic perturbation in malignant transformed cells has been implicated in the induction of various epigenetic modifications. However, it remains unclear if epigenetic modifications result directly from Cd interaction with the genome or via enhanced ROS levels during Cd exposure. In addition, targeted Cd-induced epigenetic modifications, such as hypermethylation of specific target genes, have yet to be further characterized. These open areas of exploration make the study of Cd-induced epigenetic changes during malignant transformation an attractive pursuit, and will likely elucidate many mechanisms that underlie the initiation and development of Cd-induced carcinogenesis. Further investigation of Cd-induced perturbation of gene regulation associated with ROS and epigenetic mechanisms are equally warranted.
Cd-related changes in expression levels of some proteins may be associated in part with a cellular compensatory response to increasing ROS levels. Nevertheless, the mechanisms by which Cd-induced pro-survival mechanisms or inhibition of pro-apoptotic machinery lead to increases in ROS levels remains to be further examined. Cd-induced apoptosis and expression of pro-survival genes play an important role in selection and subsequent development of Cd-resistance during Cd-induced malignant transformation. This suggests that induction of certain specific factors during chronic exposure to Cd is essential for carcinogenesis, and emphasizes the double edge sword nature of the effects of Cd on cell populations. More importantly, identification of factors/genes that dictate the molecular events responsible for Cd-induced transformation should be investigated extensively.
Another area that requires further attention is Cd-induced autophagy and autophagic cell death (ACD). Based on in vitro studies, autophagy is engaged for cell survival under stress by removing proteins and organelles. While cells can display extensive autophagic vacuolization before or during death, it remains unclear whether this represents cell death by autophagy. Thus, further investigating the possibility of Cd-induced inhibition of autophagy during Cd-exposure is required to conclude that Cd-induced cell death is ACD. Regardless, if Cd exposure directly induces autophagy, then characterizing the mechanisms governing this process would be of great importance in further understanding Cd-induced malignant transformation and the development of Cd-resistance.
All organs discussed share some overlapping mechanistic features in response to Cd exposure. These shared responses of Cd-induced gene expression are illustrated in Figure 1. For example, indicators of Cd-induced ROS stress and genomic instability involving key players like metallothionein and p53 appear consistent throughout all three Cd-exposed tissue types. On the other hand, certain mechanisms that mediate Cd exposure appear to be unique to each specific tissue type. Mechanisms involving ER-dependent pathways are important for the study of Cd-metalloestrogen driven malignancies such as breast cancers. ER-independent studies of Cd-induced malignant transformation are necessary to further understand the shared profile of Cd-induced carcinogenesis in breast tissue and other transformed organ tissues.
While this review focuses on chronic Cd exposure and resulting malignant transformations, the majority of literature on in vitro Cd exposure seems to center around acute Cd toxicity, which creates a vacuum for chronic Cd studies in vitro and in animal models. As a result, the multi-faceted mechanisms involved in chronic Cd-induced malignant transformations require much more detailed characterization. Thus, further studies using low concentrations of Cd, which mimics chronic human exposure, are warranted to achieve further insight into Cd as a carcinogen.
Footnotes
Declaration of Interests
The employment affiliation of the authors is shown on the cover page. This study was supported by the ACS-RSG-10-186-01-TBE, R01CA140605 and R01CA138797.
