

RASAL1, a RAS GTPase-activating protein (RasGAP), has been recently identified for the first time as a major tumor suppressor gene in thyroid cancer (1). RasGAP activates the intrinsic GTPase of RAS, which hydrolyzes GTP, converting the active GTP-bound RAS to inactive GDP-bound RAS and terminating the RAS signaling. For decades, RAS mutations have been the only known genetic mechanism for the activation of RAS in thyroid cancer. The study, by discovering inactivating genetic and epigenetic alterations of RASAL1, identified a new prominent genetic mechanism for the activation of RAS in thyroid cancer (1). This work is believed to add to our current understanding of thyroid tumorigenesis by providing a compelling model of how alternative RAS signaling-related genes impact thyroid tumorigenesis and, in the context of recent molecular studies in thyroid cancer (2), to help further refine the molecular taxonomy of thyroid cancer (3). The study characterized RASAL1 as a tumor suppressor gene in thyroid cancer in a comprehensive manner, involving genetic, epigenetic, and functional aspects at both in vitro and in vivo levels. It demonstrated for the first time its tumor-suppressor function and common mutually exclusive inactivating somatic genetic and epigenetic alterations, thus firmly establishing a prominent role of RASAL1 in thyroid tumorigenesis. Among a large number of RasGAP genes examined, only RASAL1 was found to be completely methylated and silenced in thyroid cancer cells (1), suggesting a unique and particularly important role of this RasGAP gene in thyroid tumorigenesis. This importance of RASAL1 in thyroid cancer is now further supported by the germline study of the RASAL1 gene from the Eng group (4) published in July issue of the JCEM, in which the authors interestingly identified deleterious RASAL1 germline mutations in PTENwild-type patients with Cowden syndrome who developed follicular thyroid cancer (FTC) and in patients who had apparent somatic follicular variant of papillary thyroid cancer (PTC). This study provides the first evidence that germline genetic alterations can occur in RASAL1 in human diseases. The finding of deleterious germline mutations in the RASAL1 gene in patients with apparent somatic follicular variant PTC is intriguing, which is expected to stimulate new studies to define further its significance and implications. The results of the Eng group (4) are strikingly consistent with the previous findings that inactivating genetic and epigenetic alterations of RASAL1 were more commonly found in FTC than in PTC (1). These somatic and germline genetic findings of RASAL1 in the two studies are typical of some other RasGAP tumor suppressor genes, such as NF1 in the development of neurofibromatosis type 1 and various human cancers (5, 6), whose inactivation can occur as a primary genetic driver of tumorigenesis of both somatic and familial forms. Being such a RasGAP tumor suppressor gene and now supported by the germline data from the Eng group (4), it is quite possible that mutations in RASAL1 may prove to be responsible for certain yet unidentified familial cancers other than Cowden syndrome. Inactivation of the RASAL1 gene seemed to play a particularly important role in the progression and aggressiveness of thyroid cancer because inactivating mutations and hypermethylation of this gene most commonly occurred in anaplastic thyroid cancer (ATC), the most aggressive type of thyroid cancer (1). As could be expected for a RasGAP, the wild-type RASAL1 was functionally shown to suppress both the MAPK and phosphatidylinositol-3-kinase (PI3K) pathways; this function was lost in RASAL1 mutants (1). Thus, inactivating mutations of RASAL1, by resulting in the activation of RAS, are dull activators of the MAPK and PI3K pathways. This may partially explain the preferential presence of RASAL1 mutations in ATC and is consistent with a previous proposal that dual activation of the MAPK and PI3K pathways is an important mechanism for the progression of thyroid cancer to aggressive forms, such as ATC (2).
As shown in the canonical scheme for the MAPK and PI3K pathways in Figure 1, the aberrant activation of the two pathways dually regulated by RAS plays a fundamental role in thyroid tumorigenesis (2). Oncogenic activation of the two pathways has been well known to be driven by activating mutations in classical proto-oncogenes in these pathways, such as RAS, BRAF, and PIK3CA. Genomic amplifications of these and other proto-oncogenes in these pathways, such as the genes for various receptor tyrosine kinases and AKT, are also common in thyroid cancer (7). All of these are activating genetic alterations that constitute the classically known mechanism for the aberrant activation of the two RAS-related signaling pathways. The one known mechanism involving inactivating genetic alterations, resulting in aberrant activation of this pathway signaling, is the PTEN alterations. This is specifically a mechanism for the aberrant activation of the PI3K pathway. PTEN functions as a lipid phosphatase to convert phosphatidylinositol (3,4,5)-trisphosphate to phosphatidylinositol (4,5)-bisphosphate, terminating the activation of the PI3K pathway. PTEN is thus a prominent tumor suppressor. Its genetic and epigenetic inactivation is an important mechanism in thyroid tumorigenesis (8). Inactivating germline mutations of the PTEN gene are the main cause of Cowden syndrome, which predisposes to the development of FTC (9). PTEN mutations also occur in somatic thyroid cancers, particularly in FTC and ATC (7, 10, 11). Hypermethylation of the PTEN gene is also common in thyroid cancer (12). RASAL1 now represents a second example in which inactivating genetic and epigenetic alterations of a tumor suppressor gene is a major mechanism in driving aberrant activation of the RAS-regulated pathways in thyroid cancer. Unlike PTEN inactivation, which only activates the PI3K pathway, RASAL1 inactivation results in the activation of both the MAPK and PI3K pathways, thus potentially having a more profound impact on tumorigenesis, such as tumor progression and aggressiveness of ATC discussed above. Both the MAPK and PI3K pathways play an important role in undifferentiated ATC, whereas the former plays a dominant role preferentially in PTC and the latter plays a dominant role preferentially in FTC (2). The fact that genetic and epigenetic alterations of RASAL1 more commonly occurred in FTC than PTC (1) suggests that alterations in RASAL1 may preferentially affect the PI3K pathway over the MAPK pathway in differentiated thyroid cancers. This is quite consistent with previous findings that RAS mutations were more commonly associated with the activation of the PI3K pathway over the MAPK pathway in thyroid cancer (7, 13).
