

Abstract
The small single-stranded DNA (ssDNA) bacteriophages of the subfamily Gokushovirinae were traditionally perceived as narrowly targeted, niche-specific viruses infecting obligate parasitic bacteria, such as Chlamydia. The advent of metagenomics revealed gokushoviruses to be widespread in global environmental samples. This study expands knowledge of gokushovirus diversity in the environment by developing a degenerate PCR assay to amplify a portion of the major capsid protein (MCP) gene of gokushoviruses. Over 500 amplicons were sequenced from 10 environmental samples (sediments, sewage, seawater and freshwater), revealing the ubiquity and high diversity of this understudied phage group. Residue-level conservation data generated from multiple alignments was combined with a predicted 3D structure, revealing a tendency for structurally internal residues to be more highly conserved than surface-presenting protein–protein or viral–host interaction domains. Aggregating this data set into a phylogenetic framework, many gokushovirus MCP clades contained samples from multiple environments, although distinct clades dominated the different samples. Antarctic sediment samples contained the most diverse gokushovirus communities, whereas freshwater springs from Florida were the least diverse. Whether the observed diversity is being driven by environmental factors or host-binding interactions remains an open question. The high environmental diversity of this previously overlooked ssDNA viral group necessitates further research elucidating their natural hosts and exploring their ecological roles.
Keywords: single-stranded, phage, virus, gokushovirus, Microviridae
Introduction
The first DNA genome to be completely sequenced belonged to the diminutive single-stranded DNA (ssDNA) phage ϕX174, initiating the genomic sequencing era in 1977 (Sanger et al., 1977). Knowledge of phage biology and ecology has expanded rapidly since that time, and phages are currently recognized as the most abundant biological entities on the planet, exerting significant driving forces on bacterial diversity and global biogeochemistry (Breitbart, 2012). Despite the early characterization of ssDNA phages, their double-stranded DNA (dsDNA) counterparts have received a disproportionate amount of attention over the past three decades. As of 2011, >80% of the completely sequenced phage genomes in Genbank belonged to tailed dsDNA phages of the Caudovirales (Krupovic et al., 2011). The Caudovirales also account for the vast majority (96%) of phages characterized by electron microscopy (Ackermann, 2007). Culture-based studies, combined with pulsed-field gel electrophoresis results (Steward et al., 2000) and early metagenomic methods that excluded ssDNA phages (Breitbart et al., 2002), created the general paradigm that dsDNA tailed phages belonging to the Caudovirales dominate in environmental communities. However, recent studies have challenged this dogma by demonstrating the abundance of nontailed viral particles and DNase-insensitive viral genomes in the oceans (Steward et al., 2012; Brum et al., 2013).
Icosahedral, ssDNA phages belonging to the family Microviridae have been present in culture collections since the 1920s, yet until 2006, this phage family had not been described in the oceans, one of the most extensively studied ecosystems in terms of microbial ecology. In 2006, next-generation 454 pyrosequencing was applied to viral metagenomics, requiring the introduction of a non-specific amplification technique (rolling circle amplification) to obtain sufficient starting quantities of DNA. The first study to utilize this approach found that the recognizable sequences from an 80 m deep viral metagenome from the Sargasso Sea were dominated by sequences similar to the Microviridae, specifically to the Gokushovirinae subfamily (Angly et al., 2006). This finding was unexpected as gokushoviruses had previously only been reported to infect parasitic bacteria (Chlamydia, Bdellovibrio, Spiroplasma) and were believed to be successful in a fairly narrow niche (Brentlinger et al., 2002; Cherwa and Fane, 2011). The study of Angly et al. (2006) relied on the use of rolling circle amplification, which is known to preferentially enrich for circular ssDNA elements (Kim and Bae, 2011). Despite this caveat it was surprising to find environmental settings with significant community composition of heretofore human- and agriculturally-associated phages.
Building upon the Angly et al. (2006) study, viral metagenomic studies employing rolling circle amplification have uncovered novel ssDNA phages in a variety of environments (reviewed in Rosario and Breitbart (2011)), including freshwater aquifers (Smith et al., 2013), freshwater lakes (López-Bueno et al., 2009; Roux et al., 2012a), stromatolites (Desnues et al., 2008), soils (Kim et al., 2008), coastal estuaries (McDaniel et al., 2008, 2013; Labonté and Suttle, 2013), sediments (Yoshida et al., 2013) seawater and reclaimed water (Rosario et al., 2009). Although metagenomic studies generate sequence fragments, two complete gokushovirus genomes (SARssϕ1 and SARssϕ2) were assembled and PCR verified from the Sargasso Sea (Tucker et al., 2011) and a data-mining study assembled 81 additional complete Microviridae genome sequences from various environments and human gut/stool samples (Roux et al., 2012b). A key finding of this latest, most comprehensive study was an intriguing emergent topology for the Gokusho- subfamily with dichotomous clading of environmental (for example, SARssφ1 and −2) vs ‘human-associated' gokushoviruses (incl. ChP's and SpV4) (Roux et al., 2012b).
The International Committee on the Taxonomy of Viruses, divides the Microviridae into two groups; the Gokushovirinae subfamily, and the enterobacteria-infecting φX174-type ‘true' Microvirus genus, for which a subfamily has not been officially adopted (Fane, 2005). As the majority of ssDNA sequences that have been identified in environmental metagenomes are similar to gokushoviruses, this study further explores their diversity and environmental distribution by amplifying and sequencing a portion of the gokushovirus major capsid protein (MCP). The selected MCP fragment is flanked by highly conserved motifs to enable efficient amplification and alignment and includes the hypervariable threefold loop believed to dictate host specificity. Results reveal diverse gokushoviruses in all environments examined, demonstrating that ssDNA phages are a pervasive but understudied component of the global environmental virome.
