

Abstract
Summary: Efficient workflows to shepherd clinically generated genomic data through the multiple stages of a next-generation sequencing pipeline are of critical importance in translational biomedical science. Here we present COSMOS, a Python library for workflow management that allows formal description of pipelines and partitioning of jobs. In addition, it includes a user interface for tracking the progress of jobs, abstraction of the queuing system and fine-grained control over the workflow. Workflows can be created on traditional computing clusters as well as cloud-based services.
Availability and implementation: Source code is available for academic non-commercial research purposes. Links to code and documentation are provided at http://lpm.hms.harvard.edu and http://wall-lab.stanford.edu.
Contact: dpwall@stanford.edu or peter_tonellato@hms.harvard.edu.
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
The growing deluge of data from next-generation sequencers leads to analyses lasting hundreds or thousands of compute hours per specimen, requiring massive computing clusters or cloud infrastructure. Existing computational tools like Pegasus (Deelman et al., 2005) and more recent efforts like Galaxy (Goecks et al., 2010) and Bpipe (Sadedin et al., 2012) allow the creation and execution of complex workflows. However, few projects have succeeded in describing complicated workflows in a simple, but powerful, language that generalizes to thousands of input files; fewer still are able to deploy workflows onto distributed resource management systems (DRMs) such as Platform Load Sharing Facility (LSF) or Sun Grid Engine that stitch together clusters of thousands of compute cores. Here we describe COSMOS, a Python library developed to address these and other needs.
2 FEATURES AND METHODS
An essential challenge for a workflow definition language is to separate the definition of tools (which represent individual analyses) from the definition of the dependencies between them. Several workflow libraries require each tool to expect specifically named input files and produce similarly specific output files; however, in COSMOS, tool I/O is instead controlled by specifying file types. For example, the BWA alignment tool (Fig. 1a) can expect FASTQ-typed inputs and produce a SAM-typed output, but does not depend on any specific file names or locations. Additionally, tool definitions do not require knowledge of the controlling DRM.
Fig. 1.
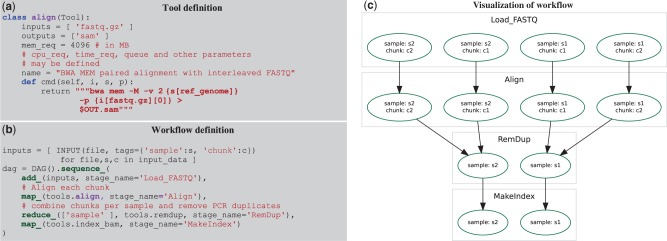
(a) Tools are defined in COSMOS by specifying input and output types, not files, and a cmd() function returning a string to be executed in a shell. cpu_req and other parameters may be inspected by a programmer-defined Python function to set DRM parameters or redirect jobs to queues. (b) Workflows are defined using map-reduce primitives: sequence_, map_ (execute the align tool from (a) on each ‘chunk’ in parallel) and reduce_ (group the aligned outputs by sample tag). (c) Directed acyclic graph of jobs generated by the workflow in (b) to be executed via the DRM for four input FASTQ files (with sample tags s1 and s2, and chunk tags of c1 and c2)
Once tools have been defined, their dependencies can be formalized via a COSMOS workflow, which is defined using Python functions that support the map-reduce paradigm (Dean and Ghemawat, 2004) (Fig. 1b). Sequential workflows are defined primarily by the sequence_ primitive, which runs tools in series. The apply_ primitive is provided to describe workflows with potentially unrelated branching by executing tools in parallel. To facilitate map-reduce in large and branching workflows, COSMOS introduces a tagging system that associates a set of key-value tags (e.g. a sample ID, chunk ID, sequencer ID or other job parameter) with specific job instances. This tagging feature enables users to formalize reductions over existing tag sets or to split by creating new combinations of tags (Supplementary Fig. S1). To execute a workflow, COSMOS creates a directed acyclic graph (DAG) of tool dependencies at runtime (Fig. 1c) and automatically links the inputs and outputs between tools by recognizing file extensions as types. All file paths generated by tool connections are managed by COSMOS, automatically assigning intermediate file names.
Another major challenge in workflow management is execution on large compute clusters, where transient errors are commonplace and must be handled gracefully. If errors cannot be automatically resolved, the framework should record exactly which jobs have failed and allow the restart of an analysis after error resolution. COSMOS uses the DRMAA library (Troger et al. 2007) to manage job submission, status checking and error handling. DRMAA supports most DRM platforms, including Condor, although our efforts used LSF and Sun Grid Engine. Users may control DRM submission parameters by overriding a Python function that is called on every job control event. COSMOS’ internal data structures are stored in an SQL database using the Django framework (https://djangoproject.com) and is distributed with a Web application for monitoring the state of both running and completed workflows, querying individual job states, visualizing DAGs and debugging failed jobs (Supplementary Figs S2–S5).
