

Abstract
Radiotracers labelled with short-lived fluorine-18 (t1/2 = 109.7 min) are keenly sought for biomedical imaging with positron emission tomography (PET). The radiotracers are mostly required at high specific radioactivities, necessitating their radiosyntheses from cyclotron-produced no-carrier-added [18F]fluoride ion. PET radiotracers encompass wide structural diversity and molecular weight. Hence, diverse 18F-labeling methodology is needed to accomplish the required radiosyntheses in a simple and rapid manner. A useful strategy is to introduce nucleophilic [18F]fluoride ion first into a labeling synthon that may then be applied to label the target radiotracer. Here, we show that various functionalized [18F]fluoroarenes may be rapidly synthesized as labeling synthons through single-step reactions of appropriate diaryliodonium salts with [18F]fluoride ion. Decay-corrected radiochemical yields (RCYs) varied with position of functional group, choice of electron-rich aryl ring in the diaryliodonium salt, and choice of anion. Under best conditions, 18F-labeled fluorobenzaldehydes, fluorobenzyl halides, fluorobenzoic acid esters and fluoro-phenyl ketones were obtained selectively in 40–73%, 20–55%, 46–89% and 81–98% RCYs, respectively. This versatile straightforward methodology will enhance the scope for producing structurally complex, yet useful, PET radiotracers.
Introduction
The use of biochemically-specific radiotracers with positron emission tomography (PET) is an increasingly versatile molecular imaging modality for biomedical research, medical diagnosis and drug development. Among radionuclides used in PET imaging, short-lived fluorine-18 (t1/2 = 109.7 min; β+ = 97%) has become widely popular because [18F]fluoride ion can be produced in high amounts and in high no-carrier-added (NCA) specific radioactivity from the 18O(p,n)18F reaction on 18O-enriched water with a moderate energy cyclotron. Moreover, this radionuclide can be incorporated covalently into radiotracers having diverse structure and molecular weight. For these and other reasons increasing efforts are being made to develop efficient 18F-based radiochemistry for PET radiotracer production. [18F]Fluoride ion may be used as a nucleophile for direct incorporation at aliphatic or aromatic carbons in many radiotracers, provided that suitably reactive precursors can be prepared for classical SN2 or SNAr reactions.1 Aryl C–18F bonds are generally favored in PET radiotracers because of their usual resistance to cleavage in vivo. However, the creation of aromatic C–18F bonds is more demanding than that of aliphatic C–18F bonds, especially if the arene is electron-rich or not well-activated for the desired nucleophilic substitution reaction. Improved methods for producing NCA [18F]fluoroarenes have emerged,2 including reactions of [18F]fluoride ion with diaryliodonium salts,3–8 triarylsulfonium salts9 and diaryl sulfoxides.10 The use of diaryliodonium salt precursors has in particular enabled direct incorporation of [18F]fluoride ion into electron-rich as well as electron-deficient arenes, and into meta- as well as into ortho- or para-positions (Fig. 1).3–8 This methodology is now being applied to the direct labeling of low molecular weight (<500 Da) radiotracers, such as [18F]DAA1106,11 [18F]SP203,12 and [18F]flumazenil.13 Radio-tracers of higher molecular weight or more complex structure may not be amenable to direct incorporation of nucleophilic [18F]fluoride ion, and may need to be labeled by use of an electrophilic reagent, a labeling synthon, derived from [18F]fluoride ion. Examples, of such labeling synthons include 18F-labeled haloalkanes, esters, benzyl halides, and benzaldehydes.1 Many of these labeling synthons have already found application for labeling useful or experimental PET radiotracers14 including various 18F-labeled amino acids receptor ligands, and peptides and proteins. Nonetheless, radiosyntheses of many of the employed synthons have been multi-step, and lacking desirable efficiency or speed.
Fig. 1.
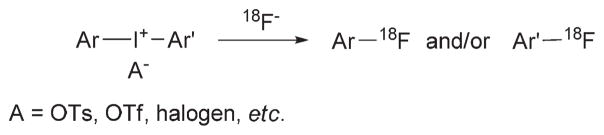
[18F]Fluoroarenes from diaryliodonium salts.
Recently, we have reported the use of diaryliodonium salts for the radiosyntheses of aryl and heteroaryl labeling synthons, such as [18F]fluorohalopyridines15 and [18F]azidomethyl-fluoro-arenes.16 Herein, we extend the use of diaryliodonium salts for single-step radiosyntheses of various other functionalized 18F-labeling synthons. Many of these synthons are new. Some are already known, but in this study were produced with comparable or greater efficiency from diaryliodonium salts than by previous methods.