Materials and methods
Sample collection, processing and DNA extraction
Samples from 10 different sites were examined: six in Florida, USA, and four from the Antarctic shelf. Several methods were used to purify viruses and concentrate DNA from these environmental samples, which were mostly samples of opportunity prepared for other projects. Surface water samples were collected in August 2012 from Wall Springs (freshwater (FW); 100 l), Wall Estuary (saline (SW); 100 l) and Bayboro Harbor (SW; 200 l). GPS coordinates, salinity and temperature data are recorded in Table 1. Water was strained through 100 μm Nitex mesh, then concentrated down to ∼100 ml using a 100 kD tangential flow filter (GE Healthcare, Pittsburg, PA, USA) as described previously (Thurber et al., 2009). The retentate was filtered through a 0.22 μm Sterivex filter (Millipore, Billerica, MA, USA) to remove bacteria and larger cells. Viral DNA was extracted from the concentrate using the MinElute Virus Spin Kit (Qiagen, Valencia, CA, USA) following the standard kit protocol and eluted into 50 μl of water.
Table 1. Description of samples processed in this study, including available metadata and results from diversity analysis (Yu et al., 2006).
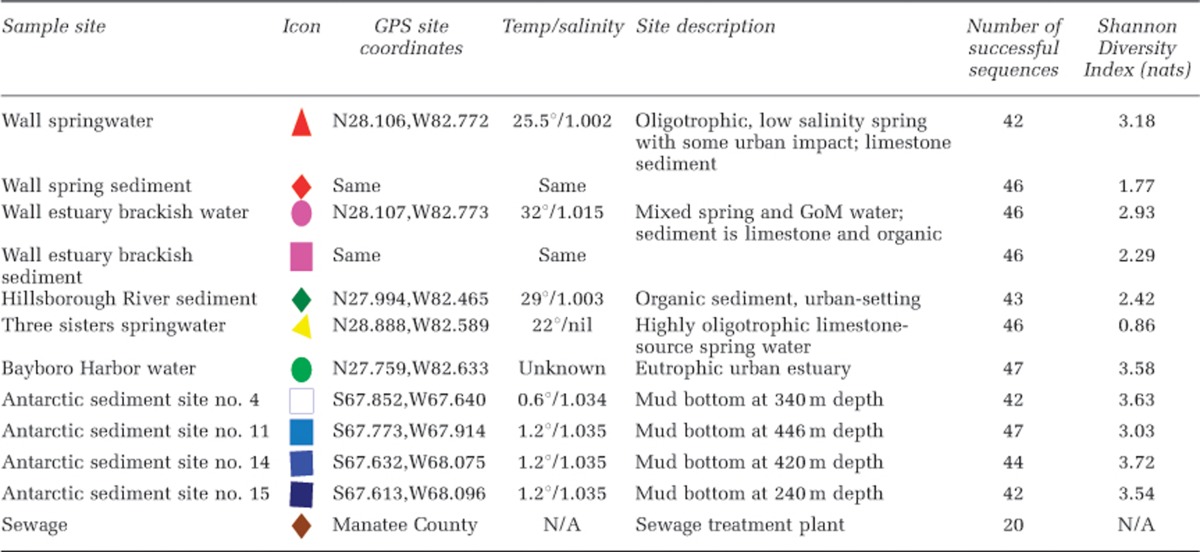
Freshwater samples were collected by snorkelers from Three Sisters Springs in Florida in May 2009. A sterile 60 ml syringe was used to collect 50 ml of water directly from the spring boil (∼3 m below the surface). Water samples were immediately filtered through a 0.22 μm Sterivex filter and then onto a 0.02 μm Anotop filter (Whatman, Maidstone, UK), which was frozen at −80 °C until extraction. DNA was extracted from the Anotop filter using the Masterpure complete DNA and RNA purification kit (Epicenter, Madison, WI, USA) as described previously (Culley and Steward, 2007, Tucker et al., 2011).
Surface sediment samples from Wall Spring, Wall Estuary and Hillsborough River were collected with conical tubes directly below their corresponding water samples. The Antarctic margin marine sediments (n=4; sites #4, 11, 14, 15) were collected in February 2012 during the British Services Antarctic Expedition (http://www.bsae2012.co.uk/science.html). Surface grab samples were taken in Marguerite Bay (∼68°S, 68°W) from water depths between 200 and 425 m and the upper 0–2 cm of sediment was subsampled immediately into conical tubes and frozen at −20 °C until processing. DNA extractions were performed from a starting mass of ∼250 mg sediment. Sediment samples with high water content were first centrifuged at 7000 × g for 4 min in order to obtain a cohesive sediment plug that could be adjusted for mass. The ∼250 mg solid was combined with reagents from the PowerSoil DNA extraction kit (MoBio, Carlsbad, CA, USA) and vigorously homogenized and disrupted with 1 min of bead beating followed by 10 min of vortexing. The extraction was performed following the manufacturer's protocol with a final elution volume of 100 μl.
The sewage sample was collected in February 2009 from a wastewater treatment plant in Manatee County, Florida. Virus particles were purified from 1.2 l of sample by filtering through 0.45 μm and 0.2 μm Sterivex filters (Millipore). Virus particles were further concentrated and purified using polyethylene glycol precipitation followed by cesium chloride gradient centrifugation with composite collection in a density range from 1.2 to 1.5 g ml−l (Thurber et al., 2009). Viral DNA was extracted using the MinElute Virus Spin Kit (Qiagen).
Degenerate PCR for amplification of Microviridae
Degenerate PCR primers were designed using the standalone version of the PhiSiGns utility (Dwivedi et al., 2012). Initially, PhiSiGns failed to generate acceptable primers for all of the extant Gokushovirinae due to the highly divergent nature of SpV4; therefore, SpV4 was excluded from the design. Degenerate PCR primers MCPf (5′-CCYKGKYYNCARAAAGG-3′) and MCPr (5′-AHCKYTCYTGRTADCC-3′) are designed to amplify an 895 nt fragment of the MCP gene from the remaining extant Gokushovirinae (Chp1, NC_001741; Chp2, NC_002194; Chp3, NC_008355; Chp4, NC_007461; CPAR39, NC_002180; φCPG1, NC_001998; BdφMH2K, NC_002643; SARssϕ1, HQ157199; SARssϕ2, HQ157198). These extant genomes from which the primers were derived are henceforth referred to as the nine ‘reference genomes'.