References
- 1.Meeting of the IARC working group on beryllium, cadmium, mercury and exposures in the glass manufacturing industry Scand J Work Environ Health. 1993;19(5):360–3. doi: 10.5271/sjweh.1461. [DOI] [PubMed] [Google Scholar]
- 2.Roy SS, Mahapatra R, Rath S, Bajpai A, Singh V, Nair N, et al. Improved neonatal survival after participatory learning and action with women's groups: a prospective study in rural eastern India. Bull World Health Organ. 2013;91(6):426–33B. doi: 10.2471/BLT.12.105171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cadmium and Certain Cadmium Compounds 8th Report on Carcinogen US Department of Health and Human Services, National Toxicology Program. 1998;II:III-405–III-8. [Google Scholar]
- 4.Kjellstrom T, Friberg L, Rahnster B. Mortality and cancer morbidity among cadmium-exposed workers. Environ Health Perspect. 1979;28:199–204. doi: 10.1289/ehp.28-1637490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nordberg GF, Goyer RA, Clarkson TW. Impact of effects of acid precipitation on toxicity of metals. Environ Health Perspect. 1985;63:169–80. doi: 10.1289/ehp.8563169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Satarug S, Moore MR. Adverse health effects of chronic exposure to low-level cadmium in foodstuffs and cigarette smoke. Environ Health Perspect. 2004;112(10):1099–103. doi: 10.1289/ehp.6751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Suwazono Y, Kido T, Nakagawa H, Nishijo M, Honda R, Kobayashi E, et al. Biological half-life of cadmium in the urine of inhabitants after cessation of cadmium exposure. Biomarkers. 2009;14(2):77–81. doi: 10.1080/13547500902730698. [DOI] [PubMed] [Google Scholar]
- 8.Satarug S. Long-term exposure to cadmium in food and cigarette smoke, liver effects and hepatocellular carcinoma. Curr Drug Metab. 2012;13(3):257–71. doi: 10.2174/138920012799320446. [DOI] [PubMed] [Google Scholar]
- 9.Takenaka S, Oldiges H, Konig H, Hochrainer D, Oberdorster G. Carcinogenicity of cadmium chloride aerosols in W rats. J Natl Cancer Inst. 1983;70(2):367–73. [PubMed] [Google Scholar]
- 10.Waalkes MP, Rehm S, Perantoni AO, Coogan TP. Cadmium exposure in rats and tumours of the prostate. IARC Sci Publ. 1992;118:391–400. [PubMed] [Google Scholar]
- 11.Julin B, Wolk A, Bergkvist L, Bottai M, Akesson A. ietary cadmium exposure and risk of postmenopausal breast cancer: a population-based prospective cohort study. Cancer Res. 2012;72(6):1459–66. doi: 10.1158/0008-5472.CAN-11-0735. [DOI] [PubMed] [Google Scholar]
- 12.Basu M, Mukhopadhyay S, Chatterjee U, Roy SS. FGF16 Promotes Invasive Behavior of SKOV-3 Ovarian Cancer Cells through Activation of Mitogen-activated Protein Kinase (MAPK) Signaling Pathway. J Biol Chem. 2014;289(3):1415–28. doi: 10.1074/jbc.M113.535427. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 13.Xu F, Wu MY, Safron NS, Roy SS, Jacobberger RM, Bindl DJ, et al. Highly Stretchable Carbon Nanotube Transistors with Ion Gel Gate Dielectrics. Nano Lett. 2014 doi: 10.1021/nl403941a. [DOI] [PubMed] [Google Scholar]
- 14.Roy SS, Chakraborty P, Bhattacharya S. Intervention in cyclophosphamide induced oxidative stress and DNA damage by a flavonyl-thiazolidinedione based organoselenocyanate and evaluation of its efficacy during adjuvant therapy in tumor bearing mice. Eur J Med Chem. 2013;73C:195–209. doi: 10.1016/j.ejmech.2013.12.015. [DOI] [PubMed] [Google Scholar]