Results and discussion
Syntheses of functionalized diaryliodonium salts
We have previously reported17 the syntheses of most of the functionalized diaryliodonium tosylates used in this study, based on two methods, A and B (Fig. 2). These methods were also used in this study to prepare several new salts (see ESI†).
Fig. 2.
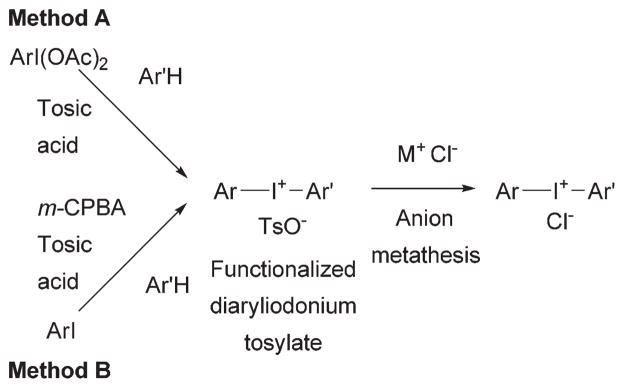
Methods used to synthesize functionalized diaryliodonium salts.
Method A was applicable to diaryliodonium salts bearing a halomethyl group (4b, 7b, 7d, 8b) and was based on treating an electron-rich arene, such as anisole, (1,3,5-trimethoxy)-benzene, thiophene, or 2-methylthiophene, with a functionalized [hydroxy(tosyloxy)iodo]arene (HTIA). In another example, the formyl compound 3b was prepared from 4-methoxy-3-(tri-n-butylstannyl)benzaldehyde and 2-(diacetoxyiodo)thiophene. The HTIAs were generated in situ from the corresponding functionalized (diacetoxyiodo)arenes by treatment with tosic acid in acetonitrile. The required (diacetoxyiodo)arenes were themselves obtained as white crystalline solids by oxidation of the corresponding iodoarenes with 32 wt% peracetic acid in acetic acid.17
Method B was based on oxidizing an iodoarene directly with m-CPBA (m-chloroperbenzoic acid) in p-tosic acid to generate an HTIA in situ for reaction with an activated arene, such as anisole. This method was suitable for preparing diaryliodonium salts bearing functionality resistant to oxidation by m-CPBA, such as the ketone 14 and the (chloromethyl)-naphthalene 10a. Method B is more convenient than method A, because readily available iodoarenes can be directly converted into diaryliodonium salts in one-pot.
Notably, these synthetic methods are generally regioselective, placing hypervalent iodine preferentially in para position to a substituent on an electron-rich benzene, such as anisole, or in α-position to sulphur in thiophene.
Both methods provide the diaryliodonium salts as tosylates. In order to explore the possible influence of the anion on the radiofluorination of these salts, many of the tosylates were conveniently converted into chlorides (1b, 2b, 2d, 3c, 6c, 7c, 7e, 10b) through high-yielding metathesis reactions.
Fig. 3 defines the structures of the full range of salts used in this study. In each of these salts, one aryl ring was made relatively electron-rich (e.g., 4-methoxyphenyl, (2,4,6-tri-methoxy)phenyl, 2-thienyl, or 5-methyl-2-thienyl) with the aim of conferring selectivity for radiofluorination at the desired functionalized partner ring.5,7,8,18
Fig. 3.

Structures of functionalized diaryliodonium salts used in this study.
Radiochemistry
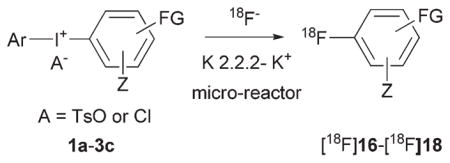
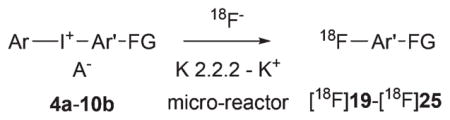
We used a commercial microfluidic apparatus (NanoTek; Advion, Ithaca, NY) to investigate the radiofluorination of the functionalized diaryliodonium salts with [18F]fluoride ion, because this platform permits fast throughput of radiolabeling experiments with small amounts of substrate under highly-controlled conditions of substrate concentration, reaction time, and temperature.7 Also, use of this apparatus excludes any possible photo-initiated decomposition of precursor iodonium salt. Full details of the configuration and operation of this apparatus in our laboratory have been described previously.7 Briefly, dry NCA [18F]fluoride ion-kryptofix 2.2.2-K+ complex (18F−-K 2.2.2-K+) and the iodonium salt were separately dissolved in either acetonitrile or DMF and each loaded into a storage loop of the apparatus. Each solution (10–20 μL) was infused into the micro-reactor (internal volume 31.4 μL) at equal set flow rates (4–10 μL min−1) and at a preset temperature. The concentration of diaryliodonium salt was 10 mM in the storage loop, and consequently the amounts of precursor used in a single microfluidic reaction were typically in the sub-micromole range. Radioactive effluents were directly quenched by dilution in H2O–MeCN (1:1, v/v; 3 mL) at room temperature and analyzed with reversed phase HPLC equipped with a radioactivity detector. Reaction temperature and time were varied over 6 to 12 runs for each salt. We report the reaction conditions that gave the highest RCYs, as determined with HPLC. Radioactivity adsorption on reactor vessel surfaces is commonly observed during radiofluorination reactions with NCA [18F]fluoride ion.19 In this study, recovery of radioactivity from the microfluidic apparatus was determined, and we also report yields corrected for the radioactivity adsorption in the instrument.