To enrich for circular, ssDNA templates, 1 μl of the extracted DNA from each sample was subjected to rolling circle amplification (Templiphi; GE Healthcare, Piscataway, NJ, USA) according to the manufacturer's instructions. This Templiphi product was diluted 10-fold and used as the target for degenerate PCR. The 50 μl PCR mixture contained 1 U Apex Taq DNA polymerase (Genesee Scientific, San Diego, CA, USA), 1X Apex Taq reaction buffer, 0.5 μM of each primer, 0.2 mM dNTPs and 1 μl of the diluted Templiphi product. The touchdown PCR conditions were (i) 3 min of initial denaturation at 94 °C, (ii) 32 cycles of 60 s of denaturation (95 °C), 45 s of annealing (47 °C with a 0.1° decrease/cycle), 90 s of extension (72 °C) and (iii) 10 min of final extension at 72 °C.
The resulting PCR products were visualized using gel electrophoresis. One sample, the Bayboro Harbor estuary concentrate, yielded multiple PCR products of different sizes, so the band most similar in size to the positive control was excised and gel-purified (Zymo, Irvine, CA, USA). The verified PCR products were given a poly-adenine tail using Sigma Taq polymerase, ligated into TOPO TA vector (Invitrogen, Grand Island, NY, USA) and subsequently transformed into OneShot competent Escherichia coli (Invitrogen) and plated with X-gal (20 mg ml−1). White colonies were picked and insert sizes were verified by PCR with M13 primers. Forty-eight clones from each sample were Sanger sequenced with the M13F primer by Beckman Genomics (Danvers, MA, USA).
Sequence analysis
Sequences were trimmed for quality and vector removal using Sequencher (Gene Codes, Ann Arbor, MI, USA). Trimmed sequences were compared against the Genbank non-redundant (nr) database using a batch BLASTX search (cutoff e=0.05) to confirm that the amplicons were similar to the MCP of known Microviridae. Sequences that did not have BLASTX similarity to Microviridae were considered to be nonspecific amplification and therefore removed from further analyses. Microviridae sequences were recovered with high efficiency from most environments, with the exception of the sewage sample, in which ∼60% of the sequenced clones were not similar to Microviridae.
Sequences with BLASTX similarity to Microviridae were dereplicated at the 97% nucleotide level using FastGroup II (Yu et al., 2006), which was also used to compute the Shannon–Weiner Diversity Index (Shannon and Weaver, 1949). The sequences were provisionally translated into amino-acid format and aligned using the ClustalW algorithm in MEGA5 with subsequent manual adjustment (Tamura et al., 2011). After obtaining optimal amino-acid alignment, the alignment was back-toggled and exported for phylogenetic construction. The phylogeny in Figure 2 was generated using the PhyML package (Guindon et al., 2010); a maximum-likelihood method employing the GTR model with support values determined by approximate likelihood-ratio test (Anisimova and Gascuel, 2006). The phylogeny in Figure 3 is a maximum-likelihood tree generated in the FastTree package (Price et al., 2009) using the Whelan-and-Goldman residue model from a maximum-likelihood training ‘intree' generated in MEGA5 from a ClustalW alignment (Tamura et al., 2011). The clade-based hidden Markov models combined in Supplementary Figure 1 were calculated using ‘hmmbuild' within the HMMER3.0 package (Eddy, 2008) and visualized using LogoMat (Schuster-Böckler et al., 2004) with the positional variability histogram generated in the web-based Protein Variability Server (Garcia-Boronat et al., 2008).
Homology modeling of gokushoviruses
Structural models of gokushovirus MCPs were built using the homology model-building package MODELER (Yang et al., 2012). The full-length MCP sequences of the reference ‘seed' gokushoviruses (six Chlamydia phages, SARssφ1&2, BdφMH2k) were aligned against the MCP of Microviridae with available structures (φX174, Bacteriophage alpha-3, G4, SpV4) using CLUSTAL-W2 (Larkin et al., 2007). Presence of large insertions (>80 amino acids) at the threefold loop region as seen in SpV4 prompted use of the pseudo-atomic model of SpV4 instead of the higher resolution coliphage (φX174, α3 and G4) models as the primary template for model building. Superposition of the homology models was carried out in the COOT package (Emsley et al., 2010). The online server, VIPERdb2 (Carrillo-Tripp et al., 2009), was used to generate a capsid composed of 60 identical copies of the MCP by icosahedral matrix multiplication (Figure 1). UCSF-CHIMERA (Pettersen et al., 2004) was used to calculate percent conservation values based on the presence of the most prevalent residue at a particular position in the alignment of all the selected gokushoviruses. These values were projected onto a ribbon representation of Chp1 using PyMOL (http://www.pymol.org/). Surface representation of Chp1 was generated in UCSF-CHIMERA.
Figure 1.

Surface representation of a Chlamydiaphage-1 (ChP1) capsid homology model is shown in gray looking down the fivefold axis of symmetry of an icosahedron (right). The inset to the left is a cartoon representation of the predicted ChP1 MCP homology model, in which the amplicon region from this study is highlighted with arrows (S: Start; E: End) and has been colored according to residue conservation of the full aligned data set from blue (most conserved; 1) to red (least conserved; 0.1). The N-terminal (N) and C-terminal (C) end of the MCP have been labeled along with the two, three and fivefold axes of symmetry for an icosahedron shown as an oval, triangle and pentagon, respectively. The β-strands of the eight stranded β-barrel that are contained within the amplicon (βF-H) and referenced in the discussion are also labeled.
Results and discussion
Building upon the initial discovery of gokushoviruses in a wide range of natural environments, this study designed a degenerate PCR assay to amplify a portion of the gokushovirus MCP. Although the amplification of genes conserved within specific viral families (that is, signature genes) is commonly used to explore the diversity and environmental distribution of dsDNA phages (for example, Filée et al. (2005)), this is the first study to examine ssDNA phage diversity with such an approach. The gokushovirus MCP amplicon contains regions of both low and high conservation, presenting an ideal target for studying the diversity of these phages in the environment. The 5′ portion of the amplicon is dominated by the hypervariable, threefold interaction loop, whereas the 3′ portion includes three of the eight β-sheets (βF-βG) that comprise the ‘β-barrel' motif common to all Microviridae (Bull et al., 2000) as well as many other viral families with T=1 capsids (McKenna et al., 1992; Agbandje-McKenna and Kleinschmidt, 2011).
