

Abstract
A comprehensive study of the cellular components of the immune system requires both deep and broad immunophenotyping of numerous cell populations in an efficient and practical manner. In this chapter, we describe the technical aspects of studying the human immunome using high-dimensional (15 color) fluorescence-based immunophenotyping. We focus on the technical aspects of polychromatic flow cytometry and the initial stages in developing a panel for comprehensive leukocyte immunophenotyping (CLIP). We also briefly discuss how this panel is being used and the challenges of encyclopedic analysis of these rich data sets.
1 Introduction
All the genes and proteins that constitute the immune system are collectively known as the immunome; the immunome is a vastly complex and highly regulated structure that protects against infection and preserves health. An ever-increasing number of cell types comprise the immunome, and these are being defined through increasingly complex patterns of antigen expression. Technological advances over the last decade now permit high-dimensional examination of the cellular components of the immunome, making possible in-depth and broad analysis of the immune system at the same time. Such integrative, comprehensive studies will permit a much better understanding of the dynamic relationships among the myriad of cell types within the immune system than is currently possible through limited studies of only specific cell lineages or more cursory assessments of the broader system. A meaningful understanding of the immunome must begin by a study of this system in healthy individuals and analyses of the variations that exist among individuals and within each individual over time. This can then be extended to studies involving defined perturbations, such as vaccination or administration of a commonly used therapeutic such as an antibiotic, and to studies involving patients with a well-defined disease. Together, these points of information can be used to construct a meaningful database of the immunome in health, which in turn can be used to develop a deep understanding of the networks involved in responses to stimuli or in disease. It is increasingly appreciated that these studies must be conducted in humans because mouse models fail to replicate all nuances of the human immune system (Davis 2008, 2012).
Given the hundreds of cell types that have been identified in the immune system, detecting and characterizing these in an encyclopedic manner in a given individual would require collection of a large amount of blood (or other specimen) if comprehensive analyses were to be performed using low-dimensional immunophenotyping technologies. It is readily apparent that as one moves toward higher multiplicity in the number of parameters examined, the volume of specimen required for an encyclopedic study decreases proportionally. Furthermore, examination of multitudes of markers simultaneously will facilitate discovery of new cellular populations not identified by lower dimensional immunophenotyping.
The technological advances that now permit high-dimensional immunophenotyping studies can be broadly categorized into two areas: those using isotopes of lanthanide and those using fluorophores (Bendall et al. 2012). The former approach is based on mass spectroscopy, thus avoiding the use of fluorochromes and the limitations occurring due to overlap of their emission spectra in polychromatic studies. This opens the door for very high-dimensional studies—arguably as many as 100 parameters simultaneously. The trade-offs for this ability are decreased throughput compared to conventional cytometry and inability to recover the cells identified. Slow throughput can be a crucial issue, as millions or tens of millions of cells need to be analyzed when immunophenotyping specific, and likely rare, cell subpopulations. Furthermore, to have a meaningful database, immunophenotyping need to be performed on scores, if not hundreds, of samples from different subjects, thus making throughput of the assay a serious concern.
New approaches involving measurements of fluorescence take advantage of improvements in both the fluorochromes and instrument hardware. Novel fluorescent molecules ranging from quantum dot and brilliant violet stains to new viability probes have been introduced covering a wide range of excitation and emission wavelengths. Hardware improvements include low-cost, high-performance lasers in a number of wavelengths, fiber optic transmission of emitted light, and novel detectors and configurations. Such improvements have made 15–18 color immunophenotyping practical in some laboratories (Perfetto et al. 2004). Recently, a spectral analyzing cytometer has been developed that deconvolutes the spectral overlap among 32 detectors, bringing fluorescence-based cytometry to higher dimensions (Sony 2013). Although more limited in the number of potential parameters capable of examination than mass cytometry, fluorescence-based methods offer higher throughput and the ability to recover cells of interest.
Here we describe the technical aspects of high-dimensional fluorescence-based immunophenotyping as well as the early stages of our approach for comprehensive leukocyte immunophenotyping (CLIP). Our goal in using CLIP is to provide both a broad and in-depth assessment of the peripheral immune system. We opted for the fluorescence-based approach because of the higher throughput and the potential to recover populations of cells defined by complex phenotyping for functional studies. Even though the dimensionality of fluorescence-based immunophenotyping is lower than that of time-of-flight cytometry, this approach permits the identification of tens of thousands of leukocyte populations.
2 Technical Issues in Polychromatic Immunophenotyping
2.1 Instrument Specifications
In the past few years, tremendous advancements have been made in flow cytometers. There are commercially available flow cytometers that are capable of measuring up to 20 parameters simultaneously, including 18 distinct fluorochromes as well as forward and side scatter parameters. Factors in selecting the configuration of the instrument for high polychromatic immunophenotyping include the number and types of lasers, the number of photomultiplier tubes (PMT) per laser, and the arrangement of filters and dichroic mirrors. In general, high polychromatic flow cytometry is greatly facilitated by using numerous lasers with their beams spatially separated with detectors somewhat evenly disbursed among the lasers to permit multiple fluorochromes to be excited from each laser. We utilize an instrument equipped with five lasers (355 nm, 406 nm, 488 nm, 532 nm, and 639 nm wavelengths) with 22 PMT detectors. Filters were selected to minimize fluorescence spillover from one detector to another. A full list of PMTs along with the appropriate dichroic and bandpass filters that were chosen for our CLIP panel can be found in Table 1.
Table 1.
Instrument configuration
| Laser wavelength (nm) | Laser color | Laser power (mW) | Laser type | Spectral range for detection (nm) | Dichroic #1 (nm) | Dichroic #2 (nm) | Band Pass (nm) | Fluorochrome |
|---|---|---|---|---|---|---|---|---|
| 639 | red | 40 | cube | 750–810 | 740LP | 780/60 | APC Cy7/APC H7 | |
| 685–735 | 740LP | 685LP | 710/50 | AF700 | ||||
| 630–670 | 685LP | 660/20 | APC/AF647 | |||||
| 532 | green | 150 | compass | 760–800 | 740LP | 780/40 | PE-Cy7 | |
| 685–735 | 740LP | 690LP | 710/50 | PE-Cy5.5 | ||||
| 640–680 | 690LP | 640LP | 660/40 | PE-Cy5 | ||||
| 600–620 | 640LP | 600LP | 610/20 | PE-TR | ||||
| 562–588 | 600LP | 575/25 | PE | |||||
| 488 | blue | 50 | saphire LP | 685–735 | 685LP | 685LP | 710/50 | PerCP Cy5.5 |
| 505–525 | 505LP | 505LP | 515/20 | FITC/AF488 | ||||
| 406 | violet | 100 | cube | 750–810 | 740LP | 780/60 | QD800 | |
| 670–740 | 740LP | 670LP | 705/70 | x | ||||
| 640–680 | 670LP | 630LP | 660/40 | QD655 | ||||
| 585–625 | 630LP | 595LP | 605/40 | QD605 | ||||
| 570–595 | 595LP | 570LP | 585/42 | x | ||||
| 540–580 | 570LP | 557LP | 560/40 | x | ||||
| 505–535 | 557LP | 505LP | 515/20 | Aqua blue | ||||
| 425–475 | 450/50 | V450 | ||||||
| 355 | ultraviolet | 60 | genesis | 500–575 | 505LP | 505LP | 525/50 | UV525 |
| 425–475 | 450/50 | UV450 |
DPSS Diode Pumped Solid State
22 PMT are in this instrument, but only 20 are useable thanks to a switch between 2 PMT on violet and ultraviolet laser
Prior to evaluating the reagents and panels to be used, it is important to examined the performance of the cytometer through the use of a three-step protocol that includes optimization, calibration, and standardization of numerous components such as optical filters, dichroic mirrors (reflection and mirror transmission), laser power, laser delays, electronic noise, and window extensions (Perfetto et al. 2006, 2012). It is important to stress that any instrument to be used in such studies must have highly stable fluidics, be capable of high throughput, and be sufficient sensitivity to simultaneously detect numerous dim antigens.
2.2 Evaluation of Marker Expression Intensity
To obtain optimal results using a multicolor panel, numerous antibody clones directed against a specific antigens and multiple antibody-fluorochrome combinations must be screened to select reagents that yield optimal detection and staining intensities (Mahnke and Roederer 2007; Biancotto et al. 2011). Not all clones against a particular antigen recognize the same epitope, and thus may yield different patterns and intensities of staining. Therefore, one should begin by selecting clones that perform in the manner desired. We tested each antibody clone using two fluorochromes with relatively good quantum yields, phycoerythrin (PE) and allophycocyanin (APC) (Fig. 1). The clone yielding the best detection of antigen based on the separation of positive from negative staining was selected for further use. Based on the intensity of marker expressions in these experiments, antigens were categorized as markers of low (dim) expression, intermediate expression, or high expression. Various fluorochromes yield various amounts of emitted light, known as a quantum yield. High quantum yield fluorochromes are considered ‘bright’, whereas low quantum yield ones are considered ‘dim’. A sound approach for designing high polychromatic immunophenotyping panels is to pair dim markers with bright fluorochromes and, conversely, to pair bright markers with dim fluorochromes (Mahnke and Roederer 2007). This helps to keep most markers in mid-scale and to avoid excessively high or low voltage from being applied to photomultiplier tubes (PMTs). This in turn helps to prevent the need for high levels of spectral compensation. We used this approach in designing the CLIP panel whenever possible, although practical necessities, such as the availability of the reagents or the presence of multiple dim markers in a single tube, made some compromise a necessity.
Fig. 1.

Testing of CD8 antibody clone 3B5. During the development of the CLIP panel, different fluorochrome-conjugates of CD8 antibodies were tested. Initially CD8 APC and PE were tested to measure the intensity of antigen expression. Based on the intensity of this staining, we performed secondary testing using red laser excitation (639 nm) or violet laser excitation (406 nm). Staining indices are shown in red and were calculated by dividing the MFI of the positive peak by the MFI of the negative peak
2.3 Evaluation of Performance of Antibody-Fluorochrome Conjugates in Multicolor Combinations
In addition to PE and APC, it is worth testing a number of other fluorochrome conjugates. This testing can reveal how effectively fluorochromes with different quantum yields permit discrimination of cell populations bearing the marker of interest and can give some indication of how much flexibility there is in assigning a specific fluorochrome for use with each antibody clone in a high polychromatic panel. For our CLIP staining (15 color tubes), once the intensity level of expression of our 14 markers was established, they were preliminarily tested in different panel combinations of 10 colors (a staining matrix) before moving to our final 15-color matrix. In these different staining mixtures, certain fluorochrome-antibody combinations were invariant, and others had different fluorochromes. Each staining tube was analyzed, and the final fluorochrome conjugates were selected for the antibodies based on discrimination of positive and negative populations, fluorescence intensity, percentages of positive cells, and ease of compensation. This allowed the identification of fluorochrome-antibody conjugates that yielded either optimal or suboptimal performance in a multicolor panel, and thus provided a strong rationale for the combinations to be included in the final panel (Fig. 1).
One of the goals in the development of the CLIP was to use commercial ‘off the shelf’ reagents whenever possible to both minimize cost and to make this panel reproducible in other laboratories. Only if commercial reagents were not available or reliable were custom-conjugated antibodies used. Before the final panel was assembled, numerous clones of antibodies and fluorochrome combinations of antibodies were tested in order to optimize the identification of cell populations and to prevent unnecessarily high amounts of spectral compensation. The feasibility of developing a panel of multiple 15-color tubes has been demonstrated in our laboratory, and the panel’s utility in describing complex alterations of the immune system in the context of disease and vaccination has been demonstrated (Biancotto et al. 2012a, b).