Radiosyntheses of [18F]fluorobenzaldehydes
[18F]4-Fluorobenzaldehyde ([18F]16) is widely used as a labeling synthon for preparing complex PET radiotracers, and also other labeling agents.1,20,21 For these purposes, [18F]16 has been prepared efficiently through SNAr reactions of NCA [18F]-fluoride ion on formyl-benzenes having either a p-nitro or p-tri-methylammonium substituent as leaving group.20,21 We explored whether (4-formylphenyl)(4′-methoxyphenyl)iodonium salts (1a and 1b) were viable alternative precursors for preparing [18F]16. We rapidly obtained [18F]16 in 34 and 73% yields from the radiofluorination of the tosylate 1a and chloride 1b in DMF, respectively (Table 1, entries 1 and 2). The latter yield is comparable to the highest yield previously reported (65%) from non-hypervalent substrates.21 We note that comparably high yields (up to 83%) have recently been obtained from (4-formylphenyl)aryliodonium triflates.22
Table 1.
RCYs of [18F]fluorobenzaldehydes from formyl-functionalized diaryliodonium salts
| ||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Substratea | Conditionsb
|
Radioactive product
|
|||||
| Temp. (°C) | Timec (s) | Solvent | # | Z | FG | RCYd (%) | ||
| 1 | 1a | 180 | 236 | DMF | [18F]16 | H | 4-CHO | 34 (29) |
| 2 | 1b | 180 | 314 | DMF | [18F]16 | H | 4-CHO | 73 (58) |
| 3 | 2a | 180 | 314 | MeCN | [18F]17 | H | 3-CHO | 17 (12) |
| 4 | 2b | 180 | 252 | DMF | [18F]17 | H | 3-CHO | 31 (28) |
| 5 | 2c | 180 | 236 | DMF | [18F]17 | H | 3-CHO | 40 (37) |
| 6 | 2d | 180 | 314 | DMF | [18F]17 | H | 3-CHO | 46 (31) |
| 7 | 3a | 200 | 236 | DMF | [18F]18 | 4-MeO | 3-CHO | 20 (15) |
| 8 | 3a | 160 | 188 | MeCN | [18F]18 | 4-MeO | 3-CHO | 5 (3) |
| 9 | 3b | 200 | 236 | DMF | [18F]18 | 4-MeO | 3-CHO | 40 (26) |
| 10 | 3c | 200 | 188 | DMF | [18F]18 | 4-MeO | 3-CHO | 10 (7) |
5 mM concentration.
Conditions giving the highest yields from 6–12 different runs on each salt.
Residence time in the micro-reactor.
Yields determined by HPLC; yields in parentheses are corrected for radioactivity adsorption. The highest yield obtained for each radioactive product is shown in bold.
Attempts to prepare [18F]3-fluorobenzaldehyde ([18F]17) through conventional SNAr reactions have given almost no yield (<1%).23 Recently, microwave-promoted radiofluorinations of (3-formylphenyl)(phenyl)iodonium salts in DMF have been shown to provide [18F]17 in useful yields (up to 62%), but only in the presence of a relatively large amount of the free-radical scavenger, 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO), and with co-production of a low proportion of [18F]-fluorobenzene.24 We were interested in whether the radio-synthesis of [18F]17 would be viable in the microfluidic apparatus in the absence of TEMPO. Also we considered that the use of 4-methoxyphenyl or 2-thienyl as the partner aryl ring in the diaryliodonium salt precursor might increase the selectivity for producing the desired [18F]17. [18F]17 was readily obtained in 17–46% yields from salts bearing a 3-formyl substituent (Table 1, entries 3–6; see ESI† for radio-HPLC chromatogram). The highest yield (46%) was obtained from the salt having a 2-thienyl ring and a chloride anion (2d) (Table 1, entry 6). Even with an electron-donating methoxy group present in 4-position, adjacent to the formyl group, as in precursors 3a–3c, the desired product [18F]3–fluoro-4-methoxybenzaldehyde ([18F]18) was obtained (Table 1, entries 7–10). [18F]18 has previously been inaccessible through conventional SNAr.23 Reaction of 3b, bearing a 2-thienyl ring partner and a tosylate anion, gave a moderately high and useful yield of [18F]18 in DMF (40%; Table 1, entry 9).