Gokushovirus MCPs were recovered from all environments tested (freshwater, estuarine, sediments, sewage). A total of 537 sequences were retained following BLASTX parsing, which were then dereplicated within each sample at 97% identity with gaps, yielding 315 sequences for downstream analyses (Genbank Accession No. KF689226–KF689540), which are represented in Figure 2 phylogeny. Notably, the average size of the aligned amplicons from the environmental samples was 636±22 nt, compared with 705±32 nt in the Chlamydia phages. The difference in length between environmental and cultured gokushoviruses was largely driven by differences in the length of the threefold loop; in the Chlamydia phages the loop-coding region had an average length of 230±23 nt, whereas in the environmental samples the same region averaged 166±19 nt. The smaller size of environmental phage amplicons as compared with cultured isolates has also been reported for dsDNA phages (Breitbart et al., 2004), although the reason for this discrepancy is unknown.
Figure 2.

Unrooted maximum likelihood phylogeny of 316 novel gokushovirus MCP sequences, unique at 97%. Statistical support values are percentages calculated by the aLRT method. Sequences are coded by color (sampling site) and shape (sample type), as shown in the legend. A primary division in the data set is shown with a dashed line and annotation, and small secondary clades of interest are indicated by shading and annotation. The icons are dereplicated, unique sequences, which in one instance represents as many as 19 recovered sequences.
A capsid homology model was generated to examine the amplicon from a structural perspective. High-resolution crystal structures with ∼3–3.5 Å resolution are available for bacteriophages φX174 (PDB ID: 1AL0), α3 (PDB ID: 1M06) and G4 (PDB ID: 1GFF), which are all members of the enterobacteria-infecting ‘true' Microvirus genus (family Microviridae). Structures of ‘true' Microvirus capsids by X-ray crystallography revealed spike (‘G'), DNA-pilot (‘H') and DNA-binding (‘J') proteins, in addition to major capsid proteins (‘F') (Dokland et al., 1997). However, the nucleotide-level sequence identity of the MCPs of the nine reference gokushoviruses to those structurally resolved ‘true' microviruses only ranged between 18 and 20%. A notable difference between the gokushoviruses targeted by this study and the ‘true' microvirus MCPs is that the ‘true' microviruses do not carry large insertions loops (>80 residues) between strands βE and βF found at the threefold axis of symmetry (see Figure 1).
Although there are no high-resolution models for gokushoviruses, a pseudo-atomic 27 Å resolution model for the gokushovirus SpV4 built into cryo-reconstructed density is available (Chipman et al., 1998). The MCP encoded by the gokushovirus SpV4 is homologous to the F proteins in enterobacterial ‘true' microviruses (like φX174, α3, φK and G4) and shares a canonical, eight-stranded β-motif (Chipman et al., 1998). The gokushovirus genomes do not encode for the pentameric G proteins, which create star-shaped spikes at each of the 12 fivefold vertices of the φX174 capsid. Instead, pseudo-atomic modeling of the SpV4 MCP, the only gokushovirus for which a structure has been solved, albeit in low resolution cryo-reconstructed density, suggested the presence of ‘mushroom-like' protrusions on the surface formed by prominent loops found at each threefold axis of symmetry of the MCP (Chipman et al., 1998). The gokushoviruses also lack external scaffolding proteins. However, in Chlamydia phage 2, it has been demonstrated that gokushoviruses do encode a VP3 capsid protein which is lost during the maturation of procapsids to infectious virions (Clarke et al., 2004). In spite of sharing a low sequence similarity, its role is considered analogous to internal scaffold protein B.
Owing to the closer sequence similarity and presence of the threefold loop, the SpV4 MCP pseudo-atomic model was used as a template to build a homology model for Chp1. A conservation percentage for our total environmental data set (n=315 sequences) was calculated using UCSF-CHIMERA based on the presence of the most prevalent residue at a particular position in the full alignment. The conservation percentage was projected onto a threaded model of Chp1 in a red-to-blue spectrum of the least-to-most conserved regions for the amplified region specifically (Figure 1).
Because of its prominent, surface-protruding location, the ‘threefold loop' has been predicted to be important for host specificity in the gokushovirus SpV4 (Chipman et al., 1998). As further evidence for this hypothesis, an experimental study demonstrated that three Chlamydia phages with the same sequence in their threefold loop motif had the same host-infectivity range (Everson et al., 2003). The reported hypervariability within this region is therefore a proposed mechanism for accessing new host types. Our aggregated conservation analysis found similar hypervariability in the threefold loops of environmental gokushovirus MCP sequences, with conservation ⩽10% for much of the length (Figure 1).
Only at the downstream base of the threefold protrusion where there is a prominent α-helix did the residue conservation rise to >50%. This helix is one of the most highly conserved motifs of these amplicons, implying that it is inherent to the environmental gokushoviruses as it is to SpV4 and φX174-type phages for which the structure is known (McKenna et al., 1992, 1996; Chipman et al., 1998).
The residues participating in the formation of the first β-strand downstream of the threefold insertion loop, βF (Chipman et al., 1998), which is part of the eight sheet β-barrel core, had conservation values of ∼50%. This conservation percentage in the aligned residues rapidly degenerated in the succeeding loop connecting strands βF and βG. The degenerate connecting loop is modeled to interact with four other protein monomers at the fivefold axis of symmetry, contravening the paradigm of multimeric interaction forcing genetic purity (Bahadur and Janin, 2008). The residues in strands βG and βH had successively higher levels of conservation, rising to levels >80%. The high level of conservation maintained at the β-core and α-helical regions suggests the importance of these residues in viral capsid assembly. Strand βH runs internally through the core of the structure, emerging to participate in a twofold interaction with an adjacent monomer at the twofold axis of symmetry, indicated by the oval in Figure 1. Residues in the loop emerging from this strand show a rapid shift from a high to low value of conservation percentage.