Figure 1.
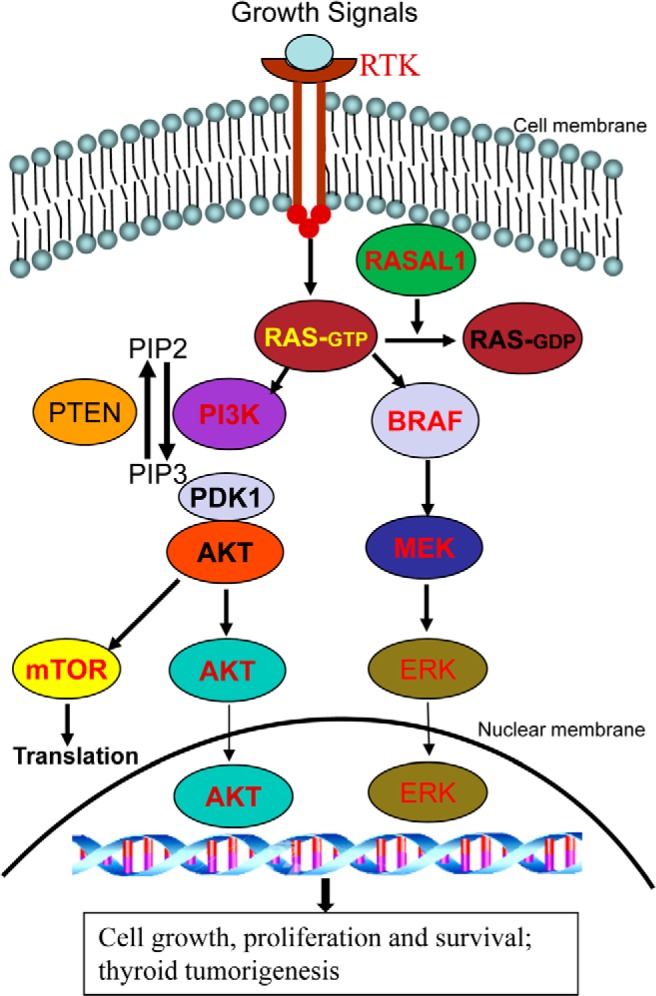
Addition of RASAL1 to the canonical scheme of the RAS-coupled MAPK and PI3K pathways. Shown are the classical RAS-coupled MAPK and PI3K signaling pathways. The normal signaling flow is typically initiated by growth factors acting on various receptor tyrosine kinases (RTKs) at the cell membrane, which is relayed to RAS, where the downstream MAPK pathway (right side) and PI3K pathway (left side) are activated. Genes for many components in the pathways, such as RTKs, RAS, BRAF, PIK3CA in PI3K, PDK1, AKT, and mTOR are proto-oncogenes, and their activating genetic alterations, such as mutations or copy gains, are the well-established genetic mechanism for the aberrant activation of these pathways in thyroid cancer. PTEN is a tumor suppressor that, by converting phosphatidylinositol (3,4,5)-trisphosphate (PIP3) to phosphatidylinositol (4,5)-bisphosphate (PIP2), normally antagonizes the signaling of the PI3K pathway. Genetic or epigenetic inactivation of PTEN results in aberrant activation of the PI3K pathway. RASAL1 as a newly documented prominent tumor suppressor can now be added to this signaling scheme as an important component. As a RAS GTPase-activating protein, RASAL1 normally terminates the RAS signaling by converting the active GTP-bound RAS to the inactive GDP-bound RAS. Genetic or epigenetic inactivation of RASAL1 results in aberrant activation of RAS signaling and the downstream two pathways; this represents a prominent new genetic mechanism for the aberrant activation of the RAS-coupled signaling pathways and thyroid tumorigenesis.