Radiosyntheses of [18F]fluorobenzyl halides
2- and 4-[18F]fluorobenzyl halides have become key labeling synthons in the preparation of complex radiotracers.1,25 Nevertheless, their radiosyntheses have generally been achieved through three steps starting with [18F]fluoride ion substitution in (N-trimethyl)amino-benzaldehydes, followed by reduction and halogenation (Fig. 4).25 The automation of such multi-step radiochemistry for radiation safety is challenging, and the complexity of the process may lead to inefficient or unreliable production of derived radiotracers. We considered that such labeling synthons might be produced adequately from a single-step radiofluorination of a diaryliodonium salt. Our experiments showed that each of the [18F]fluorobenzyl chlorides and bromides ([18F]19–24) could be rapidly produced in this manner in useful yields, ranging from 20 to 52% (Table 2). Notably, 3-[18F]fluorobenzyl bromide ([18F]21) and chloride ([18F]22) were obtained in moderately useful yields of 33 and 31%, respectively (Table 2, entries 6 and 12). These two labeling synthons have not been available from non-hypervalent precursor due to the absence of a method for making the precursor aldehyde [18F]17 for multi-step radiosynthesis. Recently, [18F]22 was prepared, from a diaryliodonium salt but in two steps.24 The rapid single-step process described here should greatly expand the utility of [18F]fluorobenzyl halides as labeling synthons.
Fig. 4.

Previous and new radiosyntheses of [18F] fluorobenzyl halides.
Table 2.
RCYs of [18F]halomethyl-fluoroarenes from diaryliodonium salts bearing halomethyl groups
| ||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Substratea | Conditionsb
|
Radioactive product
|
|||||
| Temp. (°C) | Timec (s) | Solvent | # | Ar′ | FG | RCYd (%) | ||
| 1 | 4a | 180 | 236 | DMF | [18F]19 | C6H4 | 2-CH2Br | 37 (25) |
| 2 | 4b | 160 | 188 | DMF | [18F]19 | C6H4 | 2-CH2Br | 50 (36) |
| 3 | 5a | 180 | 236 | DMF | [18F]20 | C6H4 | 2-CH2Cl | 55 (42) |
| 4 | 6a | 160 | 188 | MeCN | [18F]21 | C6H4 | 3-CH2Br | 25 (22) |
| 5 | 6b | 180 | 236 | MeCN | [18F]21 | C6H4 | 3-CH2Br | 18 (16) |
| 6 | 6c | 200 | 236 | DMF | [18F]21 | C6H4 | 3-CH2Br | 31 (28) |
| 7 | 7a | 140 | 314 | MeCN | [18F]22 | C6H4 | 3-CH2Cl | 24 (19) |
| 8 | 7b | 180 | 188 | MeCN | [18F]22 | C6H4 | 3-CH2Cl | 28 (24) |
| 9 | 7c | 200 | 236 | DMF | [18F]22 | C6H4 | 3-CH2Cl | 25 (20) |
| 10 | 7d | 180 | 236 | MeCN | [18F]22 | C6H4 | 3-CH2Cl | 20 (18) |
| 11 | 7d | 200 | 236 | DMF | [18F]22 | C6H4 | 3-CH2Cl | 25 (17) |
| 12 | 7e | 200 | 236 | DMF | [18F]22 | C6H4 | 3-CH2Cl | 33 (27) |
| 13 | 8a | 160 | 236 | DMF | [18F]23 | C6H4 | 4-CH2Br | 15 (9) |
| 14 | 8b | 140 | 236 | DMF | [18F]23 | C6H4 | 4-CH2Br | 20 (17) |
| 15 | 9a | 180 | 236 | DMF | [18F]24 | C6H4 | 4-CH2Cl | 45 (36) |
| 16 | 9b | 180 | 188 | DMF | [18F]24 | C6H4 | 4-CH2Cl | 52 (40) |
| 17 | 10a | 140 | 236 | DMF | [18F]25 | C10H6 | 2-CH2Cl | 28 (25) |
| 18 | 10b | 140 | 236 | DMF | [18F]25 | C10H6 | 2-CH2Cl | 30 (26) |
5 mM concentration.
Conditions giving the highest yields from 6–12 different runs on each salt.
Residence time in the micro-reactor.
Yields determined by HPLC; yields in parentheses are corrected for radioactivity adsorption. The highest yield obtained for each radioactive product is shown in bold.