- 15.Meier B, Dumez JN, Stevanato G, Hill-Cousins JT, Roy SS, Hakansson P, et al. Long-lived nuclear spin States in methyl groups and quantum-rotor-induced polarization. J Am Chem Soc. 2013;135(50):18746–9. doi: 10.1021/ja410432f. [DOI] [PubMed] [Google Scholar]
- 16.Mati SS, Roy SS, Chall S, Bhattacharya S, Bhattacharya SC. Unveiling the groove binding mechanism of a biocompatible naphthalimide-based organoselenocyanate with calf thymus DNA: an "ex vivo" fluorescence imaging application appended by biophysical experiments and molecular docking simulations. J Phys Chem B. 2013;117(47):14655–65. doi: 10.1021/jp4090553. [DOI] [PubMed] [Google Scholar]
- 17.Agyin JK, Santhamma B, Roy SS. Design, synthesis, and biological evaluation of bone-targeted proteasome inhibitors for multiple myeloma. Bioorg Med Chem Lett. 2013;23(23):6455–8. doi: 10.1016/j.bmcl.2013.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roy SS, Patra M, Nandy SK, Banik M, Dasgupta R, Basu T. In Vitro Holdase Activity of E. Coli Small Heat-Shock Proteins IbpA, IbpB and IbpAB: A Biophysical Study with Some Unconventional Techniques. Protein Pept Lett. 2013 doi: 10.2174/0929866521666131224094408. [DOI] [PubMed] [Google Scholar]
- 19.Achanzar WE, Diwan BA, Liu J, Quader ST, Webber MM, Waalkes MP. Cadmium-induced malignant transformation of human prostate epithelial cells. Cancer Res. 2001;61(2):455–8. [PubMed] [Google Scholar]
- 20.Waalkes MP. Cadmium carcinogenesis. Mutat Res. 2003;533(1-2):107–20. doi: 10.1016/j.mrfmmm.2003.07.011. [DOI] [PubMed] [Google Scholar]
- 21.Waisberg M, Joseph P, Hale B, Beyersmann D. Molecular and cellular mechanisms of cadmium carcinogenesis. Toxicology. 2003;192(2-3):95–117. doi: 10.1016/s0300-483x(03)00305-6. [DOI] [PubMed] [Google Scholar]
- 22.Giaginis C, Gatzidou E, Theocharis S. DNA repair systems as targets of cadmium toxicity. Toxicol Appl Pharmacol. 2006;213(3):282–90. doi: 10.1016/j.taap.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 23.Aimola P, Carmignani M, Volpe AR, Di Benedetto A, Claudio L, Waalkes MP, et al. Cadmium induces p53-dependent apoptosis in human prostate epithelial cells. PLoS One. 2012;7(3):e33647. doi: 10.1371/journal.pone.0033647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meplan C, Mann K, Hainaut P. Cadmium induces conformational modifications of wild-type p53 and suppresses p53 response to DNA damage in cultured cells. J Biol Chem. 1999;274(44):31663–70. doi: 10.1074/jbc.274.44.31663. [DOI] [PubMed] [Google Scholar]
- 25.Hartmann M, Hartwig A. Disturbance of DNA damage recognition after UV-irradiation by nickel(II) and cadmium(II) in mammalian cells. Carcinogenesis. 1998;19(4):617–21. doi: 10.1093/carcin/19.4.617. [DOI] [PubMed] [Google Scholar]
- 26.Asmuss M, Mullenders LH, Eker A, Hartwig A. Differential effects of toxic metal compounds on the activities of Fpg and XPA, two zinc finger proteins involved in DNA repair. Carcinogenesis. 2000;21(11):2097–104. doi: 10.1093/carcin/21.11.2097. [DOI] [PubMed] [Google Scholar]
- 27.Souza V, Escobar Md Mdel C, Gomez-Quiroz L, Bucio L, Hernandez E, Cossio EC, et al. Acute cadmium exposure enhances AP-1 DNA binding and induces cytokines expression and heat shock protein 70 in HepG2 cells. Toxicology. 2004;197(3):213–28. doi: 10.1016/j.tox.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 28.Freitas M, Fernandes E. Zinc, cadmium and nickel increase the activation of NF-kappaB and the release of cytokines from THP-1 monocytic cells. Metallomics. 2011;3(11):1238–43. doi: 10.1039/c1mt00050k. [DOI] [PubMed] [Google Scholar]