Overall, the environmental gokushovirus MCP amplicons show poor conservation on the surface-exposed regions, although maintaining high conservation at the interior of the capsid. This model suggests that the high sequence and possibly structural variance at these surface-exposed regions may facilitate rapid co-evolution of phages with their hosts and allow for exploration of new host space (Paterson et al., 2010; Breitbart, 2012).
Phylogenies built with the full-data set alignment yielded chaotic, irreproducible trees due to an inability to align the highly divergent threefold loop region (<10% conservation). Upon removal of the threefold loop region, more robust alignments were achieved, revealing a phylogeny with many long-branch singletons and clustered clades of varying cohesion. Despite the fact that all the Chlamydia phages were included as reference genomes when designing the degenerate primers used in this study, the environmental amplicons are only distantly related to these cultured isolates (Figure 2). The tight clading of the cultured gokushovirus sequences adjacent to those recovered in this study, combined with the aforementioned finding of aberrantly long threefold loops in the cultured isolates, reinforces the notion that the ‘type strains' for the gokushoviruses are not close representatives of the ssDNA phages that dominate in the environment. It is notable that some of the recovered sequences did cluster with SARssϕ1 and SARssϕ2, uncultured gokushoviruses that were assembled from a metagenomic survey of the Sargasso Sea (Tucker et al., 2011).
Overall, an extremely broad diversity of novel gokushovirus MCP sequences was recovered with these primers, reflecting results seen in signature gene amplification studies of dsDNA phages (for example, Filée et al., 2005; Goldsmith et al., 2011). The recovered level of gokushovirus MCP diversity varied across the different environments. This is quantified in Table 1 using the Shannon–Wiener Diversity Index (Shannon and Weaver, 1949) as computed in FastgroupII (Yu et al., 2006). This diversity metric has been criticized as providing a biologically meaningless numerical output in loge ‘nats', as well as being highly sensitive to inexhaustive ‘species' sampling (Magurran, 2004), a shortcoming as applied here since it is highly unlikely that we have exhaustively sampled all of the gokushovirus ‘species' from even the most homogenous of our sample sites. Furthermore, it is possible that certain methods used to process our samples (for example, cesium chloride centrifugation) may have biased the recovery of gokushoviruses. However, as approximately equal sequencing efforts were applied to each site, these diversity estimates are useful for comparison between sites (Soetaert and Heip, 1990). The sewage site was excluded from the diversity calculation because of the smaller number of sequences from this site. The Antarctic sediment samples were characterized by extremely high diversity with Shannon scores exceeding 3 nats. Almost all of the combined 196 amplicons from the four Antarctic sediment samples were unique (that is, <97% identical), demonstrating that far more sequencing is needed to comprehensively document the gokushovirus diversity in Antarctic margin marine sediments. The next highest diversity was recovered from the Bayboro Harbor estuary, likely due to its combination of marine and terrestrial runoff inputs. The riverine systems (Hillsborough River, Wall Springs) had an intermediate level of diversity and the lowest diversity was found in the pristine Florida spring site (Three Sisters Springs), where the 46 sequences obtained dereplicated into only 6 ‘unique' sequences.
The phylogenetic analysis reveals some clustering by sample type and location (Figure 2). The primary partition in the tree (indicated by a dashed black line) is between the Antarctic sediment sequences (blue squares in Clade 1°A, shaded by site) and the other samples, which all originated from Florida, USA (Clade 1°B). Owing to the limited number of samples analyzed in this study and the number of variables differentiating each site, it is not possible to determine which variable (for example, geography, temperature, depth, salinity) or combination of variables is responsible for this dichotomy in the data. Interestingly, the only other sample type to recruit into the right-half of the tree in any significant abundance also originated from brackish sediments (pink squares, taken from the shallow water interface of Wall Estuary), suggesting that Clade 1°A sequences may be more prevalent in saline sediments than those belonging to Clade 1°B (Figure 2).
The point-source radiation of several sequence clusters should be considered as a false attraction of several long branches; there should be underlying sequence similarity among the grouped branches but the point source rooting of the cluster is an artifact of the radiating tree diagram. However, several secondary clades with high support were identified in the data, especially from the 1°B portion of the tree. Noteworthy secondary clades include: clade 2°A, populated exclusively with sewage sequences; clade 2°B, populated almost exclusively by spring water sequences; the sprawling clade 2 °C of saltwater and Antarctic sediment sequences; clade 2°D, containing sequences from two saltwater sites; and 2°E which contains a mixture of springwater and saltwater sequences including SARssφ1, somewhat removed on a long branch (Figure 2). None of the sample sites (icon colors) or sample types (icon shapes) shows exclusive recruitment into a single monophyletic clade that would be the hallmark of pure environmental forcing. To highlight the biochemical differences driving the topology of the phylogeny in Figure 2, the sequences belonging to each of these 2° clades were aligned and HMM logos were generated using HMMbuild (Eddy, 2008) and visualized using LogoMat (Schuster-Böckler et al., 2004). The HMM profiles of each clade were manually aligned relative to each other in an effort to juxtapose homologous regions (Supplementary Figure 1).
To place the current data set in the larger context of Microviridae diversity, representative sequences from throughout the tree in Figure 2 were integrated into the comprehensive Microviridae MCP alignment created by Roux et al. (2012b). This alignment was supplemented with sequences with strong gokushoviral BLASTX hits (e-value ⩽0.01) from two recent marine metagenomic studies; a single gokushovirus capsid sequence from the data set of Labonté and Suttle (2013) and 41 sequences from the recent viral community analysis of hadopelagic sediments off of Japan (Yoshida et al., 2013). The resulting combined phylogenetic tree is visualized in Figure 3, which contains as an outgroup the Microviridae subfamily most closely related to Gokushovirinae, tentatively named the Pichovirinae (Roux et al., 2012b).
Figure 3.

Rooted neighbor-joining tree combining sequences from Roux et al. (2012b) (n=60, black typeface with no icons), sequences from Yoshida et al. (2013) (n=40, blue typeface), a sequence from Labonté and Suttle (2013) (n=1, red typeface) and sequences from this study (43 environmental, 20 sewage; black typeface with icons from Figure 2) to give a comprehensive view of the Gokushovirinae, with the Pichovirinae as an outgroup at the top. Bootstrap values were calculated out of 100 replicates. Clades referenced in the discussion are annotated with brackets to the right.
