

Abstract
An O-aminated naphthol AS-E was designed as a prodrug to achieve reductive activation and improved aqueous solubility. During the synthesis of this designed compound, a novel transformation from aryl triflates and ethyl acetohydroximate to oxazoles was discovered. Although the initially designed O-amino naphthol AS-E was not made successfully, the eventually synthesized O-tert-butylamino derivative was found to be biologically inactive, suggesting that reductive N-O cleavage in this compound was not facile due to unfavorable steric and electronic effects.
Keywords: CREB, ethyl acetohydroximate, naphthol AS-E, oxazole, O-amination, prodrug, reductive activation
INTRODUCTION
Naphthol AS-E (1, Chart 1) was recently discovered to be able to inhibit gene transcription activated by cyclic-AMP response element binding protein (CREB) in living cells [1]. Chemical inhibitors of CREB-mediated gene transcription can be potentially used as novel anticancer agents because CREB is often overactivated in various cancer tissues [2]. Indeed, compound 1 has been shown to present selective cytotoxicity against cancer cells while sparing normal cells [3]. Due to the formation of an intramolecular hydrogen bond between the −OH group and the carbonyl oxygen [4] as shown in Chart 1, compound 1 has poor aqueous solubility, which may limit its eventual in vivo application where high concentrations of the compound in aqueous environment is needed. In order to improve the aqueous solubility, an O-amino compound 2 (Chart 1) was designed. It was hypothesized that the free amino group present in 2 would significantly boost its aqueous solubility. In addition, compound 2 can be considered as a prodrug of 1 because the N-O bond can be selectively cleaved in hypoxic tumor tissues or by intracellular glutathione through reductive activation [5–7]. Herein, we report our studies on the synthesis toward 2.
Chart 1.
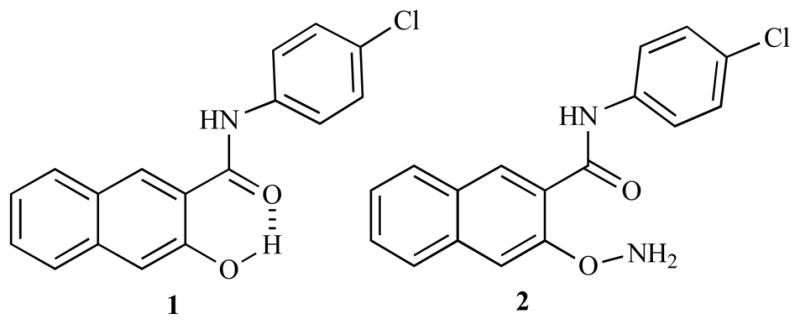
Chemical structures of 1 and 2.
RESULTS AND DISCUSSION
Recently, Buchwald’s group reported that ethyl acetohydroximate could undergo cross-coupling reaction with aryl halides in the presence of Pd(OAc)2/t-BuBrettPhos/Cs2CO3 to give aromatic hydroxylamine after acidic hydrolysis [8]. Therefore, we designed our initial synthesis to 2 based on this novel transformation (Scheme 1). To this end, naphthol 3 [4] was converted into triflate 4, which was then subjected to Pd(0)-catalyzed hydroxylamination [9]. However, no desired product 5 was observed from this reaction. Instead, two triflate hydrolysis products 3 and 6 (6%), two oxazoles 7a and 7b (10%), along with an unknown compound (23% [10]) were obtained after careful silica gel chromatographic separations. The structure of 7 was initially deduced from its NMR and mass spectra. To further confirm the structure elucidation, a sample of 7a was prepared from an independent synthesis by condensing ortho-aminonaphthol 9 and trimethyl orthoacetate [11, 12]. Intermediate 9 was in turn synthesized from reduction of 8 derived from nitration of 3 [13].
Scheme 1.

Attempted synthesis of 5.
The plausible mechanisms for the novel transformation from 4 to 7 are presented in Scheme 2. Under basic condition, the trifluoromethanesulfonyl group can be presumably transferred from 4 to ethyl acetohydroximate to give 10 and 11. A Beckmann-type rearrangement of 10 involving EtO− migration would give cation 12, trapping of which by naphthoxide 11 would yield 13 (path A). A [2,3]-sigmatropic rearrangement of 13 followed by elimination of EtO− provided 7a. The thus-generated EtO− could attack methyl esters 3 and 7a to produce ethyl esters 6 and 7b. Alternatively, naphthoxide 11 could react with 10 directly to generate 15 (path B). Elimination of EtO− from 15 gave 16, which then underwent similar sequence of reactions to that of 13 eventually yielding 7a. The mechanisms depicted in Scheme 2 suggested that the palladium catalyst played no role in forming 3, 6 and 7. Indeed, when Pd(OAc)2 and the phosphine ligand were omitted from the reaction, we observed exactly the same reaction outcome as shown in Scheme 1.