- 29.Yang Z, Yang S, Qian SY, Hong JS, Kadiiska MB, Tennant RW, et al. Cadmium-induced toxicity in rat primary mid-brain neuroglia cultures: role of oxidative stress from microglia. Toxicol Sci. 2007;98(2):488–94. doi: 10.1093/toxsci/kfm106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xie J, Shaikh ZA. Cadmium-induced apoptosis in rat kidney epithelial cells involves decrease in nuclear factor-kappa B activity. Toxicol Sci. 2006;91(1):299–308. doi: 10.1093/toxsci/kfj131. [DOI] [PubMed] [Google Scholar]
- 31.Severson PL, Tokar EJ, Vrba L, Waalkes MP, Futscher BW. Agglomerates of aberrant DNA methylation are associated with toxicant-induced malignant transformation. Epigenetics. 2012;7(11):1238–48. doi: 10.4161/epi.22163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mao WP, Zhang NN, Zhou FY, Li WX, Liu HY, Feng J, et al. Cadmium directly induced mitochondrial dysfunction of human embryonic kidney cells. Hum Exp Toxicol. 2011;30(8):920–9. doi: 10.1177/0960327110384286. [DOI] [PubMed] [Google Scholar]
- 33.Bagchi D, Vuchetich PJ, Bagchi M, Hassoun EA, Tran MX, Tang L, et al. Induction of oxidative stress by chronic administration of sodium dichromate [chromium VI] and cadmium chloride [cadmium II] to rats. Free Radic Biol Med. 1997;22(3):471–8. doi: 10.1016/s0891-5849(96)00352-8. [DOI] [PubMed] [Google Scholar]
- 34.Roy SS, Gonugunta VK, Bandyopadhyay A, Rao MK, Goodall GJ, Sun LZ, et al. Significance of PELP1/HDAC2/miR-200 regulatory network in EMT and metastasis of breast cancer. Oncogene. 2013 doi: 10.1038/onc.2013.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Manca D, Ricard AC, Tra HV, Chevalier G. Relation between lipid peroxidation and inflammation in the pulmonary toxicity of cadmium. Arch Toxicol. 1994;68(6):364–9. doi: 10.1007/s002040050083. [DOI] [PubMed] [Google Scholar]
- 36.Valko M, Izakovic M, Mazur M, Rhodes CJ, Telser J. Role of oxygen radicals in DNA damage and cancer incidence. Mol Cell Biochem. 2004;266(1-2):37–56. doi: 10.1023/b:mcbi.0000049134.69131.89. [DOI] [PubMed] [Google Scholar]
- 37.Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact. 2006;160(1):1–40. doi: 10.1016/j.cbi.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 38.Liu Y, Roy SS, Nebie RH, Zhang Y, Nair MG. Functional food quality of Curcuma caesia, Curcuma zedoaria and Curcuma aeruginosa endemic to Northeastern India. Plant Foods Hum Nutr. 2013;68(1):72–7. doi: 10.1007/s11130-013-0333-5. [DOI] [PubMed] [Google Scholar]
- 39.Basu M, Roy SS. Wnt/beta-catenin pathway is regulated by PITX2 homeodomain protein and thus contributes to the proliferation of human ovarian adenocarcinoma cell, SKOV-3. J Biol Chem. 2013;288(6):4355–67. doi: 10.1074/jbc.M112.409102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bohara V, Raut L, Badarkhe G, Roy SS, Chaudhuri U. Homozygosity for the severe beta(+)-thalassemia mutation [IVS-I-5 (G>C)] causes the phenotype of thalassemia trait: an extremely rare presentation. Hemoglobin. 2013;37(1):101–5. doi: 10.3109/03630269.2012.751395. [DOI] [PubMed] [Google Scholar]