2.4 Titration of Fluorochrome-Antibody Conjugates on Cells
One of the important steps in selection of optimal fluorochrome-antibody conjugates is the titration of the selected fluorochrome-antibody conjugates with relevant cells. Most antibodies have a broad range of concentrations in which they bind to antigens. If the concentration of antibody is too low, stained cells might not be well separated from the background or negative cells. However, if the concentration of antibody is too high, staining of the background events could increase resulting in poor separation of the positive cells from those not expressing the antigen. Therefore, the optimal concentration of antibody is that which approaches the saturation level and presents the lowest background staining, therefore giving optimal separation between positive and negative events. This will ensure a fluorescent signal that is linearly proportional to the antigen present in the sample. Thus each antibody titer was selected based on low levels of background staining, maximum separation between negative and positive populations (as measured by mean fluorescence intensity, MFI, of negative and positive populations), and for levels of positive staining close to the plateau (Fig. 2). Determining the optimal concentration of antibody to use often will decrease the cost of reagents, as titration often reveals that lower concentrations of the antibodies may be used than those recommended by the supplier. It should be stressed that titration is very much reagent and lot specific—each antibody lot that will be used in the experiment must be titered, and the use of the same clone of antibody but in a different fluorochrome conjugate requires a new titration. Titrations are usually performed as single-color assessments done independently and prior to any attempt to construct any combinations of reagents. Additionally, titration of surface markers should be performed on non-permeabilized cells whenever possible. Since this titration is not performed in combination with other antibodies or fluorochromes, titrations should be performed without compensation.
Fig. 2.

Titration of CD3 APC-Cy7. Peripheral blood mononuclear cells (PBMCs) were stained with serial dilutions of CD3 APC-Cy7 antibody. Data shown are concatenated FCS files representing serial dilution of each antibody lot used over a period of years in the CLIP panel. The cells are gated on lymphocytes (forward scatter, FSC, vs. side scatter, SSC, gates). The MFI of the positive population is marked by a red line. The MFI of the negative population is marked by a green line. Selected concentrations used in the CLIP are indicated by with an arrow
2.5 Fluorescence Compensation and Single-Color Controls
Multicolor flow cytometry uses several different fluorophores. At a specific excitation wavelength, each fluorophore emits light in a particular spectrum unique to that molecule, and most emission spectra exhibit a tail extending toward longer wavelengths, a consequence of the physics of fluorescence. In order to properly analyze multicolor flow cytometry experiments, it is necessary to assign a fluorescent signal to a particular fluorochrome and to remove any spectral spillover from all other fluorochromes. To separate fluorescence emission from the excitation light source and to resolve different fluorophores from one another, flow cytometers typically use a number of band-pass and dichroic filters, as well as spatially separated beams from excitation lasers. Even with these hardware features to separate fluorescence signals due to each fluorochrome and in spite of careful selection of the fluorochromes to be used in a panel, it is currently not possible to remove all of the spectral overlap among fluorochromes when one looks at 15 colors simultaneously. Software is used to subtract a portion of one detector’s signal from another, leaving only the desired signal, a process termed fluorescence compensation. Compensation in high polychromatic flow cytometry is complex and is only practical through the use of specialized software for the purpose of determining compensation matrices. Compensation is always specific for the particular combination of specificities and fluorophores in a multicolor staining panel and, therefore, needs to be determined for each unique panel. Compensation concerns are among the foremost constraints on how many parameters can be examined simultaneously using fluorescence and have led to development of non-fluorescence-based cytometry methods such as time-of-flight cytometry. Nonetheless, when panels are properly constructed and controlled, fluorescence compensation is practical for up to 18 colors.
A matrix of the needed spectral compensation is created by running each fluorochrome-antibody conjugate individually. Although the best way to perform accurate compensation is to use the same cells that will be used in the experiments, it is difficult to accurately titer antibodies for antigens that are expressed at a low level. For this reason, one can use beads that bind to the Fc region of antibodies, and thus obtain a non-antigen binding, positive staining with high fluorescence intensity to determine compensation (Roederer 2002). For each experiment, compensation can be performed with unstained cells and compensation bead particle sets (using only the positive beads, or using positive and negative beads) (Fig. 3). In general, compensation matrices will vary little on a day-to-day basis if the same lots of properly stored reagents are used and if the cytometer in use is tightly monitored for any change in performance. For this reason, as well as the necessity of re-titrating each lot of antibodies, it is highly recommended that large lots of each reagent are purchased when performing high-dimensional immunophenotyping, thus assuring a high level of standardization across time.
Fig. 3.
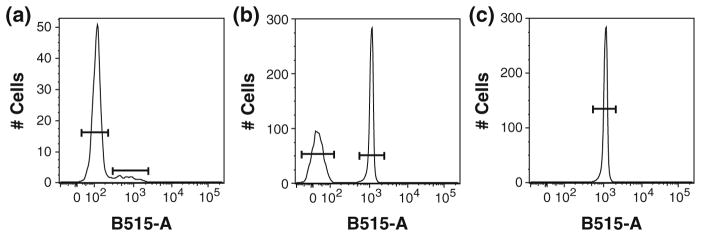
Examples of alternatives for mono-stained controls. a Use of cells. b Use of compensation beads for both the negative and positive events. c Use of positive compensation beads only
2.6 Isotype and Fluorescence-Minus-One Control
Unstained controls indicate the background or auto-fluorescence of cells in the sample, and isotype controls help to identify concerns that one may have about the specificity of antibody staining. The latter controls are particularly useful in intracellular staining where non-specific staining could be more problematic than in the staining of surface markers (Fig. 4). Isotype controls must be carefully matched to the specific primary antibody (species and isotype, including heavy and light chains) and to the precise fluorochrome conjugate (including the fluorophore: protein ratio if at all possible) in order to accurately determine the level of specific staining by the primary antibody. An additional type of control—fluorescence- minus-one (FMO) is used to assist in placement of quadrant gates and analysis regions. These controls contain every stain in the panel except for the one. The excluded marker is that one for which the determination of positive events may be difficult due to low intensity of staining and/or the lack of a discrete population of positive cells (Fig. 5).
Fig. 4.
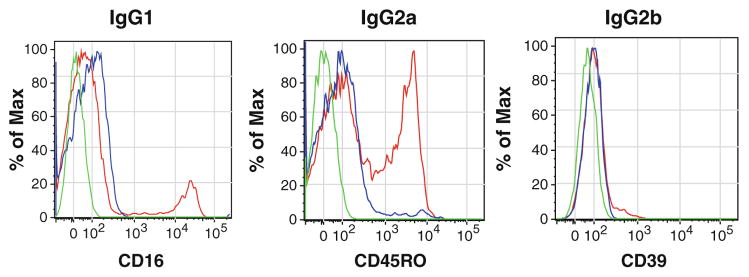
Isotype controls. Examples of overlays of unstained cells (green line), isotype control (blue line), and the antibody of interest (red line) for CD16 (IgG1), CD45-RO (IgG2a), and CD39 (IgG2b) antibodies. Isotype controls were used at the same concentration as the antibody of interest
Fig. 5.

Fluorescence-minus-one control (FMO). PBMCs from the same donor were stained. Mononuclear cells were selected using forward and side light scatter parameters together with CD45 and viability staining. CD3 expression was used to identify T cells, and subsets were identified by CD4 and CD8 expression. The boxes in red indicate the position of the gating based on FMO. a Gated on T cells, left panel shows a bivariate dot plot of the complete staining for CD4 versus CD8; middle panel shows FMO control of CD8 Qd605; and right panel shows FMO control of CD4 V450. b Gated on CD4+ T cells, left panel shows bivariate dot plot of the complete staining for regulatory T cell identification; middle panel shows FMO control of CD25 PE-Cy7; and right panel shows FMO control of Foxp3 PE. c Gated on T cells, left panel shows bivariate dot plot of the complete staining for CD27 versus CD39; middle panel shows FMO control of CD27 Qd655; and right panel shows FMO control of CD39 AF488. Note, that with CD25, the FMO identifies all positive cells, not only the bright cells characteristic of T regulatory cells, thus illustrating the subjectivity of place gates even with the use of FMO controls
2.7 Viability Staining
Many studies for clinical trials are performed on cryopreserved specimens, and the viability of cells might vary for a number of reasons. Although it is difficult to physically remove dead cells from every sample, it is essential to be able to gate out these dead cell during analysis. This is because some antibodies bind non-specifically to dead cells present in the sample, resulting in increased fluorescence from cells not truly positive for the antigen in question. Non-specific binding may result in an overestimation of the proportion of cells that are positive for the markers of interest, and thus create false results and even falsely positive cellular populations, particularly when rare events are analyzed. For this reason, viability markers are essential and permit one to gate out dead cells. For the CLIP panel, which uses intracellular staining, a fixable dye that would not leak out of the cells after permeabilization was needed. We decided to use amine-reactive dyes as viability stains since these dyes react with free amines in the cytoplasm rather than intercalating into nucleic acids as do most other viability markers. Live cells exclude amine-reactive dyes, thanks to the integrity of the cell membrane. Dead cells take up the dye and the reaction is irreversible; the dye remains bound to the amine even when the cells are permeabilized for intracellular staining (Perfetto et al. 2010). Like all antibody-fluorochrome conjugates used in the panel, the amine-reactive dye was titered, and, even though the positive cells are gated out, this color was included in the compensation matrix (Fig. 6).
Fig. 6.

Viability staining of a sample of cryopreserved PBMCs for which we observed significant loss of cell viability upon thawing. Mononuclear cells were selected using forward and side light scatter parameters, and the expression of both CD45 and CD3 was used to identify T cells, and then CD4 and CD8 for the respective subsets (not shown). CD45-RA versus CD27 and CD39 versus CD103 expressions are shown for CD4+ T cells (upper panel). The same analysis was performed with viability staining, in which dead cells were excluded prior detection of markers (lower panel). Non-specific bindings due to dead cells changed the proportion of measured populations and are indicated by a red arrow
2.8 The Conundrum of How Many Cells are Needed for Data Acquisition
Panels for high-dimensional immunophenotyping can be structured to provide a wide assessment of multiple lineages without in-depth analysis of each, with in-depth analysis of a particular lineage of cells, or using a combination of the two. How a particular panel is constructed will dictate the number of cells that must be acquired for statistical relevance. Acquisition counts should be predicated on collecting sufficient numbers of the rarest subset in order for a statistically meaningful analysis between samples to be conducted on that subset (generally a few hundred cells). For dendritic cell or plasmablast subsets, or even more scant populations such as endothelial progenitor cells, millions, perhaps tens of millions, of mononuclear cells must be evaluated. This presents concerns related to dimensionality. As the number of parameters increase, the depth in which any subset can be examined increases; hence, the number of cells that must be acquired increases. In turn, increasing the number of cells to be collected increases the time for data collection as well as the size of the data files, even if separate files are collected and later concatenated. Increasing collection time increases the odds of instrument fluctuations, a particular problem for instruments with low acquisition speeds. For example, collecting 10 million events at a collection rate of 20,000 events per second takes a little over 8 min, but collecting the same number of events at 500 cells per second takes roughly 5.5 h. The increased numbers of parameters acquired as well as acquiring higher number of cells needed can lead to data files of such enormity that transfer or analysis of these files on anything but the most powerful computers is agonizing slow. This issue pertains to all techniques for high-dimensional immunophenotyping and will ultimate prove to be a dimensionality barrier.
2.9 Reproducibility and Normalization
Even when using large lots of standardized reagents, a potential concern with high-dimensional immunophenotyping, or any immunophenotyping used for clinical or translational studies, is the reproducibility of the staining over time. In many protocols, several longitudinal samples from the same patient are run at multiple time points, and it is crucial to distinguish between differences in the data that arise from biological changes from those due to assay variability. Clearly, proper instrument maintenance and assessment of its performance is a crucial element in this process; another is strict standardization in sample preparation, staining, and analysis. These issues can be addressed by strict adherence to standard operating procedures, automation of the sample processing, and possibly the use of pre-mixed, lyophilized cocktails of the antibodies in each tube. Reproducibility of the staining panel can also be evaluated by periodically running cryopreserved aliquots from a single specimen from one donor (such as a large volume apheresis specimen that could yield hundreds of millions of cells in one specimen) over a designated period of time. These aliquots can be thawed at various intervals and stained and analyzed in a consistent manner. We have used this approach for our studies over a number of years to evaluate the reproducibility of our panel (Fig. 7). Not only does this type of analysis indicate the reproducibility of the staining, but the data collected may be used to normalize experimental data.
Fig. 7.