Each COSMOS job is continuously monitored for resource usage, and a summary of these statistics and standard output and error streams are stored in the database. This allows users to fine-tune estimated DRM parameters such as CPU and memory usage for more efficient cluster usage. Pipeline restarts are also facilitated by the persistent database, as it records both success and failure using job exit codes.
3 COMPARISON AND DISCUSSION
Projects such as Galaxy and Taverna (Wolstencroft et al., 2013) are aimed at users without programming expertize and offer graphical user interfaces (GUIs) to create workflows, but come at the expense of power. For example, it is straightforward to describe task dependencies in Galaxy’s drag-and-drop workflow creator; however, to parallelize alignment by breaking the input FASTQ into several smaller chunks to be aligned independently, input stages must be manually created for each chunk or the workflow must be applied to each chunk manually. In addition, the user must fix the number of input chunks a priori. COSMOS resolves this tedious process for the programmer by dynamically building its DAG at runtime.
Such limitations may not be a major concern for small-scale experiments where massive parallelization to reduce runtime is not critical; however, when regularly analyzing terabytes of raw data, the logistics of parallelization and job management play a central role. Snakemake (Köster and Rahmann, 2012) looks to the proven design of GNU Make to describe DAGs for complicated workflows, whereas the Ruffus project (Goodstadt, 2010) aims to create a DAG by providing a library of Python decorators. However, neither of these projects directly supports integration with a DRM. The Pegasus system offers excellent integration with DRMs and even the assembly of several independent DRMs using the Globus software; however, the description of some simple workflows can require considerably more code than the equivalent COSMOS code (Supplementary Fig. S6), and the DAG is not determined at runtime, so cannot depend on the input. Bpipe offers an elegant syntax for defining the DAG, but does not include a graphic user interface for monitoring and runtime statistics. Additionally, COSMOS’ persistent database and Web front end allow rapid diagnosis of errors in data input or workflow execution (see Supplementary Table S1 for a detailed feature comparison). COSMOS has been tested on the Ubuntu, Debian and Fedora Linux distributions. The only dependency is Python 2.6 or newer and the ability to install Python packages; we recommend a DRMAA-compatible DRM for intensive workloads.
Funding: This work was supported by the National Institutes of Health [1R01MH090611-01A1 to D.P.W, 1R01LM011566 to P.J.T., and 5T15LM007092 to P.J.T. and J.B.H.]; and a Fulbright Fellowship [to Y.S.].
Conflict of Interest: L.J.L. is also an employee with Claritas Genomics Inc., a licensee of COSMOS.
Supplementary Material
REFERENCES
- Dean J, Ghemawat S. Proceedings of the 6th Conference on Symposium on Operating Systems Design & Implementation. Berkeley, CA: USENIX Association; 2004. MapReduce: simplified data processing on large clusters; p. 10. [Google Scholar]
- Deelman E, et al. Pegasus: A framework for mapping complex scientific workflows onto distributed systems. Sci. Program. 2005;13:219–237. [Google Scholar]
- Goecks J, et al. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010;11:R86. doi: 10.1186/gb-2010-11-8-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodstadt L. Ruffus: a lightweight Python library for computational pipelines. Bioinformatics. 2010;26:2778–2779. doi: 10.1093/bioinformatics/btq524. [DOI] [PubMed] [Google Scholar]
- Köster J, Rahmann S. Snakemake—a scalable bioinformatics workflow engine. Bioinformatics. 2012;28:2520–2522. doi: 10.1093/bioinformatics/bts480. [DOI] [PubMed] [Google Scholar]
- Sadedin SP, et al. Bpipe: a tool for running and managing bioinformatics pipelines. Bioinformatics. 2012;28:1525–1526. doi: 10.1093/bioinformatics/bts167. [DOI] [PubMed] [Google Scholar]
- Troger P, et al. Seventh IEEE International Symposium on Cluster Computing and the Grid. Brazil: IEEE, Rio De Janeiro; 2007. Standardization of an API for distributed resource management systems; pp. 619–626. [Google Scholar]
- Wolstencroft K, et al. The Taverna workflow suite: designing and executing workflows of Web Services on the desktop, Web or in the cloud. Nucleic Acids Res. 2013;41:W557–W561. doi: 10.1093/nar/gkt328. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.