Within the Gokushovirinae subfamily, Roux et al. (2012b) depicted a phylogenetic topology consisting of three apparently coherent groups; two of ‘eukaryote-associated' and one of ‘environmental' strains from freshwater metagenomes as well as SARssφ2 and BdφMH2K. Of those eukaryote-associated clusters from Roux et al. (2012b), the clade containing human gut associates (as well as a turkey gut associate, ‘Microvirus CA82') was highly supported and distinct, whereas the clustering of the Chlamydia phages and other human gut associates occupied an unstable position adjacent to the Environmental clade, which suggested a more recent divergence.
This topology is broadly recapitulated in our expanded phylogeny (Figure 3), where a well-supported clade of eukaryote associates, including turkey gut-derived CA82 and human gut derivatives, continues to occur. The fact that a similar bifurcated topology has now been reproduced by both primer-based and primer-independent metagenomic methods (Roux et al., 2012b) suggests that there may be a true split between environmental and eukaryotic-associated Gokushovirinae.
The weaker eukaryotic-associated clade containing the Chlamydia phages from Roux et al. (2012b) now nests within the ‘environmentally dominated clade', which has been significantly expanded through this study. It is not surprising that the primers used in this study amplified sequences most closely related to the Chlamydia phages, as the primers were designed based largely on the Chlamydia phages (six of nine reference sequences). However, all but one of the amplicons that was generated using the primers (colored icons) belonged to the ‘environmentally dominated clade', regardless of the sample type (sewage, aquatic, sediment). Future work should test both environmental and eukaryote-derived samples with the same primer sets to determine whether this persistent split is a real feature of the gokushoviral topology.
Conclusion
The discovery of diverse ssDNA phages in all environments tested is highly significant and prompts many questions for future studies. At present, the hosts for these environmental gokushoviruses remain unknown, as do the ecological effects of these phages on their hosts and ecosystems. To date, all cultured gokushoviruses infect intracellular parasites, a possibility that must be considered when attempting to culture environmental gokushoviruses. As opposed to Chlamydia, which are obligate parasites of eukaryotic organisms, Bdellovibrio parasitizes Gram-negative bacteria that are far more abundant in the environment than their eukaryotic counterparts. If the targeting of obligate intracellular parasitic bacteria continues to hold true as a hallmark of the Gokushovirinae, parasites of abundant bacteria and single-celled eukaryotes may prove fruitful as an avenue for exploring the hosts of the environmental gokushoviruses.
This study, taken together with the data mining work of Roux et al. (2012b), demonstrates the diverse and cosmopolitan nature of the Gokushovirinae subfamily, changing the perception of this group of ssDNA phages from one with a fairly narrow, primarily eukaryote-associated niche to a group of importance for microbial ecology. Another significant implication of these data is that studies utilizing nucleic acid staining and epifluorescence microscopy to enumerate environmental viruses (Patel et al., 2007) may be underestimating total viral abundance. The small genome sizes of gokushoviruses and other ssDNA phages produce a weak fluorescence signal that is below the detection limit of most microscopes and flow cytometers (Tomaru and Nagasaki, 2007). Along with other recent work (Steward et al., 2012; Brum et al., 2013; Labonté and Suttle, 2013), this study emphasizes the need for a shift in the paradigm that dsDNA Caudovirales dominate environmental viral communities.
Acknowledgments
Funding for this study was provided by grants to MB from the National Science Foundation (Microbial Interactions and Processes MCB-0701984 and Division of Biological Infrastructure DBI-0850206) and a Proposal Enhancement Grant from the University of South Florida Internal Awards Program. MAM and SK were funded by University of Florida COM Research Funds; MH was funded by a Presidential Fellowship from the University of South Florida; AC was funded by an REU Supplement from the National Science Foundation; SR was supported by a PhD grant from the French Defense Procurement Agency (DGA); AS was funded by NSF ANT-1246378. Thanks to the members of the British Services Antarctic Expedition 2012 for collecting the Marguerite Bay marine sediment samples and to Dawn Goldsmith and Bhakti Dwivedi for assistance with bioinformatics and primer design, respectively.
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies this paper on The ISME Journal website (http://www.nature.com/ismej)
Supplementary Material
References
- Ackermann HW. 5500 phages examined in the electron microscope. Arch Virol. 2007;152:227–243. doi: 10.1007/s00705-006-0849-1. [DOI] [PubMed] [Google Scholar]
- Agbandje-McKenna M, Kleinschmidt J.2011AAV capsid structure and cell interactionsIn: Snyder R, Moullier P (eds)Methods in Molecular Biology: Adeno-Associated Virus; Methods and Protocols Springer: New York, USA; 47–92. [DOI] [PubMed] [Google Scholar]
- Angly FE, Felts B, Breitbart M, Salamon P, Edwards RA, Carlson C, et al. The marine viromes of four oceanic regions. PLoS Biol. 2006;4:e368. doi: 10.1371/journal.pbio.0040368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anisimova M, Gascuel O. Approximate likelihood-ratio test for branches: a fast, accurate, and powerful alternative. Syst Biol. 2006;55:539–552. doi: 10.1080/10635150600755453. [DOI] [PubMed] [Google Scholar]
- Bahadur RP, Janin J. Residue conservation in viral capsid assembly. Proteins. 2008;71:407–414. doi: 10.1002/prot.21710. [DOI] [PubMed] [Google Scholar]