Scheme 2.

Plausible mechanisms for the formation of 7 from 4 and ethyl acetohydroximate.
With the difficulties in obtaining 5 by Pd-catalyzed cross-coupling reactions, our attention was focused on nucleophilic amination of naphthol 1 with N-Boc-O-tosyl hydroxylamine, which has been shown to be an excellent nitrogen source for carbanions [14]. Therefore, compound 1 was deprotonated with KHMDS followed by addition of lithium t-butyl N-tosyloxycarbamate to give surprisingly acid 18. It is worthy of note that the use of lithiated derivative of N-Boc-O-tosyl hydroxylamine was essential for this O-amination reaction because no amination product was observed if TosONHBoc was not pretreated with n-BuLi. Even though acid 18 was obtained from amide 1, no amination product was observed if ester 3 or its corresponding acid was employed as the starting material. With acid 18 in hand, it was then subjected to amide formation with 4-chloroaniline. However, this seemingly trivial amide formation reaction turned out to be notoriously challenging. No amide product was generated under standard amide coupling conditions (BOP, HOBT/EDCI, HBTU/HOBt, oxalyl chloride/DMF). We hypothesized the sterically demanding environment in 18 might prevent nucleophilic attack from 4-chloroaniline at the activated carboxylic acid. It was recently reported that cyanuric fluoride was able to facilitate coupling between sterically hindered alcohol and carboxylic acid to form esters [15]. However, this reagent also failed to promote the amide coupling between 18 and 4-chloroaniline. Finally, we found that mixed anhydride 19 was obtained by treating 18 with MsCl in the presence of Et3N in 84% yield. Although it did not react with 4-chloroaniline at ambient temperature, small amount (2–3%) of desired product 20 did form in the presence of DMAP at elevated temperature (140 °C) in a sealed tube. Formation of significant amount of 21 (31%) from the reaction supported the hypothesis that nucleophilic attack at the sterically hindered carboxylic acid carbon was significantly inhibited. It was then envisioned that final acidic de-protection of Boc would deliver compound 2. Unfortunately, all attempts to remove Boc only yielded compound 22 in high yield (71%). The use of a carbocation scavenger such as triiospropylsilane (TIPS), which is commonly used in the deprotection cocktail during peptide synthesis [16], did not change the reaction outcome.
Compound 22 was evaluated for its activity in inhibiting CREB-mediated gene transcription in HEK 293T cells using a CREB reporter assay as described before [1]. In this assay, compound 1 inhibited CREB’s activity with IC50 = 2.29 μM [1]. Under the same assay conditions, no inhibitory activity was observed with compound 22 up to 50 μM, suggesting that reductive activation of 22 to release 1 did not occur under the assay conditions. These results are consistent with those obtained from Boger’s group studying duocarmycin O-amino phenol derivatives where both steric and electronic effects were contributing to the reductive activation of O-amino prodrugs [6]. In the case of 22, the t-butyl group is both electron-donating and sterically bulky, synergistically resulting in inefficient cleavage of the N-O bond.
CONCLUSION
In conclusion, an O-aminated naphthol AS-E, compound 2 was designed as a prodrug of compound 1 through reductive activation by improving its aqueous solubility. During the synthesis of 2, a novel reaction to form oxazoles from aryl triflates and ethyl acetohydroximate was discovered. The yield for this reaction remains to be optimized. The loss of CREB inhibitory activity seen with the eventually synthesized t-butylated 2 or compound 22 suggested that this N-O bond is not facile to cleavage due to unfavorable steric and electronic effects.