Genetic alterations in the classical genes in the RAS-regulated MAPK and PI3K pathways collectively occur in about 60–70% of thyroid cancers (2). It is interesting to see that the inactivating mutations and high-level hypermethylation of RASAL1 occurred mainly in the remaining 30–40% of thyroid cancers that lacked the classical genetic alterations in the RAS-regulated pathways, such as mutations in the BRAF, RAS, PIK3CA, and PTEN genes (1). This mutual exclusivity of RASAL1 alterations from the classical genetic alterations in the RAS-regulated pathways strongly supports an important independent role of RASAL1 in thyroid tumorigenesis, particularly in thyroid cancers lacking the classical genetic alterations in the MAPK and PI3K pathways. Thus, genetic or epigenetic inactivation of RASAL1 represents a novel prominent mechanism in thyroid tumorigenesis, which is likely also an important genetic mechanism in other human cancers. It is plausible to propose that the classical oncogenic RAS-coupled MAPK and PI3K pathways should now be revised to welcome RASAL1 as a prominent new component in this canonical cancer cell signaling, as illustrated in Figure 1. As a new friend to the thyroid cancer research community, RASAL1 heralds great promise for better understanding of molecular mechanisms and development of novel therapeutic strategies for thyroid cancer.
Acknowledgments
This project was supported by National Institutes of Health Grants R01CA134225 and RO1CA113507 (to M.X.).
Disclosure Summary: The author has no conflict of interest to disclose.
Footnotes
- ATC
- anaplastic thyroid cancer
- FTC
- follicular thyroid cancer
- PI3K
- phosphatidylinositol-3-kinase
- PTC
- papillary thyroid cancer
- RasGAP
- RAS GTPase-activating protein.
References
- 1. Liu D, Yang C, Bojdani E, Murugan AK, Xing M. Identification of RASAL1 as a major tumor suppressor gene in thyroid cancer. J Natl Cancer Inst. 2013;105:1617–1627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Xing M. Molecular pathogenesis and mechanisms of thyroid cancer. Nat Rev Cancer. 2013;13:184–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ngeow J, Eng C. RASAL1 in thyroid cancer: wisdom from an old foe. J Natl Cancer Inst. 2013;105:1597–1599 [DOI] [PubMed] [Google Scholar]
- 4. Ngeow J, Ni Y, Tohme R, Song Chen F, Bebek G, Eng C. Germline alterations in RASAL1 in Cowden syndrome patients presenting with follicular thyroid cancer and in individuals with apparently sporadic epithelial thyroid cancer. J Clin Endocrinol Metab. 2014;99:E1316–E1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yohay K. Neurofibromatosis type 1 and associated malignancies. Curr Neurol Neurosci Rep. 2009;9:247–253 [DOI] [PubMed] [Google Scholar]
- 6. Laycock-van Spyk S, Thomas N, Cooper DN, Upadhyaya M. Neurofibromatosis type 1-associated tumours: their somatic mutational spectrum and pathogenesis. Hum Genomics. 2011;5:623–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liu Z, Hou P, Ji M, et al. Highly prevalent genetic alterations in receptor tyrosine kinases and phosphatidylinositol 3-kinase/akt and mitogen-activated protein kinase pathways in anaplastic and follicular thyroid cancers. J Clin Endocrinol Metab. 2008;93:3106–3116 [DOI] [PubMed] [Google Scholar]
- 8. Xing M. Genetic alterations in the phosphatidylinositol-3 kinase/Akt pathway in thyroid cancer. Thyroid. 2010;20:697–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gustafson S, Zbuk KM, Scacheri C, Eng C. Cowden syndrome. Semin Oncol. 2007;34:428–434 [DOI] [PubMed] [Google Scholar]
- 10. Hou P, Liu D, Shan Y, et al. Genetic alterations and their relationship in the phosphatidylinositol 3-kinase/Akt pathway in thyroid cancer. Clin Cancer Res. 2007;13:1161–1170 [DOI] [PubMed] [Google Scholar]
- 11. Wang Y, Hou P, Yu H, et al. High prevalence and mutual exclusivity of genetic alterations in the phosphatidylinositol-3-kinase/akt pathway in thyroid tumors. J Clin Endocrinol Metab. 2007;92:2387–2390 [DOI] [PubMed] [Google Scholar]
- 12. Hou P, Ji M, Xing M. Association of PTEN gene methylation with genetic alterations in the phosphatidylinositol 3-kinase/AKT signaling pathway in thyroid tumors. Cancer. 2008;113:2440–2447 [DOI] [PubMed] [Google Scholar]
- 13. Abubaker J, Jehan Z, Bavi P, et al. Clinicopathological analysis of papillary thyroid cancer with PIK3CA alterations in a Middle Eastern population. J Clin Endocrinol Metab. 2008;93:611–618 [DOI] [PubMed] [Google Scholar]