The increasingly successful use of diaryliodonium salt precursors to introduce NCA [18F]fluoride ion into electron-rich benzenes,3–8 suggested the possibility to introduce [18F]fluoride ion into other more complex electron-rich arenes, such as naphthalenes. We found that [18F]1-fluoronaphthalene and [18F]2-fluoronaphthalene could indeed be prepared regioselectively in this manner (see ESI†). We further explored whether [18F]2-(chloromethyl)-6-fluoronaphthalene ([18F]25) might also be prepared as a potential labeling synthon through single-step radiofluorination of the diaryliodonium salts, 10a and 10b. We obtained [18F]25 in moderate but useful yields (Table 2, entries 17 and 18; see ESI† for radio-HPLC chromatogram). No radioactive by-products due to replacement of the chloro substituent appeared in the radio-HPLC analyses of the reaction mixtures. This radiochemistry opens up the possibility to label future radiotracers bearing [18F]fluoronaphthyl entities more efficiently.
Radiosyntheses of [18F]fluorobenzoic acid esters
[18F]Fluorobenzoic acid esters are potentially useful labeling synthons, as exemplified by the peptide/protein labeling agent, N-succinimidyl 4-[18F]fluorobenzoate ([18F]SFB).21,26 We explored the single-step synthesis of [18F]fluorobenzoic acid esters through the radiofluorination of ester-functionalized diaryliodonium salts. Radiofluorination of diaryliodonium tosylate bearing a methoxycarbonyl group at para position gave a remarkably high yield (89%) of methyl [18F]4-fluorobenzoate ([18F]26) at 200 °C in DMF (Table 3, entry 1). This yield well exceeds that recently reported for the synthesis of ethyl [18F]fluorobenzoate through the radiofluorination of (4-(ethoxy-carbonyl)phenyl)(2-thienyl)iodonium trifluoroacetate, under various conditions, including the use of tetraethylammonium bicarbonate as a phase transfer agent.27 The t-butyl and ethyl ester analogs of [18F]26 have themselves been prepared through SNAr on non-hypervalent substrates and used to prepare [18F]SFB, but in three-stage radiosyntheses.28 We note that a single-step radiosynthesis of [18F]SFB from a diaryliodonium salt appears feasible with a reported non-optimized yield of up to 26%.29
Table 3.
RCYs of [18F]fluorobenzoic acid esters and [18F]fluorophenyl ketones from functionalized diaryliodonium tosylates in DMF
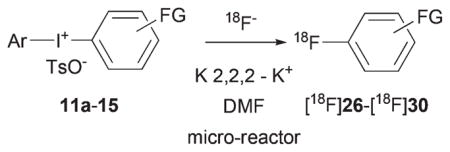
| ||||||
|---|---|---|---|---|---|---|
| Entry | Substratea | Conditionsb
|
Radioactive product
|
|||
| Temp. (°C) | Timec (s) | # | FG | RCYd (%) | ||
| 1 | 11a | 200 | 236 | [18F]26 | 4-MeOCO | 89 (74) |
| 2 | 12a | 100 | 188 | [18F]27 | 3-MeOCO | 46 (35) |
| 3 | 12b | 200 | 188 | [18F]27 | 3-MeOCO | 31 (25) |
| 4 | 13 | 200 | 236 | [18F]28 | 3-EtOCO | 80 (70) |
| 5 | 14 | 180 | 188 | [18F]29 | 4-Bz | 81 (70) |
| 6 | 15 | 200 | 188 | [18F]30 | 4-(4′-Br-Bz) | 98 (86) |
5 mM concentration.
Conditions giving the highest yields from 6–12 different runs on each salt.
Residence time in the micro-reactor.
Yields determined by HPLC; yields in parentheses are corrected for radioactivity adsorption. The highest yield obtained for each radioactive product is shown in bold.
We extended experiments to the radiofluorination of diary-liodonium salts bearing alkoxycarbonyl groups at meta position. A moderate yield (46%) of methyl [18F]3-fluorobenzoate ([18F]27) was obtained from precursor 12a (Table 3, entry 2). Change of the partner aryl ring to 2-thienyl, as in 12b, gave a lower but still useful yield (Table 3, entry 3). When an ethoxy-carbonyl group was present at meta-position in the precursor, the desired radiofluorinated ester [18F]28 was obtained in high yield (80%) (Table 3, entry 4; see ESI† for radio-HPLC chromatogram). This result appears consistent with a brief earlier report on the radiosynthesis of [18F]28,30 and in particular highlights the potential of functionalized diaryliodonium salt precursors for use in the radiosyntheses of other [18F]fluoro-benzoic acid esters, including the otherwise difficult to access meta-substituted esters.