EXPERIMENTAL SECTION
General
Glass Contout solvent purification system was used to purify all the solvents used for reactions. Melting points were determined in capillary tubes using Mel-Temp and are uncorrected. All 1H and 13C NMR spectra were obtained in a Bruker Avance 400 MHz spectrometer using CDCl3, CD3OD or DMSO-d6 as solvent and the chemical shifts of the residual CHCl3 (δ 7.24), CH3OH (δ 3.31, 4.87) or DMSO (δ 2.50) were taken as reference. The splitting pattern of a 1H NMR peak is described as follows: brs = broad singlet, s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, td = triplet of doublet. The coupling constants (J) were reported in Hertz (Hz). All the NMR peak assignments were tentative, however, the 13C signals for C and CH2 were differentiated from CH and CH3 by APT (Attached Proton Test) experiments. Silica gel flash chromatography was performed using 230–400 mesh silica gel (EMD). The mass spectra were obtained from a TSQ LC/MS system (Thermo Scientific) with electrospray operated either in positive or negative mode.
Methyl 2-Methylnaphtho[1,2-d]oxazole-4-carboxylate (7a)
To a stirred solution of 9 (32 mg, 0.14 mmol) in trimethyl orthoacetate (2.5 mL) was added TsOH (2.5 mg, 0.014 mmol). The reaction mixture was heated under reflux for 2 h under argon. The reaction mixture was cooled to room temperature and the solvent was removed to give a residue that was purified by a silica gel column, eluting with dichloromethane:ethyl acetate (20 : 1) to give product 7a (30 mg, 89%) as a white solid: m.p. 128–129 °C. 1H NMR (400 MHz, CDCl3) δ 8.46 (s, 1 H, Ar-H1), 8.44 (d, J = 8.3 Hz, 1 H, Ar-H8), 8.02 (d, J = 8.4 Hz, 1 H, Ar-H5), 7.73 (td, J = 7.6, 1.2 Hz, 1 H, Ar-H6), 7.56 (td, J = 7.6, 1.2 Hz, 1 H, Ar-H7), 4.06 (s, 3 H, OCH3), 2.81 (s, 3 H, CH3); 13C NMR (100 MHz, CDCl3) δ 164.79 (CO), 163.96 (oxazole-C), 145.74 (Ar-C3), 137.99 (Ar-C4), 129.97 (Ar-C9), 129.78 (Ar-CH8), 129.24 (Ar-CH5), 128.94 (Ar-CH1), 128.29 (Ar-C10), 125.97 (Ar-CH6), 122.04 (Ar-CH7), 114.60 (Ar-C2), 52.50 (OCH3), 14.79 (CH3).
Methyl 3-Hydroxy-4-nitro-2-naphthoate (8) [13]
To a solution of 3 [3] (202 mg, 1 mmol) in dichloromethane (5 mL) was added fuming HNO3 (55 μL, 1.1 mmol) at 0 °C. The reaction mixture was stirred at 0 °C for 15 min, when the reaction was quenched with water (10 mL). The reaction mixture was extracted with ethyl acetate (50 mL). The organic extract was then washed with brine (20 mL) and dried over Na2SO4. The solvent was removed to give a residue that was purified by a silica gel column, eluting with hexanes:ethyl acetate (3:1) to yield product 8 (134 mg, 54%) as a yellow solid: m.p. 182–183 °C. 1H NMR (400 MHz, CDCl3) δ 11.27 (s, 1 H, OH), 8.62 (s, 1 H, Ar-H1), 7.89 (d, J = 8.3 Hz, 1 H, Ar-H8), 7.75–7.67 (m, 2 H, Ar-H5 and Ar-H6), 7.48 (td, J = 7.3, 1.6 Hz, 1 H, Ar-H7), 4.08 (s, 3 H, OCH3).
Methyl 4-Amino-3-hydroxy-2-naphthoate (9) [17]
To a solution of 8 (99 mg, 0.4 mmol) in methanol-water (4 mL-1 mL) was added iron (112 mg, 2 mmol) and NH4Cl (170 mg, 3.2 mmol). The reaction mixture was heated under reflux for 4 h, then diluted with ethyl acetate (60 mL) and filtered. The filtration was concentrated to give a residue that was purified by a silica gel column, eluting with hexanes:ethyl acetate:dichloromethane (4:1:1) to yield product 9 (32 mg, 37%) as a yellow solid. 1H NMR (400 MHz, CDCl3) δ 10.49 (s, 1 H, OH), 7.99 (s, 1 H, Ar-H1), 7.79 (d, J = 8.3 Hz, 1 H, Ar-H8), 7.74 (d, J = 8.6 Hz, 1 H, Ar-H5), 7.50 (td, J = 7.6, 1.3 Hz, 1 H, Ar-H6), 7.32 (t, J = 7. 6 Hz, 1 H, Ar-H7), 4.27 (brs, 2 H, NH2), 4.02 (s, 3 H, OCH3).