Sample normalization. A Levy-Jenning plot showing iterative analysis over a 2-year period of aliquots from a single sample (CHI-002) stained with our 15-color T regulatory cell tube. a T cells and T cell subpopulations CD4+CD8−, CD4+CD8+, CD4−CD8+, CD4−CD8− are indicated. b Discrete markers such as activation markers on T CD4+ cells and T CD8+ cells and on CD4+ regulatory T cells were identified
2.10 Cell Function of Immunophenotypes Defined in High Dimension
Flow cytometry is a tool of choice for the analysis of cellular phenotypes in the immune system. Deep phenotyping can and will identify novel leukocytic subsets, and whereas the function of these novels subsets might be inferred by lineage and marker expression, proof of function ultimately requires purification of these subsets for subsequent in vivo or in vitro testing. High polychromatic assays are well suited for this purpose, as cells identified in as many as 15 or more colors can be sorted by fluorescence-activated cells sorting, if a sorter is configured in the same manner as the cytometer on which the cells were identified. Novel subsets identified in higher dimension by other technologies but not recoverable, would remain of uncertain function and therefore of uncertain significance.
3 Comprehensive Leukocyte Immunophenotyping: One Approach
To facilitate the understanding of the immune system, a CLIP panel was developed to obtain a synoptic snapshot of the cellular phenotypes within the immune system (Biancotto et al. 2011). Historically, there have been relatively few leukocyte subsets examined in clinical studies and for which reference ranges exist (McCoy and Overton 1994). Our intent was to obtain in-depth immunophenotypic profiling of as many circulating human leukocyte populations as possible, in order to build an encyclopedic database useable by us and other groups. This was performed in concert with other concomitant measurements of the immune system such as multiplex bead array measurements of plasma cytokines concentrations, gene profiling studies, Elispot assays, and antibody titers. For immunophenotyping to cover both the incredible breadth as well as fine detail of the immune system, it would be necessary to use thousands, perhaps even tens of thousands, antibodies against various cellular features. Even using the technologies for high-dimensional phenotyping that are available today, a comprehensive approach such as this would require the use of multiple individual tubes, as the number of parameters that can be measured simultaneously using any of these methods is far less than would be needed. Therefore, we reasoned that a relevant approach to construction of a CLIP panel would be to structure a series of individual tubes that each provide in-depth characterization of a specific cell type, but at the same time have a sufficient number of recurring markers among the tubes that a concatenation of the numerous phenotypes within a lineage could be assembled. For example, in our panel of 15-color tubes used to characterize T cells, there are eight redundant stains, permitting inter-tube comparisons of antigen expression among common subpopulations defined by these eight markers. Using this approach together with a literature survey of relevant markers of various leukocytes, we assembled a conceptual CLIP panel consisting of over 70 tubes of 15 parameters each. Clearly, a panel of this magnitude presents a myriad of technical concerns ranging from volume of blood to be drawn, to sample processing, to analysis of data. These will be addressed below. In spite of these concerns, our laboratory began developing tubes for this panel one at a time, in order to assess the feasibility of this approach. Here, we discuss our rationale in designing these tubes and for assembling the start of a comprehensive CLIP panel.
3.1 The CLIP Panel
The academic exercise of how to immunophenotype the entire immune system is fascinating, daunting, and frustrating. Surveying the literature for all descriptive studies of the immune system using cytometry—not only for lineage and maturation markers, but also for markers of activation, subphenotypes, cytokine or chemokines production, cytokine receptors, markers of clonality such as TCR α and β chains, and signaling pathways, to name a few—revealed hundreds, perhaps thousands of potential markers to be studied. Even using 15 fluorochromes in each tube, scores of tubes would be needed for such a comprehensive analysis. Using the approach described above, we began to build our CLIP panel one 15-color tube at a time. This was done with the full understanding that (1) each tube represented only one small part of a comprehensive panel, (2) that completely comprehensive analysis would not likely be achieved using the current approach, and (3) that new markers and technologies would likely evolve to replace the current efforts. Nonetheless, we felt that it was important to take steps toward comprehensive analysis to begin building a database of these findings and to learn the intricacies and caveats in such an endeavor.
To date, our panel consists of 12 15-color tubes, including three T lymphocyte tubes, three B lymphocyte tubes, two natural killer cell tubes, one dendritic cell tube, two monocytes tubes, and two neutrophil tubes. A complete list of the current tubes of the CLIP panel, along with the antibody clones and fluorochromes used can be found in Table 2. Together these tubes have the potential to identify over 49,000 subsets of leukocytes. This assumes that all combinations of markers are possible, that brightness of staining is not considered (i.e., staining is either positive or negative), and that viability, CD45, and primary lineage markers are solely used for gating. Our approach was for each tube to have a specified purpose. For example, within the T lymphocyte lineage, there are specific tubes for T regulatory cells (Tregs), Th17 cells, and Th1/Th2 cells. Furthermore, as discussed above, within each lineage the tubes would have a number of recurring markers, both for quality control purposes, but also for cross-tube concatenation of more detailed phenotypes. If viable, CD45 and CD3 are used for gating in each of the T cell tubes, in theory each of the T cells tubes could yield 4,096 T cell phenotypes if one assumes all marker combinations could be expressed and that marker expression is defined only as positive or negative. Thus, although certainly not comprehensive for the immunome, the existing 12-tube CLIP panel could provide a breadth and depth of immunophenotyping not reported heretofore.
Table 2.
CLIP panel
| CHI CLIP panel
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Lymphoid
|
||||||||||
| T lineage | B lineage | NK lineage | ||||||||
| Excitation | Fluorochrome name | PMT name | T1 | T2 | T3 | B1 | B2 | B3 | NK1 | NK2 |
| 407 nm Excitation | V450 | V450 | CD4 | CD4 | CD4 | Lambda | CD80 | CD80 | CD56 | CD56 |
| RPA-T4 | RPA-T4 | RPA-T4 | B39-4 | L307.4 | L307.4 | B159 | ||||
| Aquablue | V545 | Viability | Viability | Viability | Viability | Viability | Viability | Viability | Viability | |
| Qdot 605 | V605 | CD8 | CD8 | CD8 | CD10 | CD27 | CD27 | CD8 | CD8 | |
| 3B5 | 3B5 | 3B5 | MEM-78 | CLB-27/1 | CLB-27/1 | 3B5 | 3B5 | |||
| Qdot 655 | V655 | CD27 | CD27 | CD27 | CD19 | CD19 | CD19 | CD30 | NKp46 | |
| CLB-27/1 | CLB-27/1 | CLB-27/1 | SJ25-C1 | SJ25-C1 | SJ25-C1 | BerH8 | 900 | |||
| Qdot 800 | V800 | CD45 | CD45 | CD45 | CD45 | CD45 | CD45 | CD45 | CD45 | |
| HI30 | HI30 | HI30 | HI30 | HI30 | HI30 | HI30 | HI30 | |||
| 488 nm Excitation | FITC/Alexa488 | B515 | CD39 | IL-23R | CD69 | CD40 | IgA | IgA (intra) | CD57 | CD57 |
| A1 | 218213 | FN50 | 5C3 | Polyclonal Goat | Polyclonal Goat | TB01 | TB01 | |||
| PcPcy5.5 | B710 | CD38 | CD196 | IL-4 | CD138 | CD86 | CD86 | CD161 | IFNg | |
| HIT2 | 11A9 | MP4-25D2 | MI15 | IT2.2 | IT2.2 | HP-3G10 | 4S.B3 | |||
| 532 nm Excitation | PE | G560 | Foxp3 | CD146 | perforin | Kappa | CD21 | IgE (intra) | CD337 | Perforin |
| PCH101 | 142928 | δG9 | TB28-2 | BL13 | MB10-5C4 | P30-15 | δG9 | |||
| PE-TR | G610 | CD45-RA | CD45-RO | CD45-RO | CD38 | CD38 | CD38 | CD94 | CD25 | |
| 2H4LDH11LDB9 | UCHL1 | UCHL1 | HIT2 | HIT2 | HIT2 | DX22 | B1.49.9 | |||
| PE-cy5 | G660 | CD103 | CD161 | CD40L | CD103 | IgG | IgG (intra) | CD16 | CD16 | |
| LF61 | DX12 | TRAP1 | LF61 | G18-145 | G18-145 | 3G8 | 3G8 | |||
| PE-cy5.5/PE-A700 | G710 | HLA-DR | TNFa | TNFa | CD20 | CD20 | CD20 | CD244 | CD69 | |
| Tu36 | MP9-20A4 | MP9-20A4 | HI47 | HI47 | HI47 | C1.7 | CH/4 | |||
| PE-cy7 | G780 | CD25 | IL-17A | IFNg | CD5 | CD23 | CD23 | CD5 | CD5 | |
| M-A251 | BL168 | 4S.B3 | L17F12 | EBVCS-5 | EBVCS-5 | L17F12 | L17F12 | |||
| 633 nm Excitation | APC/Alexa647 | R660 | CD127 | IL-22 | IL-2 | CD22 | IgD | IgD (intra) | CD336 | CD127 |
| hIL-7R-M21 | 142928 | MQ1-17H12 | S-HCL-1 | IgD26 | IgD26 | P44-8 | ||||
| Alexa700/APC-cy5.5 | R710 | CD197 | CD197 | CD197 | CD11c | IgM | IgM (intra) | CD158e1 | CD158e1 | |
| 150503 | 150503 | 150503 | 3.9 | CH2 | CH2 | DX9 | DX9 | |||
| APC-cy7/APC-H7 | R780 | CD3 | CD3 | CD3 | CD25 | CD10 | CD10 | CD3 | CD3 | |
| Sk7 | Sk7 | Sk7 | M-A251 | HI10a | HI10a | Sk7 | Sk7 | |||
| Myeloid
|
|||||||
|---|---|---|---|---|---|---|---|
| Dendritic lineage | Mono lineage | Neutrophil lineage | |||||
| Excitation | Fluorochrome name | PMT name | DC1 | M1 | M2 | N1 | N2 |
| 407 Excitation | V450 | V450 | CD123 | HLA-DR | CD31 | CD62L | CD14 |
| 6H6 | L243 (G46-6) | WM59 | DREG-56 | TuK4 | |||
| Aquablue | V545 | Viability | Viability | Viability | Viability | Viability | |
| Qdot 605 | V605 | CD27 | CD163 | CD163 | CD3 | CD8 | |
| CLB-27/1 | eBioGHI/61 | eBioGHI/61 | sk7 | 3B5 | |||
| Qdot 655 | V655 | CD4 | CD14 | CD14 | CD14 | CD19 | |
| S3.5 | TuK4 | TuK4 | TuK4 | SJ25-C1 | |||
| Qdot 800 | V800 | CD45 | CD45 | CD45 | CD16 | CD45 | |
| HI30 | HI30 | HI30 | 3G8 | HI30 | |||
| 488 Excitation | FITC/Alexa488 | B515 | IFNa | CD77 | CD77 | CD185 | CD123 |
| MMHA-11 | 5B5 | GA-R2 (HIR2) | 2G8 | ||||
| PcPcy5.5 | B710 | TNFa | CD192 | CD34 | CD203c | CD4 | |
| MAb11 | TG5/CCR2 | 8G12 | NP4D6 | S3.5 | |||
| 532 nm Excitation | PE | G560 | CD40 | CD64 | CD64 | CD303 | CD56 |
| HB14 | 10.1 | 10.1 | 201A | NCAM16.2 | |||
| PE-TR | G610 | HLA-DR | CD16 | CD16 | CD123 | CD16 | |
| Tu36 | 3G8 | 3G8 | 6H6 | 3G8 | |||
| PE-cy5 | G660 | CD83 | CD15 | CD15 | CD15 | CD15 | |
| HB15e | 80H5 | 80H5 | 80H5 | 80H5 | |||
| PE-cy5.5 /PE-A700 | G710 | CD11c | CD4 | CD4 | HLA-DR | HLA-DR | |
| Bu15 | S3.5 | S3.5 | Tu36 | Tu36 | |||
| PE-cy7 | G780 | lineage | CD13 | CD13 | CD13 | CD34 | |
| see below | L138 | L138 | L138 | ||||
| 633 nm Excitation | APC/Alexa647 | R660 | BDCA-2 | CD36 | CD36 | FcεRI | CD11c |
| AC144 | CB38 (NL07) | CB38 (NL07) | BC4 | Bu15 | |||
| Alexa700/APC-cy5.5 | R710 | CD33 | CD33 | CD33 | CD33 | CD33 | |
| WM-53 | WM-53 | WM-53 | WM-53 | WM-53 | |||
| APC-cy7/APC-H7 | R780 | CD86 | CD206 | HLA-DR | CD10 | CD3 | |
| IT2.2 | 15-2 | L243 (G46-6) | HI10a | Sk7 | |||
Lineage mix =
CD3 SK7
CD56 NCAM16.2
CD20 L27
CD19 SJ25C1
CD14 M5E2
CD16 3G8
3.2 Applications of the CLIP Panel
The NIH Center for Human Immunology is currently using this CLIP panel for a number of translational research protocols. One early study conducted while this panel was still in development was for the characterization of Tregs in chronic lymphocytic leukemia (CLL) (Biancotto et al. 2012b). This was performed using only the one Treg tube to assess the feasibility of running and analyzing such high-dimensional data. Although several previous reports existed in the literature demonstrating elevated number of Tregs in CLL, none of these provided an in-depth characterization of Tregs found in the cancer patients compared to healthy individuals (Beyer et al. 2005; Giannopoulos et al. 2008; Deutsch et al. 2009; Jak et al. 2009; D’Arena et al. 2011). Using manual analysis, Treg populations, defined by the presence of FoxP3 in T lymphocytes, were identified not only in CD4+CD25+ T cells, but also in CD4+CD25− cells and CD8 lymphocytes either with or without CD25 co-expression. Furthermore, through the use of CD45RA, CD197 (CCR7), and CD27, these Treg populations were classified as naïve, effector, effector memory, or central memory cells. Within these Treg subpopulations, further subclassification was made based on the presence of CD38, CD39, CD103, CD127, and HLA-DR. A large number of these populations were present in significantly different levels in CLL patients compared to controls. Among the many differences were a population of CD39+CCR7+CD4+ Tregs that were present in CLL patients but rare in healthy individuals. Because fluorescent markers were used to identify this population, it was possible to use the same staining matrix (without Foxp3 since viable Tregs were required for functional studies) to sort these cells for functional studies. This subpopulation unique to CLL patients had a lower suppressive capability than other Treg subpopulations (Fig. 8). This study revealed the power of a single tube of the CLIP panel to identify unique phenotypes within a specific cell lineage and to reveal significant differences among these detailed phenotypic subsets in health and disease and the ability of the approach to recover these phenotypes for subsequent functional studies. At the same time this study demonstrated the limitations of manual analysis of even just a single 15-color tube, and the near impossibility of thorough data mining without automated approaches to analysis.