Radiosyntheses of [18F]fluorophenyl ketones
[18F]4-Fluorobenzophenone ([18F]29) has been prepared in 52% yield by SNAr in 4-nitrobenzophenone and applied, for example, to produce the dopamine transporter radiotracer, [18F]GBR13119 ([18F]1-(2-((4-fluorophenyl)(phenyl)methoxy)-ethyl)-4-(3-phenylpropyl)piperazine).31 A very recent method, based on oxidative radiofluorination with a transition-metal catalyst, gave [18F]29 in low yield (~17%).32 Here we were able to produce [18F]29 from the diaryliodonium tosylate 14 in very high yield (81%) (Table 3, entry 5).
[18F]4-Fluoroiodobenzene33 and [18F]fluorobromobenzene34 have been prepared from the radiofluorination of diaryliodonium salts without noticeable concomitant halogen displacement. We have also previously observed resistance to aryl halogen displacement by SNAr when in well-activated positions in (halopyridinyl)aryliodonium salts during their radio-fluorinations to produce [18F]fluorohalopyridines.15 We tested if this resistance might also occur in the homoaryl diaryliodonium salt (15), which bears a bromo substituent in a position that is well-activated for SNAr by a p-keto group. Remarkably, [18F]4-bromo-4′-fluorobenzophenone ([18F]30) was obtained in excellent yield (98%) from 15 (Table 3, entry 6; see ESI† for radio-HPLC chromatogram). Such efficient access to bi-functionalized [18F]fluoroarenes expands potential for preparing complex PET radiotracers.
General observations
Electron-rich aryl ring partner and anion were varied in the diaryliodonium salts used in this study. Whereas a 4-methoxy-phenyl ring partner gave [18F]fluoroanisole by-product in yields between zero and 4%, negligible radiofluorination occurred at 2-thienyl, 5-methyl-2-thienyl or (2,4,6-trimethoxy)phenyl rings in any of the studied precursors. These rings are therefore confirmed to be excellent aryl ring partners for conferring ring-selectivity in the radiofluorination of diaryliodonium salts. Salts with these ring partners are readily prepared, and hence the use of even more complex ring partners, such as cyclo-phane,35 to confer selectivity can generally be avoided. Diary-liodonium salts having (2,4,6-trimethoxy)phenyl ring partners gave higher yields of the desired [18F]fluoroarene than their 4-methoxyphenyl analogs. This was not generally true for salts having 2-thienyl or 5-methyl-2-thienyl rings.
Tosylate or chloride ion was the anion for the salts used in this study. Our results showed that there was no clear preference for either of these anions with regard to attaining high radiofluorination yield; in some cases tosylate ion seemed preferred and in others chloride ion. The manner in which the anion influences radiochemical yield remains unclear.
In the two examples in which DMF and acetonitrile were directly compared as reaction solvents, DMF gave considerably higher yield of desired [18F]fluoroarene, but at a higher temperature than acetonitrile. In practice, the ability to produce these labeling synthons in either DMF or MeCN as reaction solvents should facilitate their subsequent use, either directly without purification or after relatively simple purification by evaporation of solvent or by solid phase adsorption techniques.
Striking features of the described radiofluorination reactions are that potentially oxidizable groups (i.e., formyl or ketone) survive from precursor to labeled product, as do groups that might be considered vulnerable to nucleophilic substitution (i.e., aliphatic or aromatic-halogen). A further favorable characteristic of these reactions is their high selectivity or specificity for the desired product as almost no appreciable yields of unknown radiofluorinated by-products were observed in the analyses of reaction mixtures.
Conclusion
The radiofluorination of appropriately functionalized diarylio-donium salts with cyclotron-produced NCA [18F]fluoride ion provides versatile rapid single-step access to various functionalized [18F]fluoroarenes as useful labeling synthons for the preparation of complex PET radiotracers.
Supplementary Material
Acknowledgments
This research was funded by the Intramural Research Program of the National Institutes of Health (NIMH). Authors are grateful to the NIH Clinical PET Department (Chief, Dr Peter Herscovitch) for the cyclotron production of fluorine-18.