3-{[(tert-Butoxycarbonyl)amino]oxy}-2-naphthoic Acid (18)
To a stirred solution of TosONHBoc (1.148 g, 4 mmol) in THF (20 mL) was added n-BuLi (1.6 mL, 2.5 M, 4 mmol) at −78 °C. The reaction mixture was stirred for 15 min. On a separate flask, KHMDS (8 mL, 0.5 M, 4 mmol) was added to a stirred suspension of 1 (596 mg, 2 mmol) in THF (30 mL) at 0 °C. This reaction mixture was stirred for 15 min to form a clear solution. Then the TosONLiBoc solution was added dropwise to the above reaction mixture containing 1 at −78 °C. The resulting mixture was warmed to room temperature and stirred for overnight. The reaction mixture was then queched with saturated aqueous NH4Cl solution (20 mL) and the organic phase was seperated. The aqueous phase was further extracted with ethyl acetate (30 mL). The organic extracts were combined and dried over Na2SO4. The solvent was removed and the solid was washed with chloroform (40 mL) and the starting material 1 was removed by filtration. The chloroform solution was then concentrated to give a residue that was purified by a silica gel column, eluting with dichloromethane:ethyl acetate (20 : 1) to give acid 18 (190 mg, 31%) as an off-white solid: m.p. 162–163 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.15 (s, 1 H, COOH), 11.08 (s, 1 H, NH), 8.27 (s, 1 H, Ar-H1), 7.87 (d, J = 7.9 Hz, 1 H, Ar-H8), 7.74 (d, J = 8.4 Hz, 1 H, Ar-H5), 7.49 (t, J = 7.5 Hz, 1 H, Ar-H6), 7.34 (t, J = 7.4 Hz, 1 H, Ar-H7), 7.26 (s, 1 H, Ar-H4), 1.28 (s, 9 H, Boc-CH3); ESIMS m/z (rel intensity) 258.03 [100, (M-CO2-H)−].
3-{[(tert-Butoxycarbonyl)amino]oxy}-2-naphthoic Methanesulfonic Anhydride (19)
To a solution of acid 18 (179 mg, 0.59 mmol) in THF (5 mL) was added Et3N (91 μL, 0.65 mmol) and MsCl (51 μL, 0.65 mmol) at 0 °C. The resulting mixture was warmed to room temperature and stirred for 4 h. The reaction mixture was diluted with brine (10 mL) and extracted with ethyl acetate (40 mL). The organic extract was dried over Na2SO4. The solvent was removed to give a residue that was purified by a silica gel column, eluting with dichloromethane:ethyl acetate (10 : 1) to give 19 as a colorless oil (190 mg, 84%): 1H NMR (400 MHz, CDCl3) δ 8.50 (s, 1 H, NH), 8.22 (s, 1 H, Ar-H1), 7.93 (s, 1 H, Ar-H4), 7.92 (d, J = 8.5 Hz, 1 H, Ar-H8), 7.88 (d, J = 8.2 Hz, 1 H, Ar-H5), 7.63 (t, J = 7.0 Hz, 1 H, Ar-H6), 7.59 (t, J = 6.9 Hz, 1 H, Ar-H7), 3.29 (s, 3 H, Ms-CH3), 1.40 (s, 9 H, Boc-CH3).