- 41.Ghosh P, Roy SS, Chakraborty P, Ghosh S, Bhattacharya S. Effects of organoselenium compound 2-(5-selenocyanato-pentyl)-benzo[de]isoquinoline 1,3-dione on cisplatin induced nephrotoxicity and genotoxicity: an investigation of the influence of the compound on oxidative stress and antioxidant enzyme system. Biometals. 2013;26(1):61–73. doi: 10.1007/s10534-012-9594-y. [DOI] [PubMed] [Google Scholar]
- 42.Arriazu R, Duran E, Pozuelo JM, Santamaria L. Expression of lysophosphatidic acid receptor 1 and relation with cell proliferation, apoptosis, and angiogenesis on preneoplastic changes induced by cadmium chloride in the rat ventral prostate. PLoS One. 2013;8(2):e57742. doi: 10.1371/journal.pone.0057742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lin CE, Chen SU, Lin CC, Chang CH, Lin YC, Tai YL, et al. Lysophosphatidic acid enhances vascular endothelial growth factor-C expression in human prostate cancer PC-3 cells. PLoS One. 2012;7(7):e41096. doi: 10.1371/journal.pone.0041096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Benbrahim-Tallaa L, Waterland RA, Dill AL, Webber MM, Waalkes MP. Tumor suppressor gene inactivation during cadmium-induced malignant transformation of human prostate cells correlates with overexpression of de novo DNA methyltransferase. Environ Health Perspect. 2007;115(10):1454–9. doi: 10.1289/ehp.10207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morrissey C, Buser A, Scolaro J, O'Sullivan J, Moquin A, Tenniswood M. Changes in hormone sensitivity in the ventral prostate of aging Sprague-Dawley rats. J Androl. 2002;23(3):341–51. [PubMed] [Google Scholar]
- 46.Arunkumar A, Vijayababu MR, Venkataraman P, Senthilkumar K, Arunakaran J. Chemoprevention of rat prostate carcinogenesis by diallyl disulfide, an organosulfur compound of garlic. Biol Pharm Bull. 2006;29(2):375–9. doi: 10.1248/bpb.29.375. [DOI] [PubMed] [Google Scholar]
- 47.Pollard M, Wolter W, Sun L. Diet and the duration of testosterone-dependent prostate cancer in Lobund-Wistar rats. Cancer Lett. 2001;173(2):127–31. doi: 10.1016/s0304-3835(01)00673-5. [DOI] [PubMed] [Google Scholar]
- 48.Bosland MC. Use of animal models in defining efficacy of chemoprevention agents against prostate cancer. Eur Urol. 1999;35(5-6):459–63. doi: 10.1159/000019879. [DOI] [PubMed] [Google Scholar]
- 49.Waalkes MP, Rehm S, Riggs CW, Bare RM, Devor DE, Poirier LA, et al. Cadmium carcinogenesis in male Wistar [Crl:(WI)BR] rats: dose-response analysis of tumor induction in the prostate and testes and at the injection site. Cancer Res. 1988;48(16):4656–63. [PubMed] [Google Scholar]
- 50.Waalkes MP, Rehm S, Riggs CW, Bare RM, Devor DE, Poirier LA, et al. Cadmium carcinogenesis in male Wistar [Crl:(WI)BR] rats: dose-response analysis of effects of zinc on tumor induction in the prostate, in the testes, and at the injection site. Cancer Res. 1989;49(15):4282–8. [PubMed] [Google Scholar]
- 51.Waalkes MP, Anver M, Diwan BA. Carcinogenic effects of cadmium in the noble (NBL/Cr) rat: induction of pituitary, testicular, and injection site tumors and intraepithelial proliferative lesions of the dorsolateral prostate. Toxicol Sci. 1999;52(2):154–61. doi: 10.1093/toxsci/52.2.154. [DOI] [PubMed] [Google Scholar]
- 52.Waalkes MP, Anver MR, Diwan BA. Chronic toxic and carcinogenic effects of oral cadmium in the Noble (NBL/Cr) rat: induction of neoplastic and proliferative lesions of the adrenal, kidney, prostate, and testes. J Toxicol Environ Health A. 1999;58(4):199–214. doi: 10.1080/009841099157296. [DOI] [PubMed] [Google Scholar]