Footnotes
Electronic supplementary information (ESI) available: Experimental details for synthesis and characterization of diaryliodonium salts. 1H and 13C NMR spectra of functionalized diaryliodonium salts and selected radio-HPLC chromatograms for radiofluorination of diaryliodonium salts. See DOI: 10.1039/c3ob41353e
Notes and references
- 1.Cai L, Lu S, Pike VW. Eur J Org Chem. 2008:2853–2873. [Google Scholar]
- 2.Tredwell M, Gouverneur V. Angew Chem, Int Ed. 2012;51:11426–11437. doi: 10.1002/anie.201204687. [DOI] [PubMed] [Google Scholar]
- 3.Pike VW, Aigbirhio FI. J Chem Soc, Chem Commun. 1995:2215–2216. [Google Scholar]
- 4.Shah A, Pike VW, Widdowson DA. J Chem Soc, Perkin Trans 1. 1998:2043–2046. [Google Scholar]
- 5.Ross TL, Ermert J, Hocke C, Coenen HH. J Am Chem Soc. 2007;129:8018–8025. doi: 10.1021/ja066850h. [DOI] [PubMed] [Google Scholar]
- 6.Carroll MA, Nairne J, Smith G, Widdowson DA. J Fluorine Chem. 2007;128:127–132. [Google Scholar]
- 7.Chun JH, Lu S, Lee YS, Pike VW. J Org Chem. 2010;75:3332–3338. doi: 10.1021/jo100361d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chun J-H, Lu S, Pike VW. Eur J Org Chem. 2011:4439–4447. doi: 10.1002/ejoc.201100382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mu L, Fischer CR, Holland JP, Becaud J, Schubiger PA, Schibli R, Ametamey SM, Graham K, Stellfeld T, Dinkelborg LM, Lehmann L. Eur J Org Chem. 2011:889–892. [Google Scholar]
- 10.Chun JH, Morse CL, Chin FT, Pike VW. Chem Commun. 2013;49:2151–2153. doi: 10.1039/c3cc37795d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang MR, Kumata K, Suzuki K. Tetrahedron Lett. 2007;48:8632–8635. [Google Scholar]
- 12.Telu S, Chun J-H, Siméon FG, Lu S, Pike VW. Org Biomol Chem. 2011;9:6629–6638. doi: 10.1039/c1ob05555k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moon BC, Kil HS, Park JH, Kim JS, Park J, Chi DY, Lee BC, Kim SE. Org Biomol Chem. 2011;9:8346–8355. doi: 10.1039/c1ob06277h. [DOI] [PubMed] [Google Scholar]
- 14.(a) Coenen HH. In: PET Chemistry – The Driving Force in Molecular Imaging. Schubiger PA, Lehman L, Friebe M, editors. Vol. 2. Springer; Berlin: 2007. pp. 15–50. [Google Scholar]; (b) Wuest F. In: PET Chemistry – The Driving Force in Molecular Imaging. Schubiger PA, Lehman L, Friebe M, editors. Vol. 3. Springer; Berlin: 2007. pp. 51–78. [Google Scholar]; (c) Wester HJ, Schottelius M. In: PET Chemistry – The Driving Force in Molecular Imaging. Schubiger PA, Lehman L, Friebe M, editors. Vol. 4. Springer; Berlin: 2007. pp. 79–111. [Google Scholar]
- 15.Chun J-H, Pike VW. Chem Commun. 2012;48:9921–9923. doi: 10.1039/c2cc35005j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chun J-H, Pike VW. Eur J Org Chem. 2012:4541–4547. doi: 10.1002/ejoc.201200695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chun J-H, Pike VW. J Org Chem. 2012;77:1931–1938. doi: 10.1021/jo202517v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martín-Santamaría S, Carroll MA, Carroll CM, Carter CD, Pike VW, Rzepa HS, Widdowson DA. Chem Commun. 2000:649–650. [Google Scholar]
- 19.Brodack JW, Kilbourn MR, Welch MJ, Katzenellenbogen JA. Appl Radiat Isot. 1986;37:217–221. doi: 10.1016/0883-2889(86)90174-7. [DOI] [PubMed] [Google Scholar]