tert-Butyl {3-[(4-Chlorophenyl)carbamoyl]naphthalen-2-yl}oxycarbamate (20)
A solution of 19 (190 mg, 0.5 mol), 4-chloroaniline (64 mg, 0.5 mmol) and DMAP (61 mg, 0.5 mmol) in toluene (5 mL) was heated in a sealed tube at 140°C for overnight. The reaction mixture was dilluted with ethyl acetate (30 mL) and washed with 2 N HCl (15 mL). The organic phase was dried over Na2SO4. The solvent was removed to give a residue that was purified by a silica gel column, eluting with hexanes:ethyl acetate (8 : 1) to give compound 20 (5 mg, 2.4%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 10.25 (s, 1 H, CONH), 7.66 (d, J = 8.4 Hz, 1 H, Ar-H8), 7.57 (s, 1 H, Ar-H1), 7.46 (d, J = 8.4 Hz, 1 H, Ar-H5), 7.41 (td, J = 7.5, 1.2 Hz, 1 H, Ar-H6), 7.36 (s, 1 H, Ar-H4), 7.21 (td, J = 7.4, 1.2 Hz, 1 H, Ar-H7), 7.13–7.09 (m, 3 H, NHBoc and Ar′-H2′), 6.76 (d, J = 8.7 Hz, 2 H, Ar′-H3′), 1.42 (s, 9 H, Boc-CH3); 13C NMR (100 MHz, CDCl3) δ 153.15 (CONH), 149.09 (Boc-CO), 138.78 (Ar-C3), 134.46 (Ar′-C1′), 129.69 (Ar-CH1), 128.55 (2, Ar′-CH2′), 127.94 (Ar-C10), 127.81 (Ar-CH8), 127.11 (Ar-CH6), 126.68 (2, Ar-C9 and Ar′-C4′), 125.57 (Ar-CH5), 123.04 (Ar-CH7), 122.26 (2, Ar′-CH3′), 115.39 (Ar-C2), 111.14 (Ar-CH4), 79.65 (Boc-C), 27.13 (3, Boc-CH3).
3-[(tert-Butylamino)oxy]-N-(4-chlorophenyl)-2-naphthamide (22)
An HCl solution in Et2O (2 M, 1 mL) was added to a stirred solution of 20 (10 mg, 0.024 mmol) in CHCl3-MeOH (1 mL–0.5 mL). The resulting mixture was stirred at room temperature for overnight. The solvent was removed and the solid was treated with chloroform, filtered and washed with ethyl ether to give product 22 (7 mg, 71%) as a white solid: m.p. 170–171 °C. 1H NMR (400 MHz, CD3OD) δ 8.05 (s, 1 H, Ar-H1), 7.83 (d, J = 7.7 Hz, 1 H, Ar-H8), 7.67 (d, J = 7.7 Hz, 1 H, Ar-H5), 7.51 (td, J = 7.6, 1.1 Hz, 1 H, Ar-H6), 7.36 (td, J = 7.5, 1.2 Hz, 1 H, Ar-H7), 7.23 (d, J = 8.8 Hz, 2 H, Ar′-H2′), 7.16 (s, 1 H, Ar-H4), 7.14 (d, J = 8.8 Hz, 2 H, Ar′-H3′), 1.55 (s, 9 H, But-CH3); 13C NMR (100 MHz, CD3OD) δ 151.31 (CONH), 136.28 (2, Ar-C3 and Ar′-C1′), 134.04 (Ar-C10), 132.58 (Ar-C9), 131.18 (Ar-CH1), 128.23 (2, Ar′-CH2′), 128.13 (Ar-CH8), 127.66 (Ar-CH6), 126.66 (Ar′-C4′), 126.32 (2, Ar′-CH3′), 125.40 (Ar-CH5), 123.73 (Ar-CH7), 113.76 (Ar-C2), 109.47 (Ar-CH4), 86.01 (But-C), 24.64 (3, But-CH3); ESIMS m/z (rel intensity) 369.13 (100, 35 Cl-MH+), 371.09 (33, 37Cl-MH+).
Supplementary Material
Scheme 3.
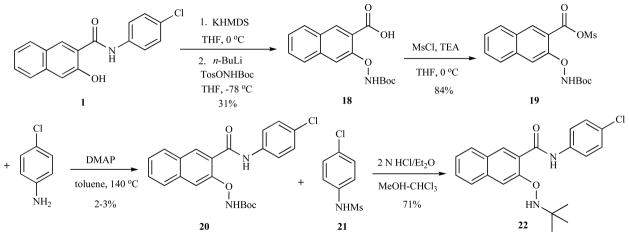
Synthesis of 22.
Acknowledgments
This work was made possible by financial support from NIH (R01GM087305). We thank Ms. Jenny Luo at Oregon Health & Science University for mass spectroscopic analyses.
Footnotes
CONFLICT OF INTEREST
The author(s) confirm that this article content has no conflict of interest.