Fig. 8.

Sorting of populations identified by CLIP. a Based on immunophenotyping findings we stained cells in an identical manner, but without permeabilization and without FOXp3 staining, to study suppressive activity of these cells. Five populations were sorted and are identified by numbers: (1) CD4+CD25− (effector T cells), (2) CD4+CD25high (Treg), (3) CD39+ Treg, (4) CD39+CCR7+ Treg, and (5) CD39−CCR7−Treg. b Suppressive capacity of Treg cells on effector T cells was expressed as relative inhibition of the percentage of CFSElow cells according to the following formula [100 × (1 − (x/y))] where x is the percentage of CFSElow cells in the effector T cell population alone culture and y is percentage of CFSElow cells in co-culture of Treg and effector T cells
Following this initial study, a second more expansive study was conducted on immune responses to an H1N1 influenza vaccine in which five of the 15-color tubes of the CLIP panel were used to assess immune responses. The tubes developed at that point included the Treg tube and tubes for Th17, Th1/2, B cells, monocytes, and dendritic cells. Cryopreserved samples from 63 patients who had received the vaccine were analyzed. Samples were collected at five time points from each patient—two pre-vaccination and three post-vaccination. As automated data analysis approaches were not yet available at the time of this study, a decision was made to select 128 subsets, out of potentially thousands, that might be the most informative for this study and perform manual analysis of these (Table 3). The guided analysis of the B cell tube alone provided 40 different populations (Fig. 9). These data were then integrated into a larger systems analysis that included gene expression data, serum cytokine levels, virus titers, and Elispot data from these same patients in order to provide a truly comprehensive assessment of immune responses to this vaccination. The results of this study have been reported elsewhere (Tsang et al. 2013). The raw flow cytometry data from this study was archived and now is being more thoroughly analyzed using a computational, rather than manual, approach (JS Tsang, personal communication).
Table 3.
Flow population reported for CHI Flu study
| Population ID |
Tube | Markers | Category (Page) | Parent population | Subset name | Name for paper | Comments |
|---|---|---|---|---|---|---|---|
| ID1 | T1 | CD45+CD3+ | T cells | (as of viable CD45+) | T cells | % CD3+of viable CD45+cells (Total T cells) | |
| ID2 | T1 | CD45+CD3+CD4+CD8− | T cells | (as of viable CD45+CD3+) quadrant | CD4+ T helper | % CD4+ of total T cells | |
| ID3 | T1 | CD45+CD3+CD4+CD8+ | T cells | (as of viable CD45+CD3+) | DOUBLE POSITIVE | % CD4+CD8+ of total T cells | |
| ID4 | T1 | CD45+CD3+CD4− CD8+ | T cells | (as of viable CD45+CD3+) quadrant | CD8+ T cytotoxic | % CD8+ of total T cells | |
| ID5 | T1 | CD45+CD3+CD4− CD8− | T cells | (as of viable CD45+CD3+) | DN immature T | % CD4−CD8− of total T cells | |
| ID6 | T1* | CD45+CD3+CD4+CD8− | T helper cells | (as of viable CD45+CD3+) gated region | CD4+ T helper | % CD4+ of total T cells | Duplicate of ID2 |
| ID7 | T3 | CD45+CD3+CD4+CD8−CD69+ | T helper cells | (as of viable CD45+CD3+CD4+CD8−) | Activated CD4+ T helper cells CD69+ | % CD69+ of CD4+ T cells | |
| ID8 | T1 | CD45+CD3+CD4+CD8−CD25+ | T helper cells | (as of viable CD45+CD3+CD4+CD8−) | Activated CD4+ T helper cells CD25+ | % CD25+ of CD4+ T cells | |
| ID9 | T1 | CD45+CD3+CD4+CD8−CD38+ | T helper cells | (as of viable CD45+CD3+CD4+CD8−) | Activated CD4+ T helper cells CD38+ | % CD38+ of CD4+ T cells | |
| ID10 | T1 | CD45+CD3+CD4+CD8−HLA−DR+ | T helper cells | (as of viable CD45+CD3+CD4+CD8−) | Activated CD4+ T helper cells HLA–DR+ | % HLA–DR+ of CD4+ T cells | |
| ID11 | T3 | CD45+CD3+CD4+CD8−CD40L+ | T helper cells | (as of viable CD45+CD3+CD4+CD8−) | Activated T helper cells CD40L+ | % CD40+ of CD4+ T cells | |
| ID12 | T2 | CD45+CD3+CD4+CD8−CD161+ | T helper cells | (as of viable CD45+CD3+CD4+CD8−) | T helper Th17 CD161+ | % CD161+ of CD4+ T cells | |
| ID13 | T2 | CD45+CD3+CD4+CD8−CD196+ | T helper cells | (as of viable CD45+CD3+CD4+CD8−) | T helper Th17 CD196+ (CCR6) | % CD196+ of CD4+ T cells | |
| ID14 | T2 | CD45+CD3+CD4+CD8−IL17a+ | T helper cells | (as of viable CD45+CD3+CD4+CD8−) | T helper Th17 IL–17A+ | % IL17+ of CD4+ T cells | |
| ID15 | T2 | CD45+CD3+CD4+CD8−IL21+ | T helper cells | (as of viable CD45+CD3+CD4+CD8−) | T helper IL–21+ | % IL21+ of CD4+ T cells | |
| ID16 | T2 | CD45+CD3+CD4+CD8−IL22+ | T helper cells | (as of viable CD45+CD3+CD4+CD8−) | T helper IL–22+ | % IL22+ of CD4+ T cells | |
| ID17 | T2 | CD45+CD3+CD4+CD8−IL23r+ | T helper cells | (as of viable CD45+CD3+CD4+CD8−) | T helper IL–23R+ | % IL23+ of CD4+ T cells | |
| ID18 | T2 | CD45+CD3+CD4+CD8−CD161+ | T helper cells | (as of viable CD45+CD3+CD4+CD8−) histogram | Th17 | % CD161+ of CD4+ T cells | Duplicate of ID12 |
| ID19 | T2 | CD45+CD3+CD4+CD8− CD161+IL17a+ | T helper cells | (as of viable CD45+CD3+CD4+CD8− CD161+) | Th17 CD161+ IL17A+ | % IL17+ of CD161+CD4+ T cells | |
| ID20 | T2 | CD45+CD3+CD4+CD8− CD161+IL21+ | T helper cells | (as of viable CD45+CD3+CD4+CD8− CD161+) | Th17 CD161+IL21+ | % IL21+ of CD161+CD4+ T cells | |
| ID21 | T2 | CD45+CD3+CD4+CD8− CD161+IL22+ | T helper cells | (as of viable CD45+CD3+CD4+CD8− CD161+) | Th17 CD161+IL22+ | % IL22+ of CD161+CD4+ T cells | |
| ID22 | T2 | CD45+CD3+CD4+CD8−CD161− | T helper cells | (as of viable CD45+CD3+CD4+CD8−) histogram | Th17 CD161− | % CD161− of CD4+ T cells | |
| ID23 | T2 | CD45+CD3+CD4+CD8− CD161−L17a+ | T helper cells | (as of viable CD45+CD3+CD4+CD8− CD161−) | Th17 CD161− IL17A+ | % IL17+ of CD161−CD4+ T cells | |
| ID24 | T2 | CD45+CD3+CD4+CD8− CD161−IL21+ | T helper cells | (as of viable CD45+CD3+CD4+CD8− CD161−) | Th17 CD161−IL21+ | % IL21+ of CD161−CD4+ T cells | |
| ID25 | T2 | CD45+CD3+CD4+CD8− CD161−IL22+ | T helper cells | (as of viable CD45+CD3+CD4+CD8− CD161−) | Th17 CD161− IL22+ | % IL22+ of CD161−CD4+ T cells | |
| ID26 | T3 | CD45+CD3+CD4+CD8−IL2+ | T helper cells | (as of viable CD45+CD3+CD4+CD8−) | IL–2 T helper cells | % IL2+ of CD4+ T cells | |
| ID27 | T3 | CD45+CD3+CD4+CD8−IFNg+ | T helper cells | (as of viable CD45+CD3+CD4+CD8−) | Th1 | % IFNg+ of CD4+ T cells | |
| ID28 | T3 | CD45+CD3+CD4+CD8−IL4+ | T helper cells | (as of viable CD45+CD3+CD4+CD8−) | Th2 | % IL4+ of CD4+ T cells | |
| ID29.T2 | T2 | CD45+CD3+CD4+CD8−TNFa+ | T helper cells | (as of viable CD45+CD3+CD4+CD8−) | TNFa T helper cells | % TNFa+ of CD4+ T cells | |
| ID29.T3 | T3 | CD45+CD3+CD4+CD8−TNFa+ | T helper cells | (as of viable CD45+CD3+CD4+CD8−) | TNFa T helper cells | % TNFa+ of CD4+ T cells | |
| ID30 | T1 | CD45+CD3+CD4+CD8−CD39+ | T helper cells | (as of viable CD45+CD3+CD4+CD8−) | Activated T helper cells CD39+ (Treg) | % CD39+ of CD4+ T cells | |
| ID31 | T1 | CD45+CD3+CD4+CD8−CD103+ | T helper cells | (as of viable CD45+CD3+CD4+CD8−) | Integrin aE T helper cells (Treg) | % CD103+ of CD4+ T cells | |
| ID32 | T1 | CD45+CD3+CD4+CD8−CD127+ | T helper cells | (as of viable CD45+CD3+CD4+CD8−) | IL7R on T helper cells (activated and Treg) | % CD127(IL7R)+ of CD4+ T cells | |
| ID33.T1 | T1 | CD45+CD3+CD4+CD8−CD27+ | T helper cells | (as of viable CD45+CD3+CD4+CD8−) | T helper CD27+ | % CD27+ of CD4+ T cells | |
| ID33.T2 | T2 | CD45+CD3+CD4+CD8−CD27+ | T helper cells | (as of viable CD45+CD3+CD4+CD8−) | T helper CD27+ | % CD27+ of CD4+ T cells | |
| ID33.T3 | T3 | CD45+CD3+CD4+CD8−CD27+ | T helper cells | (as of viable CD45+CD3+CD4+CD8−) | T helper CD27+ | % CD27+ of CD4+ T cells | |
| ID34 | T1 | CD45+CD3+CD4+CD8−CD45RA+ | T helper cells | (as of viable CD45+CD3+CD4+CD8−) | Naïve T helper cells | % CD45RA+ of CD4+ T cells (Naïve T) | |
| ID35 | T1 | CD45+CD3+CD4+CD8−CD45RA− | T helper cells | (as of viable CD45+CD3+CD4+CD8−) | Total Memory T helper cells | % CD45RA− of CD4+ T cells (Total memory CD4+ T) | |