- Breitbart M. Marine viruses: truth or dare. Ann Rev Mar Sci. 2012;4:425–448. doi: 10.1146/annurev-marine-120709-142805. [DOI] [PubMed] [Google Scholar]
- Breitbart M, Miyake JH, Rohwer F. Global distribution of nearly identical phage-encoded DNA sequences. FEMS Microbiol Lett. 2004;236:249–256. doi: 10.1016/j.femsle.2004.05.042. [DOI] [PubMed] [Google Scholar]
- Breitbart M, Salamon P, Andresen B, Mahaffy JM, Segall AM, Mead D, et al. Genomic analysis of uncultured marine viral communities. PNAS. 2002;99:14250–14255. doi: 10.1073/pnas.202488399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brentlinger KL, Hafenstein S, Novak CR, Fane BA, Borgon R, McKenna R, et al. Microviridae, a family divided: isolation, characterization, and genome sequence of ϕMH2K, a bacteriophage of the obligate intracellular parasitic bacterium Bdellovibrio bacteriovorus. J Bacteriol. 2002;184:1089–1094. doi: 10.1128/jb.184.4.1089-1094.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brum JR, Schenck RO, Sullivan MB. Global morphological analysis of marine viruses shows minimal regional variation and dominance of non-tailed viruses. ISME J. 2013;7:1738–1751. doi: 10.1038/ismej.2013.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bull J, Badgett M, Wichman H. Big-benefit mutations in a bacteriophage inhibited with heat. Mol Biol Evol. 2000;17:942–950. doi: 10.1093/oxfordjournals.molbev.a026375. [DOI] [PubMed] [Google Scholar]
- Carrillo-Tripp M, Shepherd CM, Borelli IA, Venkataraman S, Lander G, Natarajan P, et al. VIPERdb2: an enhanced and web API enabled relational database for structural virology. Nucleic Acids Res. 2009;37:D436–D442. doi: 10.1093/nar/gkn840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherwa JE, Fane BA.2011Microviridae: Microviruses and Gokushoviruses eLSe-pub ahead of print 16 May 2011doi: 10.1002/9780470015902.a0000781.pub2 [DOI]
- Chipman PR, Agbandje-McKenna M, Renaudin J, Baker TS, McKenna R. Structural analysis of the spiroplasma virus, SpV4: implications for evolutionary variation to obtain host diversity among the Microviridae. Structure. 1998;6:135–145. doi: 10.1016/s0969-2126(98)00016-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke IN, Cutcliffe LT, Everson JS, Garner SA, Lambden PR, Pead PJ, et al. Chlamydiaphage Chp2, a skeleton in the ϕX174 closet: scaffolding protein and procapsid identification. J Bacteriol. 2004;186:7571–7574. doi: 10.1128/JB.186.22.7571-7574.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culley AI, Steward GF. New genera of RNA viruses in subtropical seawater, inferred from polymerase gene sequences. Appl Environ Microbiol. 2007;73:5937–5944. doi: 10.1128/AEM.01065-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desnues C, Rodriguez-Brito B, Rayhawk S, Kelley S, Tran T, Haynes M, et al. Biodiversity and biogeography of phages in modern stromatolites and thrombolites. Nature. 2008;452:340–343. doi: 10.1038/nature06735. [DOI] [PubMed] [Google Scholar]
- Dokland T, McKenna R, Ilag LL, Bowman BR, Incardona NL, Fane BA, et al. Structure of a viral procapsid with molecular scaffolding. Nature. 1997;389:308–313. doi: 10.1038/38537. [DOI] [PubMed] [Google Scholar]
- Dwivedi B, Schmieder R, Goldsmith DB, Edwards RA, Breitbart M. PhiSiGns: an online tool to identify signature genes in phages and design PCR primers for examining phage diversity. BMC Bioinformatics. 2012;13:37. doi: 10.1186/1471-2105-13-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy SR. A probabilistic model of local sequence alignment that simplifies statistical significance estimation. PLoS Comp Biol. 2008;4:e1000069. doi: 10.1371/journal.pcbi.1000069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everson J, Garner S, Lambden P, Fane B, Clarke I. Host range of chlamydiaphages ΦCPAR39 and Chp3. J Bacteriol. 2003;185:6490–6492. doi: 10.1128/JB.185.21.6490-6492.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fane B.2005Family microviridaeIn: Fauquet CM, Mayo MA, Maniloff J, Desselberger U, Ball LA (eds)Virus Taxonomy, Classification and Nomenclature of Viruses, 8th ICTV Report of the International Committee on Taxonomy of Viruses Elsevier/Academic Press: San Diego, USA [Google Scholar]
- Filée J, Tétart F, Suttle CA, Krisch H. Marine T4-type bacteriophages, a ubiquitous component of the dark matter of the biosphere. PNAS. 2005;102:12471–12476. doi: 10.1073/pnas.0503404102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Boronat M, Diez-Rivero CM, Reinherz EL, Reche PA. PVS: a web server for protein sequence variability analysis tuned to facilitate conserved epitope discovery. Nucleic Acids Res. 2008;36:W35–W41. doi: 10.1093/nar/gkn211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldsmith DB, Crosti G, Dwivedi B, McDaniel LD, Varsani A, Suttle CA, et al. Development of phoH as a novel signature gene for assessing marine phage diversity. Appl Environ Microbiol. 2011;77:7730–7739. doi: 10.1128/AEM.05531-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S, Dufayard J-F, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- Kim K-H, Bae J-W. Amplification methods bias metagenomic libraries of uncultured single-stranded and double-stranded DNA viruses. Appl Environ Microbiol. 2011;77:7663–7668. doi: 10.1128/AEM.00289-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K-H, Chang H-W, Nam Y-D, Roh SW, Kim M-S, Sung Y, et al. Amplification of uncultured single-stranded DNA viruses from rice paddy soil. Appl Environ Microbiol. 2008;74:5975–5985. doi: 10.1128/AEM.01275-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krupovic M, Prangishvili D, Hendrix RW, Bamford DH. Genomics of bacterial and archaeal viruses: dynamics within the prokaryotic virosphere. Microbiol Mol Biol Rev. 2011;75:610–635. doi: 10.1128/MMBR.00011-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labonté JM, Suttle CA. Previously unknown and highly divergent ssDNA viruses populate the oceans. ISME J. 2013;7:2169–2177. doi: 10.1038/ismej.2013.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin M, Blackshields G, Brown N, Chenna R, McGettigan PA, McWilliam H, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- López-Bueno A, Tamames J, Velázquez D, Moya A, Quesada A, Alcamí A. High diversity of the viral community from an Antarctic lake. Science. 2009;326:858–861. doi: 10.1126/science.1179287. [DOI] [PubMed] [Google Scholar]