References
- 1.Li BX, Xiao X. Discovery of a Small-Molecule Inhibitor of the KIX-KID Interaction. ChemBioChem. 2009;10:2721–2724. doi: 10.1002/cbic.200900552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xiao X, Li BX, Mitton B, Ikeda A, Sakamoto KM. Targeting CREB for Cancer Therapy: Friend or Foe. Curr Cancer Drug Targets. 2010;10:384–391. doi: 10.2174/156800910791208535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li BX, Yamanaka K, Xiao X. Structure-activity relationship studies of naphthol AS-E and its derivatives as anticancer agents by inhibiting CREB-mediated gene transcription. Bioorg Med Chem. 2012;20:6811–6820. doi: 10.1016/j.bmc.2012.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jiang M, Li BX, Xie F, Delaney F, Xiao X. Design, synthesis, and biological evaluation of conformationally constrained analogues of naphthol AS-E as inhibitors of CREB-mediated gene transcription. J Med Chem. 2012;55:4020–4024. doi: 10.1021/jm300043c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jin W, Trzupek JD, Rayl TJ, Broward MA, Vielhauer GA, Weir SJ, Hwang I, Boger DL. A unique class of duocarmycin and CC-1065 analogues subject to reductive activation. J Am Chem Soc. 2007;129:15391–15397. doi: 10.1021/ja075398e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lajiness JP, Robertson WM, Dunwiddie I, Broward MA, Vielhauer GA, Weir SJ, Boger DL. Design, synthesis, and evaluation of duocarmycin O-amino phenol prodrugs subject to tunable reductive activation. J Med Chem. 2010;53:7731–7738. doi: 10.1021/jm1010397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Perry RR, Mazetta JA, Levin M, Barranco SC. Glutathione levels and variability in breast tumors and normal tissue. Cancer. 1993;72:783–787. doi: 10.1002/1097-0142(19930801)72:3<783::aid-cncr2820720325>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 8.Maimone TJ, Buchwald SL. Pd-catalyzed O-arylation of ethyl acetohydroximate: synthesis of O-arylhydroxylamines and substituted benzofurans. J Am Chem Soc. 2010;132:9990–9991. doi: 10.1021/ja1044874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.A different ligand from reference 8 was used in our initial synthesis. Since the reaction outcome was the same even in the absence of Pd(OAc)2 and a phosphine ligand (see below), no attempts were made to use t-BuBrettPhos.
- 10.This yield calculation was based on its molecular weight of 358 and it contained one unit of naphthyl ring. See Figure S1 in supplementary information for its 1H NMR.
- 11.Katritzky AR, Musgrave RP, Rachwal B, Zaklika C. Synthesis of 4-Hydroxy-1-methylbenzimidazole and 7-Hydroxy-1-methylbenzimidazole. Heterocycles. 1995;41:345–352. [Google Scholar]
- 12.Khajavi MS, Mohammadi AA. Reaction of 4-Amino-3-hydroxy-1-naphthalenesulfonic Acid with Orthoesters: A New Facile One-Pot Synthesis of 2-Substituted Naphth[1,2-d]oxazole-5-sulfonates. J Chem Res. 2002:136–138. [Google Scholar]
- 13.Ramesh VV, Roy A, Vijayadas KN, Kendhale AM, Prabhakaran P, Gonnade R, Puranik VG, Sanjayan GJ. Conformationally rigid aromatic amino acids as potential building blocks for abiotic foldamers. Org Biomol Chem. 2011;9:367–369. doi: 10.1039/c0ob00593b. [DOI] [PubMed] [Google Scholar]
- 14.Greck CBL, Girard A, Hajicek J, Genet JP. Electrophilic Amination of Carbanions with Metalated t-Butyl-N-Tosyloxycarbamate. Bull Soc Chim Fr. 1994;131:429–433. [Google Scholar]
- 15.Pospisil J, Muller C, Furstner A. Total synthesis of the aspercyclides. Chem Eur J. 2009;15:5956–5968. doi: 10.1002/chem.200802681. [DOI] [PubMed] [Google Scholar]
- 16.Xiao X, Yu P, Lim HS, Sikder D, Kodadek T. Design and synthesis of a cell-permeable synthetic transcription factor mimic. J Comb Chem. 2007;9:592–600. doi: 10.1021/cc070023a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bhumgara SK, Seshadri S. Studies in the Vilsmeier-Haack reaction. Part XXI. A novel synthesis of fluorescent 2-hetarylindene derivatives. Indian J Chem Sect B. 1985;24B:703–706. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.