- 20.(a) Haka SM, Kilbourn MR, Watkins GL, Toorongian SA. J Labelled Compd Radiopharm. 1989;27:823–833. [Google Scholar]; (b) Ding YS, Shiue CY, Fowler JS, Wolf AP, Plenevaux A. J Fluorine Chem. 1990;48:189–205. [Google Scholar]; (c) Ekaeva I, Barré L, Lasne MC, Gourand F. Appl Radiat Isot. 1995;46:777–782. [Google Scholar]; (d) Khan NUH, Lee BC, Lee SY, Choe YS, Jun CH, Chi DY. J Labelled Compd Radiopharm. 2002;45:1045–1053. [Google Scholar]; (e) Shen B, Löffler D, Zeller KP, Übele M, Reischl G, Machulla HJ. J Fluorine Chem. 2007;128:1461–1468. [Google Scholar]; (f) Lagisetty P, Vilekar P, Awasthi V. Bioorg Med Chem Lett. 2009;19:4764–4767. doi: 10.1016/j.bmcl.2009.06.056. [DOI] [PubMed] [Google Scholar]; (g) Li L, Hopkinson MN, Yona RL, Bejot R, Gee AD, Gouverneur V. Chem Sci. 2011;2:123–131. [Google Scholar]; (h) Aberg O, Pisaneschi F, Smith G, Nguyen QD, Stevens E, Aboagye EO. J Fluorine Chem. 2012;135:200–206. [Google Scholar]
- 21.Glaser M, Arstad E, Luthra SK, Robbins EG. J Labelled Compd Radiopharm. 2009;52:327–330. [Google Scholar]
- 22.Charlton M, Carroll MA. J Labelled Compd Radiopharm. 2013;56:S152. (Abstract) [Google Scholar]
- 23.(a) Suzuki H, Yazawa N, Yoshida Y, Furusawa O, Kimura Y. Bull Chem Soc Jpn. 1990;63:2010–2017. [Google Scholar]; (b) Hwang DR, Dence CS, McKinnon ZA, Mathias CJ, Welch MJ. Nucl Med Biol. 1991;18:247–252. doi: 10.1016/0883-2897(91)90086-z. [DOI] [PubMed] [Google Scholar]; (c) Lemaire C, Damhaut P, Plenevaux A, Cantineau R, Christiaens L, Guillaume M. Appl Radiat Isot. 1992;43:485–494. doi: 10.1016/0883-2889(92)90205-s. [DOI] [PubMed] [Google Scholar]
- 24.Basuli F, Wu H, Griffiths GL. J Labelled Compd Radiopharm. 2011;54:224–228. doi: 10.1002/jlcr.1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.(a) Iwata R, Pascali C, Bogni A, Horvath G, Kovács Z, Yanai K, Ido T. Appl Radiat Isot. 2000;52:87–92. doi: 10.1016/s0969-8043(99)00117-7. [DOI] [PubMed] [Google Scholar]; (b) Donohue SR, Halldin C, Schou M, Hong J, Phebus L, Chernet E, Hitchcock SA, Gardinier KM, Ruley KM, Krushinski JH, Schaus J, Pike VW. J Labelled Compd Radiopharm. 2008;51:146–152. [Google Scholar]; (c) Thonon D, Kech C, Paris J, Lemaire C, Luxen A. Bioconjugate Chem. 2009;20:817–823. doi: 10.1021/bc800544p. [DOI] [PubMed] [Google Scholar]; (d) Lemaire C, Libert L, Plenevaux A, Aerts J, Franci X, Luxen A. J Fluorine Chem. 2012;138:48–55. [Google Scholar]
- 26.(a) Vaidyanathan G, Zalutsky MR. Nucl Med Biol. 1992;19:275–281. [Google Scholar]; (b) Scott PJH, Shao X. J Labelled Compd Radiopharm. 2010;53:586–591. [Google Scholar]
- 27.Reed CD, Guillaume GL, Carroll MA. J Fluorine Chem. 2012;143:231–237. [Google Scholar]
- 28.(a) Wester HJ, Hamacher K, Stöcklin G. Nucl Med Biol. 1996;23:365–372. doi: 10.1016/0969-8051(96)00017-0. [DOI] [PubMed] [Google Scholar]; (b) Tang G, Zeng WB, Yu MX, Kabalka G. J Labelled Compd Radiopharm. 2008;51:68–71. [Google Scholar]; (c) Wüst F, Hulltsch C, Bergmann R, Johannsen B, Henle T. Appl Radiat Isot. 2003;59:43–48. doi: 10.1016/s0969-8043(03)00161-1. [DOI] [PubMed] [Google Scholar]
- 29.Yan R, Brichard L, Soloviev D, Aigbirhio FI, Carroll MA. J Labelled Compd Radiopharm. 2009;52:208–222. (Abstract) [Google Scholar]
- 30.Carroll M, Yan R, Aigbirhio F, Solviev D, Brichard L. J Nucl Med. 2008;49(Suppl 1):303P. (Abstract) [Google Scholar]
- 31.Kilbourn MR, Haka MS. Appl Radiat Isot. 1988;39:279–282. doi: 10.1016/0883-2889(88)90015-9. [DOI] [PubMed] [Google Scholar]
- 32.Lee E, Hooker JM, Ritter T. J Am Chem Soc. 2012;134:17456–17458. doi: 10.1021/ja3084797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wüst FR, Kniess T. J Labelled Compd Radiopharm. 2003;46:699–713. [Google Scholar]
- 34.Emert J, Hocke C, Ludwig T, Gail R, Coenen HH. J Labelled Compd Radiopharm. 2004;47:429–441. [Google Scholar]
- 35.Wang B, Graskemper JW, Qin L, DiMagno SG. Angew Chem, Int Ed. 2010;49:4079–4083. doi: 10.1002/anie.201000695. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.