- Magurran AE. Measuring Biological Diversity. Blackwell Science Ltd: Malden MA; 2004. [Google Scholar]
- McDaniel L, Breitbart M, Mobberley J, Long A, Haynes M, Rohwer F, et al. Metagenomic analysis of lysogeny in Tampa Bay: implications for prophage gene expression. PLoS One. 2008;3:e3263. doi: 10.1371/journal.pone.0003263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDaniel LD, Rosario K, Breitbart M, Paul JH. Comparative metagenomics: Natural populations of induced prophages demonstrate highly unique, lower diversity viral sequences. Environ Microbiol. 2013;16:570–585. doi: 10.1111/1462-2920.12184. [DOI] [PubMed] [Google Scholar]
- McKenna R, Bowman BR, Ilag LL, Rossmann MG, Fane BA. Atomic structure of the degraded procapsid particle of the bacteriophage G4: induced structural changes in the presence of calcium ions and functional implications. J Mol Biol. 1996;256:736–750. doi: 10.1006/jmbi.1996.0121. [DOI] [PubMed] [Google Scholar]
- McKenna R, Xia D, Willingmann P, Hag LL, Krishnaswamy S, Rossmann MG, et al. Atomic structure of single-stranded DNA bacteriophage Φ174 and its functional implications. Nature. 1992;355:137. doi: 10.1038/355137a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel A, Noble RT, Steele JA, Schwalbach MS, Hewson I, Fuhrman JA. Virus and prokaryote enumeration from planktonic aquatic environments by epifluorescence microscopy with SYBR Green I. Nat Protoc. 2007;2:269–276. doi: 10.1038/nprot.2007.6. [DOI] [PubMed] [Google Scholar]
- Paterson S, Vogwill T, Buckling A, Benmayor R, Spiers AJ, Thomson NR, et al. Antagonistic coevolution accelerates molecular evolution. Nature. 2010;464:275–278. doi: 10.1038/nature08798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Price MN, Dehal PS, Arkin AP. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol. 2009;26:1641–1650. doi: 10.1093/molbev/msp077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosario K, Breitbart M. Exploring the viral world through metagenomics. Curr Opin Virol. 2011;1:289–297. doi: 10.1016/j.coviro.2011.06.004. [DOI] [PubMed] [Google Scholar]
- Rosario K, Nilsson C, Lim YW, Ruan Y, Breitbart M. Metagenomic analysis of viruses in reclaimed water. Environ Microbiol. 2009;11:2806–2820. doi: 10.1111/j.1462-2920.2009.01964.x. [DOI] [PubMed] [Google Scholar]
- Roux S, Enault F, Robin A, Ravet V, Personnic S, Theil S, et al. Assessing the diversity and specificity of two freshwater viral communities through metagenomics. PLoS One. 2012;7:e33641. doi: 10.1371/journal.pone.0033641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux S, Krupovic M, Poulet A, Debroas D, Enault F. Evolution and diversity of the Microviridae viral family through a collection of 81 new complete genomes assembled from virome reads. PLoS One. 2012;7:e40418. doi: 10.1371/journal.pone.0040418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanger F, Air G, Barrell B, Brown N, Coulson A, Fiddes J, et al. Nucleotide sequence of bacteriophage PhiX174 DNA. Nature. 1977;265:687–695. doi: 10.1038/265687a0. [DOI] [PubMed] [Google Scholar]
- Schuster-Böckler B, Schultz J, Rahmann S. HMM logos for visualization of protein families. BMC Bioinformatics. 2004;5:7. doi: 10.1186/1471-2105-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon CE, Weaver W. The Mathematical Theory of Communication. University of Illinois Press: Urbana, IL; 1949. [Google Scholar]
- Smith RJ, Jeffries TC, Roudnew B, Seymour JR, Fitch AJ, Simons KL, et al. Confined aquifers as viral reservoirs. Environ Microbiol Rep. 2013;5:725–730. doi: 10.1111/1758-2229.12072. [DOI] [PubMed] [Google Scholar]
- Soetaert K, Heip C. Sample-size dependence of diversity indices and the determination of sufficient sample size in a high-diversity deep-sea environment. Mar Ecol Prog Ser. 1990;59:305–307. [Google Scholar]
- Steward GF, Culley AI, Mueller JA, Wood-Charlson EM, Belcaid M, Poisson G. Are we missing half of the viruses in the ocean. ISME J. 2012;7:672–679. doi: 10.1038/ismej.2012.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steward GF, Montiel JL, Azam F. Genome size distributions indicate variability and similarities among marine viral assemblages from diverse environments. Limnol Oceanogr. 2000;45:1697–1706. [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurber RV, Haynes M, Breitbart M, Wegley L, Rohwer F. Laboratory procedures to generate viral metagenomes. Nat Protoc. 2009;4:470–483. doi: 10.1038/nprot.2009.10. [DOI] [PubMed] [Google Scholar]
- Tomaru Y, Nagasaki K. Flow cytometric detection and enumeration of DNA and RNA viruses infecting marine eukaryotic microalgae. J Oceanogr. 2007;63:215–221. [Google Scholar]
- Tucker KP, Parsons R, Symonds EM, Breitbart M. Diversity and distribution of single-stranded DNA phages in the North Atlantic Ocean. ISME J. 2011;5:822–830. doi: 10.1038/ismej.2010.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Lasker K, Schneidman-Duhovny D, Webb B, Huang CC, Pettersen EF, et al. UCSF Chimera, MODELLER, and IMP: an integrated modeling system. J Struct Biol. 2012;179:269–278. doi: 10.1016/j.jsb.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida M, Takaki Y, Eitoku M, Nunoura T, Takai K. Metagenomic analysis of viral communities in (hado) pelagic sediments. PLoS One. 2013;8:e57271. doi: 10.1371/journal.pone.0057271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Breitbart M, McNairnie P, Rohwer F. FastGroupII: a web-based bioinformatics platform for analyses of large 16S rDNA libraries. BMC Bioinformatics. 2006;7:57. doi: 10.1186/1471-2105-7-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.