- 53.Bertin G, Averbeck D. Cadmium: cellular effects, modifications of biomolecules, modulation of DNA repair and genotoxic consequences (a review) Biochimie. 2006;88(11):1549–59. doi: 10.1016/j.biochi.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 54.Beyersmann D, Hechtenberg S. Cadmium, gene regulation, and cellular signalling in mammalian cells. Toxicol Appl Pharmacol. 1997;144(2):247–61. doi: 10.1006/taap.1997.8125. [DOI] [PubMed] [Google Scholar]
- 55.Yamashita H, Tripathi M, Jourquin J, Kam Y, Liu S, Weidow B, et al. Lysophosphatidic Acid Upregulates Laminin-332 Expression during A431 Cell Colony Dispersal. J Oncol. 2010 doi: 10.1155/2010/107075. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen SU, Chou CH, Chao KH, Lee H, Lin CW, Lu HF, et al. Lysophosphatidic acid up-regulates expression of growth-regulated oncogene-alpha, interleukin-8, and monocyte chemoattractant protein-1 in human first-trimester trophoblasts: possible roles in angiogenesis and immune regulation. Endocrinology. 2010;151(1):369–79. doi: 10.1210/en.2009-0779. [DOI] [PubMed] [Google Scholar]
- 57.Guo R, Kasbohm EA, Arora P, Sample CJ, Baban B, Sud N, et al. Expression and function of lysophosphatidic acid LPA1 receptor in prostate cancer cells. Endocrinology. 2006;147(10):4883–92. doi: 10.1210/en.2005-1635. [DOI] [PubMed] [Google Scholar]
- 58.Achanzar WE, Achanzar KB, Lewis JG, Webber MM, Waalkes MP. Cadmium induces c-myc, p53, and c-jun expression in normal human prostate epithelial cells as a prelude to apoptosis. Toxicol Appl Pharmacol. 2000;164(3):291–300. doi: 10.1006/taap.1999.8907. [DOI] [PubMed] [Google Scholar]
- 59.Achanzar WE, Webber MM, Waalkes MP. Altered apoptotic gene expression and acquired apoptotic resistance in cadmium-transformed human prostate epithelial cells. Prostate. 2002;52(3):236–44. doi: 10.1002/pros.10106. [DOI] [PubMed] [Google Scholar]
- 60.Chao DT, Korsmeyer SJ. BCL-2 family: regulators of cell death. Annu Rev Immunol. 1998;16:395–419. doi: 10.1146/annurev.immunol.16.1.395. [DOI] [PubMed] [Google Scholar]
- 61.Nunez G, Benedict MA, Hu Y, Inohara N. Caspases: the proteases of the apoptotic pathway. Oncogene. 1998;17(25):3237–45. doi: 10.1038/sj.onc.1202581. [DOI] [PubMed] [Google Scholar]
- 62.Qu W, Ke H, Pi J, Broderick D, French JE, Webber MM, et al. Acquisition of apoptotic resistance in cadmium-transformed human prostate epithelial cells: Bcl-2 overexpression blocks the activation of JNK signal transduction pathway. Environ Health Perspect. 2007;115(7):1094–100. doi: 10.1289/ehp.10075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gaddipati JP, Rajeshkumar NV, Grove JC, Maharaj SV, Centeno JA, Maheshwari RK, et al. Low-Dose Cadmium Exposure Reduces Human Prostate Cell Transformation in Culture and Up-Regulates Metallothionein and MT-1G mRNA. Nonlinearity Biol Toxicol Med. 2003;1(2):199–212. doi: 10.1080/15401420391434333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Albrecht AL, Singh RK, Somji S, Sens MA, Sens DA, Garrett SH. Basal and metal-induced expression of metallothionein isoform 1 and 2 genes in the RWPE-1 human prostate epithelial cell line. J Appl Toxicol. 2008;28(3):283–93. doi: 10.1002/jat.1277. [DOI] [PubMed] [Google Scholar]
- 65.Misra UK, Gawdi G, Pizzo SV. Induction of mitogenic signalling in the 1LN prostate cell line on exposure to submicromolar concentrations of cadmium+ Cell Signal. 2003;15(11):1059–70. doi: 10.1016/s0898-6568(03)00117-7. [DOI] [PubMed] [Google Scholar]