| ID36 | T1 | CD45+CD3+CD4+CD8−CD45RA−CD27+CCR7− | T helper cells | (as of viable CD45+CD3+CD4+CD8− CD45RA−) | Effector memory T helper cells | % CD27+ CCR7− of memory CD4+ T cells (Effector memory CD4+T) | |
| ID37 | T1 | CD45+CD3+CD4+CD8−CD45RA−CD27+CCR7+ | T helper cells | (as of viable CD45+CD3+CD4+CD8− CD45RA−) | Central memory T helper cells | % CD27+CCR7− of memory CD4+ T cells (Central memory CD4+ T) | |
| ID38 | T1 | CD45+CD3+CD8+CD4− | T cytotoxic cells | (as of viable CD45+CD3+) gated region | T cytotoxic | % CD8+ of total T cells | duplicate of ID4 |
| ID39 | T3 | CD45+CD3+CD8+CD4−CD69+ | T cytotoxic cells | (as of viable CD45+CD3+CD8+CD4−) | Activated T cytotoxic CD69+ | % CD69+ of CD8+ T cells | |
| ID40 | T1 | CD45+CD3+CD8+CD4−CD25+ | T cytotoxic cells | (as of viable CD45+CD3+CD8+CD4−) | Activated T cytotoxic CD25+ | % CD25+ of CD8+ T cells | |
| ID41 | T1 | CD45+CD3+CD8+CD4−CD38+ | T cytotoxic cells | (as of viable CD45+CD3+CD8+CD4−) | Activated T cytotoxic CD38+ | % CD38+ of CD8+ T cells | |
| ID42 | T1 | CD45+CD3+CD8+CD4−HLA−DR+ | T cytotoxic cells | (as of viable CD45+CD3+CD8+CD4−) | Activated T cytotoxic HLA–DR+ | % HLA–DR+ of CD8+ T cells | |
| ID43 | T1 | CD45+CD3+CD8+CD4−CD39+ | T cytotoxic cells | (as of viable CD45+CD3+CD8+CD4−) | Activated T cytotoxic CD39+ | % CD39+ of CD8+ T cells | |
| ID44 | T1 | CD45+CD3+CD8+CD4−CD103+ | T cytotoxic cells | (as of viable CD45+CD3+CD8+CD4−) | Activated T cytotoxic CD103+ | % CD103+ of CD8+ T cells | |
| ID45 | T1 | CD45+CD3+CD8+CD4−CD127+ | T cytotoxic cells | (as of viable CD45+CD3+CD8+CD4−) | Activated T cytotoxic CD 127+ | % TNFa+ of CD8+ T cells | |
| ID46 | T2 | CD45+CD3+CD8+CD4−IL17a+ | T cytotoxic cells | (as of viable CD45+CD3+CD8+CD4−) | Tc17 IL–17A+ | % IL17A+ of CD8+ T cells (Tc17) | |
| ID47 | T2 | CD45+CD3+CD8+CD4−IL23r+ | T cytotoxic cells | (as of viable CD45+CD3+CD8+CD4−) | Tc17IL–23R+ | % IL23R+ of CD8+ T cells | |
| ID48 | T3 | CD45+CD3+CD8+CD4−IL2+ | T cytotoxic cells | (as of viable CD45+CD3+CD8+CD4−) | T Cytotoxic IL2+ | % IL2+ of CD8+ T cells | |
| ID49 | T3 | CD45+CD3+CD8+CD4−IFNg+ | T cytotoxic cells | (as of viable CD45+CD3+CD8+CD4−) | T Cytotoxic CD8+ IFNg+ | % INFg+ of CD8+T cells | |
| ID50 | T3 | CD45+CD3+CD8+CD4−Perforin+ | T cytotoxic cells | (as of viable CD45+CD3+CD8+ CD4−) | T Cytotoxic CD8+ Perforin+ | % Perforin+ of CD8+ T cells | |
| ID51.T2 | T2 | CD45+CD3+CD8+CD4−TNFa+ | T cytotoxic cells | (as of viable CD45+CD3+CD8+CD4−) | T cytotoxicCD8+ TNFa+ | % TNFa+ of CD8+ T cells | |
| ID51.T3 | T3 | CD45+CD3+CD8+CD4−TNFa+ | T cytotoxic cells | (as of viable CD45+CD3+CD8+CD4−) | T cytotoxicCD8+ TNFa+ | % TNFa+ of CD8+ T cells | |
| ID52.T1 | T1 | CD45+CD3+CD8+CD4−CD27+ | T cytotoxic cells | (as of viable CD45+CD3+CD8+CD4−) | T cytotoxic CD8+ CD27+ | % CD27+ of CD8+ T cells | |
| ID52.T2 | T2 | CD45+CD3+CD8+CD4−CD27+ | T cytotoxic cells | (as of viable CD45+CD3+CD8+CD4−) | T cytotoxic CD8+ CD27+ | % CD27+ of CD8+ T cells | |
| ID52.T3 | T3 | CD45+CD3+CD8+CD4−CD27+ | T cytotoxic cells | (as of viable CD45+CD3+CD8+CD4−) | T cytotoxic CD8+ CD27+ | % CD27+ of CD8+ T cells | |
| ID53 | T1 | CD45+CD3+CD8+CD4−CD45RA+ | T cytotoxic cells | (as of viable CD45+CD3+CD8+CD4−) | T cytotoxic CD8+ RA+ | % CD45RA+ of CD8+ T cells | |
| ID54 | T1 | CD45+CD3+CD8+CD4−CD45RA+CD27+ | T cytotoxic cells | (as of viable CD45+CD3+CD8+CD4−CD45RA+) | T cytotoxic Naïve | % CD27+ of CD45RA+ CD8+ T cells (Naïve CD8+T) | |
| ID55 | T1 | CD45+CD3+CD8+CD4−CD45RA+CD27− | T cytotoxic cells | (as of viable CD45+ CD3+CD8+CD4−CD45RA+) | T cytotoxic EMRA | % CD27− of CD45RA+ CD8+ T cells (EMRA CD8+ T) | |
| ID56 | T1 | CD45+CD3+CD8+CD4−CD45RA− | T cytotoxic cells | (as of viable CD45+CD3+CD8+CD4−) | T cytotoxic Total memory | % CD45RA− of CD8+ T cells (CD45RA− memory CD8+ T) | |
| ID57 | T1 | CD45+CD3+CD8+CD4−CD45RA−CD27+CCR7+ | T cytotoxic cells | (as of viable CD45+CD3+CD8+CD4−CD45RA−) | T cytotoxic central memory | % CD27+ CCR7− of CD45RA− memory CD8+ T cells (Central memory CD8+ T) | |
| ID58 | T1 | CD45+CD3+CD8+CD4−CD45RA−CD27−CCR7− | T cytotoxic cells | (as of viable CD45+CD3+CD8+CD4−CD45RA−) | T cytotoxic effector memory | % CD27−CCR7− of CD45RA− memory CD8+ T cells (Effector memory CD8+ T) | |
| ID59 | T1 | CD45+CD3+CD4+CD8−CD25highFOXp3+ | T regulatory cells | (as of viable CD45+CD3+CD4+CD8−) | Treg | % CD25hi FoxP3+ of CD4+ T cells (Treg) | |
| ID60 | T1 | CD45+CD3+CD4+CD8−CD25highFOXp3+CD38+ | T regulatory cells | (as of viable CD45+CD3+CD4+CD8−CD25hiFOXp3+) | Treg CD38+ | % CD38+ of Treg cells | |
| ID61 | T1 | CD45+CD3+CD4+CD8−CD25highFOXp3+CD39+ | T regulatory cells | (as of viable CD45+CD3+CD4+CD8−CD25hiFOXp3+) | Treg CD39+ | % CD39+of Treg cells | |
| ID62 | T1 | CD45+CD3+CD4+CD8−CD25highFOXp3+CD103+ | T regulatory cells | (as of viable CD45+CD3+CD4+CD8−CD25hiFOXp3+) | Treg CD103+ | % CD103+ of Treg cells | |
| ID63 | T1 | CD45+CD3+CD4+CD8−CD25highFOXp3+HLADR+ | T regulatory cells | (as of viable CD45+CD3+CD4+CD8−CD25hiFOXp3+) | Treg HLA–DR+ | % HLA–DR+ of Treg cells | |
| ID64 | Mono/DC | CD45+CD14+ | Monocytes | (as of viable CD45+) | Monocytes | % CD14+ of viable CD45+ cells (Total Monocytes) | |
| ID65 | Mono/DC | CD45+CD14+CD40+ | Monocytes | (as of viable CD45+CD14+) | activated monocyte CD40+ | % CD40+ of total monocytes | |
| ID66 | Mono/DC | CD45+CD14+CD83+ | Monocytes | (as of viable CD45+CD14+) | activated monocyte CD83+ | % CD83+ of total monocytes | |
| ID67 | Mono/DC | CD45+CD14+CD86+ | Monocytes | (as of viable CD45+CD14+) | activated monocyte CD86+ | % CD86+ of total monocytes | |
| ID68 | Mono/DC | CD45+CD14+HLADR+ | Monocytes | (as of viable CD45+CD14+) | activated monocyte HLA–DR+ | % HLA–DR+ of total monocytes | |
| ID69 | Mono/DC | CD45+CD14+TNFa+ | Monocytes | (as of viable CD45+CD14+) | activated monocyteTNFa+ | % TNFa+ of total monocytes | |
| ID70 | Mono/DC | CD45+CD3−CD1−CD20−CD16−CD56−CD14−HLADR+ | Dendritic cells | (as of viable CD45+CD3−CD19−CD20−CD16−CD56−CD14−) | Dendritic cells | % HLA–DR+ of Lin–CD45+ (Total Dendritic cells) | |
| ID71 | Mono/DC | CD45+CD3−CD19−CD20−CD16−CD56−CD14−HLADR+TNFa+ | Dendritic cells | (as of viable CD45+CD3−CD19−CD20−CD16−CD56−CD14−HLADR+) | TNFa secreting DCs | % TNFa+ of total DCs | |
| ID72 | Mono/DC | CD45+CD3−CD19−CD20−CD16−CD56−CD14−HLADR+IFNa+ | Dendritic cells | (as of viable CD45+CD3−CD19−CD20−CD16−CD56−CD14−HLADR+) | IFNa secreting DCs | % INFa+ of total DCs | |
| ID73 | Mono/DC | CD45+CD3−CD19−CD20−CD16−CD56−CD14−HLADR+CD11c+CD123− | Dendritic cells | (as of viable CD45+CD3−CD19−CD20−CD16−CD56−CD14−HLADR+) | Myeloid DC | % CD11c+ CD123− of total DCs (Myeloid DCs) | |
| ID74 | Mono/DC | CD45+CD3−CD19−CD20−CD16−CD56−CD14−HLADR+CD11c+CD123−TNFa+ | Dendritic cells | (as of viable CD45+CD3−CD19−CD20−CD16−CD56−CD14−HLADR+CD11c+CD123−) | TNFa secreting mDCs | % TNFa+ of mDCs | |
| ID75 | Mono/DC | CD45+CD3−CD19−CD20−CD16−CD56−CD14−HLADR+CD11c+CD123−IFNa+ | Dendritic cells | (as of viable CD45+CD3−CD19−CD20−CD16−CD56−CD14−HLADR+ CD11c+CD123−) | IFNa secreting mDCs | % IFNa+ of mDCs | |
| ID76 | Mono/DC | CD45+CD3−CD19−CD20−CD16−CD56−CD14−HLADR+CD11c−CD123+ | Dendritic cells | (as of viable CD45+CD3−CD19−CD20−CD16−CD56−CD14−HLADR+) | Plasmacytoid dendritic cells | % CD11c−CD123+ of total DCs (Plasmacytoid DCs) | |
| ID77 | Mono/DC | CD45+CD3−CD19−CD20−CD16−CD56−CD14−HLADR+CD11c−CD123+TNFa+ | Dendritic cells | (as of viable CD45+CD3−CD19−CD20−CD16−CD56−CD14−HLADR+CD11c−CD123+) | TNFa secreting pDCs | % TNFa+ of pDCs | |
| ID78 | Mono/DC | CD45+CD3−CD19−CD20−CD16−CD56−CD14−HLADR+CD11c−CD123+IFNa+ | Dendritic cells | (as of viable CD45+CD3−CD19−CD20−CD16−CD56−CD14−HLADR+ | IFNa secreting pDCs | % INFa+ of pDCs | |
| ID79 | Mono/DC | CD45+CD3−CD19−CD20−CD16−CD56−CD14−HLADR+CD11c+CD123+ | Dendritic cells | (as of viable CD45+CD3−CD19−CD20−CD16−CD56−CD14−HLADR+) | do not know this population | % CD11c+ CD123+ of total DCs | |
| ID80 | B2 | CD45+CD19+ | B cells | (as of viable CD45+) | Total B | % CD19+ of viable CD45+ (Total B cells) | |
| ID81 | B2 | CD45+CD19+CD20+CD80+ | B cells | (as of viable CD45+CD19+CD20+) | Mature Activated B CD80+ | % CD80+ of CD20+ B cells (CD80+ activated mature B) | |
| ID82 | B2 | CD45+CD19+CD20+CD86+ | B cells | (as of viable CD45+CD19+CD20+) | Mature Activated B CD86+ | % CD86+ of CD20+ B cells (CD86+ activated mature B) | |
| ID83 | B2 | CD45+CD19+CD20+IgA+ | B cells | (as of viable CD45+CD19+CD20+) | Mature B IgA+ | % IgA+ of CD20+ B cells (IgA+ mature B) | |
| ID84 | B2 | CD45+CD19+CD20+IgG+ | B cells | (as of viable CD45+CD19+CD20+) | Mature B IgG+ | % IgG+ of CD20+ B cells (IgG+ mature B) | |
| ID85 | B2 | CD45+CD19+CD20+IgM+IgD+ | B cells | (as of viable CD45+CD19+CD20+) | Mature B IgM+ IgD+ | % IgM+ IgD+ of CD20+ B cells (IgM+ IgD+ mature B) | |
| ID86 | B2 | CD45+CD19+CD20+IgM−IgG− | B cells | (as of viable CD45+CD19+CD20+) | Mature B IgM−IgD− | % IgM−IgD− of CD20+ B cells (IgM−IgD− mature B) | |
| ID87 | B2 | CD45+CD19+CD20−CD27highCD38 ++ | Plasmablast | (as of viable CD45+CD19+) | plasmablast | % CD27hi CD38hi of CD20− B cells (Plasmablasts) | |
| ID88 | B2 | CD45+CD19+CD20−CD27highCD38 ++CD23+ | Plasmablast | (as of viable CD45+CD19+CD20−CD27highCD38++) | activated plasmablast CD23+ | % CD23+ of plasmablasts | |
| ID89 | B2 | CD45+CD19+CD20−CD27highCD38 ++CD21+ | Plasmablast | (as of viable CD45+CD19+CD20−CD27highCD38++) | activated plasmablast CD21+ | % CD21+ of plasmablasts | |
| ID90 | B2 | CD45+CD19+CD20+CD27−CD10+ | Transitional B cells | (as of viable CD45+CD19 + CD20+) | Transitional B cells | % CD10+ CD27− of CD20+ B cells (Transitional B) | |
| ID91 | B2 | CD45+CD19+CD20+CD27−CD10+CD38+ | Transitional B cells | (as of viable CD45+CD19+CD20+CD27−CD10+) | Transitional B cell subset CD38+ | % CD38+ of transitional B cells | |
| ID92 | B2 | CD45+CD19+CD20+CD27−CD10+CD23+ | Transitional B cells | (as of viable CD45+CD19+CD20+CD27−CD10+) | Transitional B cell activated CD23+ | % CD23+ of transitional B cells | |
| ID93 | B2 | CD45+CD19+CD20+CD27−CD10+CD21+ | Transitional B cells | (as of viable CD45+CD19+CD20+CD27−CD10+) | Transitional B cell activated CD21+ | % CD21+ of transitional B cells | |
| ID94 | B2 | CD45+CD19+CD20+IgD−CD27+ | Mature B cells | (as of viable CD45+CD19+CD20+ (minus viable CD45+CD19+CD27−CD10+)) | Memory B cells IgD− | % IgD−CD27+ of CD20+ B cells* (IgD−CD27+ memory B) | |
| ID95 | B2 | CD45+CD19+CD20+IgD−CD27+CD23+ | Mature B cells | (as of viable CD45+CD19+CD20+IgD−CD27+ (minus viable CD45+CD19+CD27−CD10+) | Memory B cells IgD−CD23+ | % CD23+ of IgD−CD27+ memory B cells | |
| ID96 | B2 | CD45+CD19+CD20+IgD−CD27+CD38+ | Mature B cells | (as of viable CD45+CD19+CD20+IgD−CD27+ (minus viable CD45+ CD19+CD27−CD10+) | Memory B cells IgD−CD38+ | % CD38+ of IgD−CD27+ memory B cells** | |
| ID97 | B2 | CD45+CD19+CD20+IgD−CD27+CD80+ | Mature B cells | (as of viable CD45+CD19+CD20+IgD−CD27+ (minus viable CD45+CD19+CD27−CD10+) | Memory B cells IgD−CD80+ | % CD80+ of IgD−CD27+ memory B cells | |
| ID98 | B2 | CD45+CD19+CD20+IgD−CD27+CD86+ | Mature B cells | (as of viable CD45+CD19+CD20+IgD−CD27+ (minus viable CD45+CD19+CD27−CD10+) | Memory B cells IgD−CD86+ | % CD86+ of IgD−CD27+ memory B cells | |
| ID99 | B2 | CD45+CD19+CD20+IgD−CD27+IgA+ | Mature B cells | (as of viable CD45+CD19+CD20+IgD−CD27+ (minus viable CD45+CD19+CD27−CD10+) | Memory B cells IgD− IgA+ | % IgA+ of IgD−CD27+ memory B cells | |
| ID100 | B2 | CD45+CD19+CD20+IgD−CD27+IgG+ | Mature B cells | (as of viable CD45+CD19+CD20+IgD−CD27+ (minus viable CD45+CD19+CD27−CD10+) | Memory B cells IgD− IgG+ | % IgG+ of IgD−CD27+ memory B cells | |
| ID101 | B2 | CD45+CD19+CD20+IgD+CD27+ | Mature B cells | (as of viable CD45+CD19+CD20+ (minus viable CD45+CD19+CD27−CD10+) | Memory B cells IgD+ | % IgD+ CD27+ of CD20+ B cells* (IgD+ CD27+ memory B) | |
| ID102 | B2 | CD45+CD19+CD20+IgD+CD27+CD23+ | Mature B cells | (as of viable CD45+CD19+CD20+IgD+CD27+ (minus viable CD45+CD19+ CD27−CD10+) | Memory B cells IgD+CD23+ | % CD23+ of IgD+ CD27+ memory B cells | |
| ID103 | B2 | CD45+CD19+CD20+IgD+CD27+CD38+ | Mature B cells | (as of viable CD45+CD19+CD20+IgD+CD27+ (minus viable CD45+CD19+CD27−CD10+) | Memory B cells IgD+CD38+ | % CD38+of IgD+CD27+ memory B cells*** | |
| ID104 | B2 | CD45+CD19+CD20+IgD+CD27+CD80+ | Mature B cells | (as of viable CD45+CD19+CD20+IgD+CD27+ (minus viable CD45+CD19+CD27−CD10+) | Memory B cells IgD+ CD80+ | % CD80+ of IgD+ CD27+ memory B cells | |
| ID105 | B2 | CD45+CD19+CD20+IgD+CD27+CD86+ | Mature B cells | (as of viable CD45+CD19+CD20+IgD+CD27+(minus viable CD45+CD19+ CD27−CD10+) | Memory B cells IgD+CD86+ | % CD86+ of IgD+CD27+ memory B cells | |
| ID106 | B2 | CD45+CD19+CD20+IgD+CD27− | Mature B cells | (as of viable CD45+CD19+CD20+(minus viable CD45+CD19+CD27−CD10+) | Naïve B cells | % IgD+ CD27− of CD20+ B cells* (Naïve B) | |
| ID107 | B2 | CD45+CD19+CD20+IgD+CD27−CD21+ | Mature B cells | (as of viable CD45+CD19+CD20+IgD+CD27− (minus viable CD45+CD19+CD27−CD10+) | Naïve B cells activated CD21+ | % CD21+of Naïve B cells | |
| ID108 | B2 | CD45+CD19+CD20+IgD+CD27−CD38+ | Mature B cells | (as of viable CD45+CD19+CD20+IgD+CD27− (minus viable CD45+CD19+CD27−CD10+) | Naïve B cells activated CD38+ | % CD38+ of Naïve B cells | |
| ID109 | B2 | CD45+CD19+CD20+IgD+CD27−CD80+ | Mature B cells | (as of viable CD45+CD19+CD20+IgD+CD27− (minus viable CD45+CD19+CD27−CD10+) | Naïve B cells activated CD80+ | % CD80+ of Naïve B cells | |
| ID110 | B2 | CD45+CD19+CD20+IgD+CD27−CD86+ | Mature B cells | (as of viable CD45+CD19+CD20+IgD+CD27− (minus viable CD45+CD19+CD27−CD10+) | Naïve B cells activated CD86+ | % CD86+ of Naïve B cells | |
| ID111 | B2 | CD45+CD19+CD20+IgD+CD27−IgA+ | Mature B cells | (as of viable CD45+CD19+CD20+IgD+CD27− (minus viable CD45+CD19+CD27−CD10+) | Naïve B cells IgA+ | % IgA+ of Naïve B cells | |
| ID112 | B2 | CD45+CD19+CD20+IgD+CD27−IgG+ | Mature B cells | (as of viable CD45+CD19+CD20+IgD+CD27− (minus viable CD45+CD19+CD27−CD10+) | Naïve B cells IgG+ | % IgG+ of Naïve B cells | |
| ID113 | B2 | CD45+CD19+CD20+IgD−CD27− | Mature B cells | (as of viable CD45+CD19+CD20+(minus viable CD45+CD19+CD27−CD10+) | Memory CD27−B cells | % IgD−CD27− of CD20+ B cells* (IgD−CD27−memory B) | |
| ID114 | B2 | CD45+CD19+CD20+IgD−CD27−CD21+ | Mature B cells | (as of viable CD45+CD19+CD20+IgD−CD27− (minus viable CD45+CD19+CD27−CD10+) | Memory CD27−B cells CD21+ | % CD21+ of IgD−CD27−memory B cells | |
| ID115 | B2 | CD45+CD19+CD20+IgD−CD27−CD38+ | Mature B cells | (as of viable CD45+CD19+CD20+IgD−CD27− (minus viable CD45+CD19+CD27−CD10+) | Memory CD27− B cells CD38+ | % CD38+ of IgD−CD27−memory B cells | |
| ID116 | B2 | CD45+CD19+CD20+IgD−CD27−CD80+ | Mature B cells | (as of viable CD45+CD19+CD20+IgD−CD27− (minus viable CD45+CD19+CD27−CD10+) | Memory CD27−B cells CD80+ | % CD80+ of IgD−CD27−memory B cells | |
| ID117 | B2 | CD45+CD19+CD20+IgD−CD27−CD86+ | Mature B cells | (as of viable CD45+CD19+CD20+IgD−CD27− (minus viable CD45+CD19+CD27−CD10+) | Memory CD27−B cells CD86+ | % CD86+ of IgD−CD27−memory B cells | |
| ID118 | B2 | CD45+CD19+CD20+IgD−CD27−CD23+ | Mature B cells | (as of viable CD45+CD19+CD20+IgD−CD27− (minus viable CD45+CD19+CD27−CD10+) | Memory CD27−B cells CD23+ | % CD23+ of IgD−CD27−memory B cells | |
| ID119 | B2 | CD45+CD19+CD20+IgD−CD27−IgA+ | Mature B cells | (as of viable CD45+CD19+CD20+IgD−CD27− (minus viable CD45+CD19+CD27−CD10+) | Memory CD27−B cells IgA+ | % IgA+ of IgD−CD27−memory B cells | |
| ID120 | B2 | CD45+CD19+CD20+IgD−CD27−IgG+ | Mature B cells | (as of viable CD45+CD19+CD20+IgD−CD27− (minus viable CD45+CD19+CD27−CD10+) | Memory CD27−B cells IgG+ | % IgG+ of IgD−CD27−memory B cells | |
Fig. 9.