- 66.Martin MB, Reiter R, Pham T, Avellanet YR, Camara J, Lahm M, et al. Estrogen-like activity of metals in MCF-7 breast cancer cells. Endocrinology. 2003;144(6):2425–36. doi: 10.1210/en.2002-221054. [DOI] [PubMed] [Google Scholar]
- 67.Choe SY, Kim SJ, Kim HG, Lee JH, Choi Y, Lee H, et al. Evaluation of estrogenicity of major heavy metals. Sci Total Environ. 2003;312(1-3):15–21. doi: 10.1016/S0048-9697(03)00190-6. [DOI] [PubMed] [Google Scholar]
- 68.Antila E, Mussalo-Rauhamaa H, Kantola M, Atroshi F, Westermarck T. Association of cadmium with human breast cancer. Sci Total Environ. 1996;186(3):251–6. doi: 10.1016/0048-9697(96)05119-4. [DOI] [PubMed] [Google Scholar]
- 69.Strumylaite L, Mechonosina K, Tamasauskas S. Environmental factors and breast cancer. Medicina (Kaunas) 2010;46(12):867–73. [PubMed] [Google Scholar]
- 70.Stoica A, Katzenellenbogen BS, Martin MB. Activation of estrogen receptor-alpha by the heavy metal cadmium. Mol Endocrinol. 2000;14(4):545–53. doi: 10.1210/mend.14.4.0441. [DOI] [PubMed] [Google Scholar]
- 71.Johnson MD, Kenney N, Stoica A, Hilakivi-Clarke L, Singh B, Chepko G, et al. Cadmium mimics the in vivo effects of estrogen in the uterus and mammary gland. Nat Med. 2003;9(8):1081–4. doi: 10.1038/nm902. [DOI] [PubMed] [Google Scholar]
- 72.Ponce E, Aquino NB, Louie MC. Chronic Cadmium Exposure Stimulates SDF-1 Expression in an ERalpha Dependent Manner. PLoS One. 2013;8(8):e72639. doi: 10.1371/journal.pone.0072639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Siewit CL, Gengler B, Vegas E, Puckett R, Louie MC. Cadmium promotes breast cancer cell proliferation by potentiating the interaction between ERalpha and c-Jun. Mol Endocrinol. 2010;24(5):981–92. doi: 10.1210/me.2009-0410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Benbrahim-Tallaa L, Tokar EJ, Diwan BA, Dill AL, Coppin JF, Waalkes MP. Cadmium malignantly transforms normal human breast epithelial cells into a basal-like phenotype. Environ Health Perspect. 2009;117(12):1847–52. doi: 10.1289/ehp.0900999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sinha Roy S, Mukherjee S, Mukhopadhyay S, Das SK. Differential effect of cadmium on cholinephosphotransferase activity in normal and cancerous human mammary epithelial cell lines. Mol Cancer Ther. 2004;3(2):199–204. [PubMed] [Google Scholar]
- 76.Field RW, Withers BL. Occupational and environmental causes of lung cancer. Clin Chest Med. 2012;33(4):681–703. doi: 10.1016/j.ccm.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Waalkes MP. Cadmium carcinogenesis in review. J Inorg Biochem. 2000;79(1-4):241–4. doi: 10.1016/s0162-0134(00)00009-x. [DOI] [PubMed] [Google Scholar]
- 78.Person RJ, Tokar EJ, Xu Y, Orihuela R, Ngalame NN, Waalkes MP. Chronic cadmium exposure in vitro induces cancer cell characteristics in human lung cells. Toxicol Appl Pharmacol. 2013 doi: 10.1016/j.taap.2013.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lei YX, Wei L, Wang M, Wu GR, Li M. Malignant transformation and abnormal expression of eukaryotic initiation factor in bronchial epithelial cells induced by cadmium chloride. Biomed Environ Sci. 2008;21(4):332–8. doi: 10.1016/S0895-3988(08)60051-3. [DOI] [PubMed] [Google Scholar]