Example of gating procedure for B cell populations identified by CLIP. Single-cell suspensions from healthy donors were stained with a combination of viability dye and 14 antibodies from our B2 staining panel. Lymphocytes were identified based on their FSC-SSC properties. Dead cells were excluded through the use of a viability dye. CD45 and CD19 were used to identify B cells (CD45+CD19+) among the previously selected living lymphocytes. This figure illustrates the gating strategy that allowed us to identify 40 B cell populations
Since the initial two studies, the CLIP panel has grown to include 12 tubes allowing study of neutrophils (Fig. 10). During this time various iterations of this panel have been used for analysis of samples from a number of clinical trials, including studies of monogenic diseases, rare undiagnosed diseases, fungal infections, and coronary artery disease (Dickler et al. 2013). A database is also being constructed using the full 12-tube CLIP panel to immunophenotype peripheral blood from healthy volunteers for use as a reference for future studies.
Fig. 10.

Example of the gating procedure for neutrophils identified by CLIP. Ammonium chloride-lysed single-cell suspensions from healthy donors were stained with a combination of viability dye and 14 antibodies from our N1 staining panel. Neutrophils were identified based on their FSC-SSC properties and CD15 was used as a lineage marker. Mature or immature neutrophils were identified by CD10 versus CD64 expression, and overlay of the expression of the homing marker CD62L identified mature CD10+CD64− (red line), mature activated CD10+CD64+ (green line), and immature CD10−CD64− (blue line) neutrophils
3.3 Analysis of High-Dimensional Data
In the development of our CLIP panel, it rapidly became apparent that our ability to generate high-dimensional data exceeded our capacity to analyze the data in their entirety. Data analysis can be approached in either a ‘guided’ manner or in a ‘discovery’ mode. In the guided approach it is predetermined which leukocytes subsets will be analyzed, and gating strategies are devised based on the known expression of markers on various lineages and subpopulations. This can readily be performed manually if the number of subsets is fairly limited. In the discovery mode, a priori assumptions concerning marker expression are either ignored or limited in number, thus permitting the identification of novel, unexpected subsets. Manual analysis is of very limited use in this approach, as the numbers of potential subsets is far too great for discovery to take place in any depth.
Similar to the need for compensation software ‘wizards’ rather than manual setting of compensation for high polychromatic data, it is increasingly appreciated that computational approaches need to be used for the rigorous analysis of these data. If one assumes that all marker combinations are possible, and that marker expression is graded as only positive or negative, immunophenotyping with 14 antibodies could define over 8,000 leukocyte subsets. A panel of 12 tubes of 14 antibodies each could define nearly 100,000 subsets if there were no redundancies. Clearly we do not have the capacity to readily elucidate these subsets using traditional manual gating and analysis. Thus any serious attempt to enter the realm of comprehensive, high-dimensional immunophenotyping of the immunome must include automated approaches to define phenotypes in multidimensional space.
A number of computational approaches have been published for automation of the analysis of flow cytometric data and for better understanding and presentation of high-dimensional data (Pyne et al. 2009; Rogers and Holyst 2009; Qian et al. 2010; Bagwell 2011; Dabdoub et al. 2011; Qiu et al. 2011; Aghaeepour et al. 2012; Candia in press). Most notable are the efforts of FlowCAP (Flow Cytometry Critical Assessment of Population Identification Methods), a consortium of investigators working together to advance the development of computational models for the analysis of flow cytometry data (Aghaeepour et al. 2013; Biehl et al. 2013). Such approaches are necessary not only for data mining of high-dimensional data, but also to remove the subjectivity inherent to manual gating to increase the reproducibility of these complex assays. As is discussed in a separate chapter of this edition, these approaches for automated analysis are rapidly evolving and show great promise. One recent study, using data derived during the development of our CLIP panel, demonstrated the ability to accurately classify two autoimmune diseases from a 16-parameter (14-color) immunophenotyping of peripheral blood from these patients (Candia in press). Perhaps the biggest obstacle to development or acceptance of these automated approaches is the need to compare them during the development phase to existing ‘gold standards’ for routine analyses. Unfortunately, the gold standard in this instance is manual gating of cellular populations—a process that is fraught with subjectivity, and hence great variation.
4 Future Directions and Summary
Clearly, any attempt to immunophenotype the entire immunome simultaneously remains in its infancy. One can think of nearly endless possibilities to better characterize the immune system, including pursuits such as studying signaling pathways in various lineages in response to stimuli. Furthermore, new markers are continually being discovered that identify new cell subpopulations or that correlate with specific cellular functions. Thus, immunophenotyping of the immune will be a constantly evolving process due to both biological as well as technical innovations. Nonetheless, it is important to begin efforts toward this pan-immunophenotyping as these data will prove to be a valuable resource, and lessons learned from these early steps will help to guide future efforts.
Since the inception of our CLIP panel, modifications made for biological and technical reasons. Our initial panel relied on the use of quantum dot (QDot) conjugates for three antibodies (QD605, QD655, and QD800) in each tube. QDots permitted substantial use of the violet laser (406 nm) for increasing the number of detectable parameters in the CLIP panel and are widely used in multicolor cytometry. They are generally more stable than tandem conjugates, allowing good reproducibility. Unfortunately, QDot conjugates containing unbound particles can interact with each other to form aggregates—a problem that increases as the reagents age. These aggregates appear with dual fluorescence from each QDot spectrum and can make determination of proper compensation extremely difficult. The presence of these aggregates can be minimized by subjecting the antibody dilutions to a quick high-speed spin, preventing the non-specific labeling of some cells (Chattopadhyay et al. 2006, 2010), or by gating out obvious aggregates. The new generation of Brilliant Violet (BV) stains (π-conjugated polymers) has proven to be a practical alternative to QDots as BV stains have high quantum yields, good stability, and are available in emission spectra similar to those of QDots (Chattopadhyay et al. 2012). BV-conjugated antibodies have now replaced the QDots in our CLIP panel and have completely eliminated compensation problems that arose from QDots aggregates.
Changes have also been made to the original panel based on marker discovery. For example, CD146 (MCAM) was recently identified as a marker of committed Th17 cells in humans. CD146 was introduced into the Th17 tube as a replacement for IL-21, which had produced rather uninformative data (Dagur et al. 2011). CD146 appears to be involved in the extravasation and migration of these cells, and data suggest that Th17 cells are important in a number of autoimmune diseases (Amadi-Obi et al. 2007; Kebir et al. 2007; Brucklacher-Waldert et al. 2009; Kleinschek et al. 2009; Dagur et al. 2011). In one tube CD45-RA was replaced with CD45-RO because of dim staining of the former when used in conjunction with a specific fixation and permeabilization procedure. Additional changes are also under study.
It is clear that the comprehensiveness of our current panel is limited and does not begin to cover the entire immunome. A vast array of additional features and markers could be studied, and there remains a need to add tubes designed to study additional cell types such as T follicular helper cells and peripheral blood progenitor cells. Our approach has been to design and implement one tube at a time and then to integrate it into the larger CLIP panel after validation. Development is anticipated to continue in this manner. It is also entirely possible that the design of the CLIP panel could be changed in order to add more parameters. Hardware improvements such as the spectral flow cytometry together with the development of novel fluorochromes with tight emission spectra would permit the practical examination of more fluorescent probes in a single tube and thus make this panel more comprehensive.
The original CLIP panel was designed for use on specimens from clinical trials, the overwhelming majority of which are cryopreserved and thus contain only mononuclear cells. Thus the initial panel is focused on mononuclear cell subsets to the exclusion of others. More recently, the CLIP panel has become truly more comprehensive by inclusion of two tubes designed specifically for granulocytic cells. Expansion of the CLIP assay to whole blood is necessary but will be accompanied by the need to perform studies ‘on demand’. This consideration, together with the need for a high degree of reproducibility over time in these studies, makes a strong argument against the manual preparation of the antibody cocktails each time the CLIP phenotyping is to be performed. To circumvent this necessity, there are three options: (1) to make use of automated devices for antibody dispensing, (2) to make large batches of antibody cocktails, and (3) to lyophilized pre-titered antibody cocktails. As automated liquid handlers tend to be expensive, require laboratory space, and are subject to period breakdown, the first approach would be our least favored alternative. The most attractive option is lyophilization of the panel. This approach has been pioneered as a means of widespread standardization of immunophenotyping for clinical trial samples and offers significant advantages in terms of reagent stability, storage, and ease of use (Maecker et al. 2012). A previous attempt to lyophilize our CLIP panel did not prove successful, exclusively, due to the failure of the QDots to maintain their performance characteristics after lyophilization. The replacement of QDots with the BV stains in the current iteration of our panel may make lyophilization of these reagents feasible. A remaining obstacle to this process is the intracellular staining of cytokines that requires cells to be fixed and permeabilized prior to staining. To address this, either the lyophilized reagent cocktail would need to be limited to only the cell surface markers or methods would need to be devised for permeabilization of the cells prior to adding them to the antibody cocktail.
As mentioned previously, one of the tremendous advantages of using fluorescence-based high-dimensional immunophenotyping rather than the time-of-flight cytometry is the ability to recover any subsets identified for function analyses. As discussed above, the current CLIP panel can be used to purify most subsets identified, although modifications may be needed for those populations identified by secretion of cytokines (Fitzgerald and Grivel 2013). With the imminent possibilities for lyophilization of pre-titered, premixed reagents and computational methods for setting compensation as well as for data analysis, performance of high polychromatic flow cytometry will become a practical approach for the comprehensive immunophenotyping of the immune system. In addition to permitting a rapid, in-depth study of the immunome, this approach should also prove to be robust with low variance over time. Such work, when combined with ex vivo functional studies of novel populations and other data such as gene expression studies, antibody titers, Elispot data, serum cytokine analysis, epigenetic studies, and proteomic stand to revolutionize our understanding of human immunology.
Contributor Information
Angélique Biancotto, Center for Human Immunology, Autoimmunity, and Inflammation, National Institutes of Health, Bethesda, MD 20892, USA.
J. Philip McCoy, Email: mccoyj@nhlbi.nih.gov, Center for Human Immunology, Autoimmunity, and Inflammation, National Institutes of Health, 10 Center Dr, MSC 1357 Bldg 10, Rm 8C103D, Bethesda, MD 20892, USA.
References
- Aghaeepour N, Finak G, Flow CAPC, Consortium D, Hoos H, Mosmann TR, Brinkman R, Gottardo R, Scheuermann RH. Critical assessment of automated flow cytometry data analysis techniques. Nat Methods. 2013;10:228–238. doi: 10.1038/nmeth.2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aghaeepour N, Jalali A, O’Neill K, Chattopadhyay PK, Roederer M, Hoos HH, Brinkman RR. RchyOptimyx: cellular hierarchy optimization for flow cytometry. Cytometry A. 2012;81:1022–1030. doi: 10.1002/cyto.a.22209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amadi-Obi A, Yu CR, Liu X, Mahdi RM, Clarke GL, Nussenblatt RB, Gery I, Lee YS, Egwuagu CE. TH17 cells contribute to uveitis and scleritis and are expanded by IL-2 and inhibited by IL-27/STAT1. Nat medicine. 2007;13:711–718. doi: 10.1038/nm1585. [DOI] [PubMed] [Google Scholar]
- Bagwell CB. Breaking the dimensionality barrier. Methods Mol Biol. 2011;699:31–51. doi: 10.1007/978-1-61737-950-5_2. [DOI] [PubMed] [Google Scholar]
- Bendall SC, Nolan GP, Roederer M, Chattopadhyay PK. A deep profiler’s guide to cytometry. Trends Immunol. 2012;33:323–332. doi: 10.1016/j.it.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyer M, Kochanek M, Darabi K, Popov A, Jensen M, Endl E, Knolle PA, Thomas RK, von Bergwelt-Baildon M, Debey S, Hallek M, Schultze JL. Reduced frequencies and suppressive function of CD4 + CD25hi regulatory T cells in patients with chronic lymphocytic leukemia after therapy with fludarabine. Blood. 2005;106:2018–2025. doi: 10.1182/blood-2005-02-0642. [DOI] [PubMed] [Google Scholar]
- Biancotto A, Dagur PK, Fuchs JC, Langweiler M, McCoy JP., Jr OMIP-004: in-depth characterization of human T regulatory cells. Cytometry A. 2012a;81:15–16. doi: 10.1002/cyto.a.21158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biancotto A, Dagur PK, Fuchs JC, Wiestner A, Bagwell CB, McCoy JP., Jr Phenotypic complexity of T regulatory subsets in patients with B-chronic lymphocytic leukemia. Mod Pathol. 2012b;25:246–259. doi: 10.1038/modpathol.2011.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biancotto A, Fuchs JC, Williams A, Dagur PK, McCoy JP., Jr High dimensional flow cytometry for comprehensive leukocyte immunophenotyping (CLIP) in translational research. J Immunol Methods. 2011;363:245–261. doi: 10.1016/j.jim.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biehl M, Bunte K, Schneider P. Analysis of flow cytometry data by matrix relevance learning vector quantization. PLoS ONE. 2013;8:e59401. doi: 10.1371/journal.pone.0059401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brucklacher-Waldert V, Stuerner K, Kolster M, Wolthausen J, Tolosa E. Phenotypical and functional characterization of T helper 17 cells in multiple sclerosis. Brain. 2009;132:3329–3341. doi: 10.1093/brain/awp289. [DOI] [PubMed] [Google Scholar]
- Candia JMR, Driscoll M, Biancotto A, Dagur P, McCoy JP, Sen HN, Wei L, Maritan A, Cao K, Nussenblatt RB, Banavar JR, Losert W. From cellular characteristics to disease diagnosis: uncovering phenotypes with supercells. PLOS Comput Biol. doi: 10.1371/journal.pcbi.1003215. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyay PK, Gaylord B, Palmer A, Jiang N, Raven MA, Lewis G, Reuter MA, Nur-ur Rahman AK, Price DA, Betts MR, Roederer M. Brilliant violet fluorophores: a new class of ultrabright fluorescent compounds for immunofluorescence experiments. Cytometry A. 2012;81:456–466. doi: 10.1002/cyto.a.22043. [DOI] [PubMed] [Google Scholar]
- Chattopadhyay PK, Perfetto SP, Yu J, Roederer M. The use of quantum dot nanocrystals in multicolor flow cytometry. Nanomed nanobiotechnol. 2010;2:334–348. doi: 10.1002/wnan.75. [DOI] [PubMed] [Google Scholar]
- Chattopadhyay PK, Price DA, Harper TF, Betts MR, Yu J, Gostick E, Perfetto SP, Goepfert P, Koup RA, De Rosa SC, Bruchez MP, Roederer M. Quantum dot semiconductor nanocrystals for immunophenotyping by polychromatic flow cytometry. Nat Medicine. 2006;12:972–977. doi: 10.1038/nm1371. [DOI] [PubMed] [Google Scholar]
- D’Arena G, Laurenti L, Minervini MM, Deaglio S, Bonello L, De Martino L, De Padua L, Savino L, Tarnani M, De Feo V, Cascavilla N. Regulatory T-cell number is increased in chronic lymphocytic leukemia patients and correlates with progressive disease. Leuk Res. 2011;35:363–368. doi: 10.1016/j.leukres.2010.08.010. [DOI] [PubMed] [Google Scholar]
- Dabdoub SM, Ray WC, Justice SS. FIND: a new software tool and development platform for enhanced multicolor flow analysis. BMC Bioinformatics. 2011;12:145. doi: 10.1186/1471-2105-12-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagur PK, Biancotto A, Wei L, Sen HN, Yao M, Strober W, Nussenblatt RB, McCoy JP., Jr MCAM-expressing CD4(+) T cells in peripheral blood secrete IL-17A and are significantly elevated in inflammatory autoimmune diseases. J Autoimmun. 2011;37:319–327. doi: 10.1016/j.jaut.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MM. A prescription for human immunology. Immunity. 2008;29:835–838. doi: 10.1016/j.immuni.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MM. Immunology taught by humans. Sci transl med. 2012;4:117f, s112. doi: 10.1126/scitranslmed.3003385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch V, Perry C, Polliack A. Expansion of regulatory T cells in B chronic lymphocytic leukemia: enhanced ‘brakes’ on host immunity. Leuk Lymphoma. 2009;50:687–688. doi: 10.1080/10428190902954389. [DOI] [PubMed] [Google Scholar]
- Dickler HB, McCoy JP, Nussenblatt R, Perl S, Schwartzberg PA, Tsang JS, Wang E, Young NS. The national institutes of health center for human immunology, autoimmunity, and inflammation: history and progress. Ann N Y Acad Sci. 2013;1285:133–147. doi: 10.1111/nyas.12101. [DOI] [PubMed] [Google Scholar]
- Fitzgerald W, Grivel JC. A universal nanoparticle cell secretion capture assay. Cytometry A. 2013;83:205–211. doi: 10.1002/cyto.a.22199. [DOI] [PubMed] [Google Scholar]
- Giannopoulos K, Schmitt M, Kowal M, Wlasiuk P, Bojarska-Junak A, Chen J, Rolinski J, Dmoszynska A. Characterization of regulatory T cells in patients with B-cell chronic lymphocytic leukemia. Oncol Rep. 2008;20:677–682. [PubMed] [Google Scholar]
- Jak M, Mous R, Remmerswaal EB, Spijker R, Jaspers A, Yague A, Eldering E, Van Lier RA, Van Oers MH. Enhanced formation and survival of CD4 + CD25hi Foxp3 + T-cells in chronic lymphocytic leukemia. Leuk Lymphoma. 2009;50:788–801. doi: 10.1080/10428190902803677. [DOI] [PubMed] [Google Scholar]
- Kebir H, Kreymborg K, Ifergan I, Dodelet-Devillers A, Cayrol R, Bernard M, Giuliani F, Arbour N, Becher B, Prat A. Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat Medicine. 2007;13:1173–1175. doi: 10.1038/nm1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinschek MA, Boniface K, Sadekova S, Grein J, Murphy EE, Turner SP, Raskin L, Desai B, Faubion WA, de Waal Malefyt R, Pierce RH, McClanahan T, Kastelein RA. Circulating and gut-resident human Th17 cells express CD161 and promote intestinal inflammation. J Exp Med. 2009;206:525–534. doi: 10.1084/jem.20081712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maecker HT, McCoy JP, Nussenblatt R. Standardizing immunophenotyping for the Human Immunology Project. Nat Rev Immunol. 2012;12:191–200. doi: 10.1038/nri3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahnke YD, Roederer M. Optimizing a multicolor immunophenotyping assay. Clin lab med. 2007;27:469–485. doi: 10.1016/j.cll.2007.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy JP, Jr, Overton WR. Quality control in flow cytometry for diagnostic pathology: II. a conspectus of reference ranges for lymphocyte immunophenotyping. Cytometry. 1994;18:129–139. doi: 10.1002/cyto.990180304. [DOI] [PubMed] [Google Scholar]
- Perfetto SP, Ambrozak D, Nguyen R, Chattopadhyay P, Roederer M. Quality assurance for polychromatic flow cytometry. Nat Protocols. 2006;1:1522–1530. doi: 10.1038/nprot.2006.250. [DOI] [PubMed] [Google Scholar]
- Perfetto SP, Ambrozak D, Nguyen R, Chattopadhyay PK, Roederer M. Quality assurance for polychromatic flow cytometry using a suite of calibration beads. Nat Protocols. 2012;7:2067–2079. doi: 10.1038/nprot.2012.126. [DOI] [PubMed] [Google Scholar]
- Perfetto SP, Chattopadhyay PK, Lamoreaux L, Nguyen R, Ambrozak D, Koup RA, Roederer M. Amine-reactive dyes for dead cell discrimination in fixed samples. Curr Protoc Cytom. 2010;Chapter 9(Unit 9.34) doi: 10.1002/0471142956.cy0934s53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perfetto SP, Chattopadhyay PK, Roederer M. Seventeen-colour flow cytometry: unravelling the immune system. Nat Rev Immunol. 2004;4:648–655. doi: 10.1038/nri1416. [DOI] [PubMed] [Google Scholar]
- Pyne S, Hu X, Wang K, Rossin E, Lin TI, Maier LM, Baecher-Allan C, McLachlan GJ, Tamayo P, Hafler DA, De Jager PL, Mesirov JP. Automated high-dimensional flow cytometric data analysis. PNAS. 2009;106:8519–8524. doi: 10.1073/pnas.0903028106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Y, Wei C, Eun-Hyung Lee F, Campbell J, Halliley J, Lee JA, Cai J, Kong YM, Sadat E, Thomson E, Dunn P, Seegmiller AC, Karandikar NJ, Tipton CM, Mosmann T, Sanz I, Scheuermann RH. Elucidation of seventeen human peripheral blood B-cell subsets and quantification of the tetanus response using a density-based method for the automated identification of cell populations in multidimensional flow cytometry data. Cytometry B Clin cytom. 2010;78(1):S69–82. doi: 10.1002/cyto.b.20554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu P, Simonds EF, Bendall SC, Gibbs KD, Jr, Bruggner RV, Linderman MD, Sachs K, Nolan GP, Plevritis SK. Extracting a cellular hierarchy from high-dimensional cytometry data with SPADE. Nat Biotechnology. 2011;29:886–891. doi: 10.1038/nbt.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roederer M. Compensation in flow cytometry. Curr protoc cytom. 2002;Chapter 1(Unit 1.14) doi: 10.1002/0471142956.cy0114s22. [DOI] [PubMed] [Google Scholar]
- Rogers WT, Holyst HA. FlowFP: a bioconductor package for fingerprinting flow cytometric data. Adv bioinform. 2009;193947:11. doi: 10.1155/2009/193947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sony. Sony announces the development of the ‘Spectral’ cell analyzer. sony; 2013. http://www.sony.net/SonyInfo/News/Press/201206/12-082E/index.html. [Google Scholar]
- Tsang JS, Schwartzberg PL, Kotliarov Y, Biancotto A, Xie Z, Wang EMO, Narayanan M, Cheung F, Golding H, Moir S, Ho J, Khurana S, Shum P, Perl S, Fuchs JC, Langweiler M, Nussenblatt R, Kastner D, Trinchieri G, Marincola F, Young NS, Dickler H, McCoy JP, Germain RNJST. Global analyses of human immune states utilizing natural variations and vaccine perturbation reveals baseline predictors of the post-vaccination response. Cell. 2013 submitted. [Google Scholar]