- 80.Demirhan O, Tastemir D, Hasturk S, Kuleci S, Hanta I. Alterations in p16 and p53 genes and chromosomal findings in patients with lung cancer: fluorescence in situ hybridization and cytogenetic studies. Cancer Epidemiol. 2010;34(4):472–7. doi: 10.1016/j.canep.2010.03.018. [DOI] [PubMed] [Google Scholar]
- 81.Suzuki Y, Oonishi T, Kudo T, Doi H. LKB1, TP16, EGFR, and KRAS somatic mutations in lung adenocarcinomas from a Chiba Prefecture, Japan cohort. Drug Discov Ther. 2012;6(1):24–30. [PubMed] [Google Scholar]
- 82.Jin M, Inoue S, Umemura T, Moriya J, Arakawa M, Nagashima K, et al. Cyclin D1, p16 and retinoblastoma gene product expression as a predictor for prognosis in non-small cell lung cancer at stages I and II. Lung Cancer. 2001;34(2):207–18. doi: 10.1016/s0169-5002(01)00225-2. [DOI] [PubMed] [Google Scholar]
- 83.Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455(7216):1069–75. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kholodnyuk ID, Kozireva S, Kost-Alimova M, Kashuba V, Klein G, Imreh S. Down regulation of 3p genes, LTF, SLC38A3 and DRR1, upon growth of human chromosome 3-mouse fibrosarcoma hybrids in severe combined immunodeficiency mice. Int J Cancer. 2006;119(1):99–107. doi: 10.1002/ijc.21794. [DOI] [PubMed] [Google Scholar]
- 85.Qu W, Fuquay R, Sakurai T, Waalkes MP. Acquisition of apoptotic resistance in cadmium-induced malignant transformation: specific perturbation of JNK signal transduction pathway and associated metallothionein overexpression. Mol Carcinog. 2006;45(8):561–71. doi: 10.1002/mc.20185. [DOI] [PubMed] [Google Scholar]
- 86.Eckschlager T, Adam V, Hrabeta J, Figova K, Kizek R. Metallothioneins and cancer. Curr Protein Pept Sci. 2009;10(4):360–75. doi: 10.2174/138920309788922243. [DOI] [PubMed] [Google Scholar]
- 87.Jing Y, Liu LZ, Jiang Y, Zhu Y, Guo NL, Barnett J, et al. Cadmium increases HIF-1 and VEGF expression through ROS, ERK, and AKT signaling pathways and induces malignant transformation of human bronchial epithelial cells. Toxicol Sci. 2012;125(1):10–9. doi: 10.1093/toxsci/kfr256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Son YO, Wang L, Poyil P, Budhraja A, Hitron JA, Zhang Z, et al. Cadmium induces carcinogenesis in BEAS-2B cells through ROS-dependent activation of PI3K/AKT/GSK-3beta/beta-catenin signaling. Toxicol Appl Pharmacol. 2012;264(2):153–60. doi: 10.1016/j.taap.2012.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhou Z, Wang C, Liu H, Huang Q, Wang M, Lei Y. Cadmium Induced Cell Apoptosis, DNA Damage, Decreased DNA Repair Capacity, and Genomic Instability during Malignant Transformation of Human Bronchial Epithelial Cells. Int J Med Sci. 2013;10(11):1485–96. doi: 10.7150/ijms.6308. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 90.Roy SS, Sengupta S. A rare case of congenital cytomegalovirus infection in pregnancy. J Obstet Gynaecol India. 2012;62(1):70–2. doi: 10.1007/s13224-012-0143-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang SH, Shih YL, Kuo TC, Ko WC, Shih CM. Cadmium toxicity toward autophagy through ROS-activated GSK-3beta in mesangial cells. Toxicol Sci. 2009;108(1):124–31. doi: 10.1093/toxsci/kfn266. [DOI] [PubMed] [Google Scholar]
- 92.Park CH, Lee BH, Ahn SG, Yoon JH, Oh SH. Serine 9 and Tyrosine 216 Phosphorylation of GSK-3beta Differentially Regulates Autophagy in Acquired Cadmium Resistance. Toxicol Sci. 2013 doi: 10.1093/toxsci/kft158. [DOI] [PubMed] [Google Scholar]