

Abstract
Primary pigmented nodular adrenocortical disease (PPNAD) is associated with inactivating mutations of the PRKAR1A tumor suppressor gene that encodes the regulatory subunit R1α of the cAMP-dependent protein kinase (PKA). In human and mouse adrenocortical cells, these mutations lead to increased PKA activity, which results in increased resistance to apoptosis that contributes to the tumorigenic process. We used in vitro and in vivo models to investigate the possibility of a crosstalk between PKA and mammalian target of rapamycin (mTOR) pathways in adrenocortical cells and its possible involvement in apoptosis resistance. Impact of PKA signaling on activation of the mTOR pathway and apoptosis was measured in a mouse model of PPNAD (AdKO mice), in human and mouse adrenocortical cell lines in response to pharmacological inhibitors and in PPNAD tissues by immunohistochemistry. AdKO mice showed increased mTOR complex 1 (mTORC1) pathway activity. Inhibition of mTORC1 by rapamycin restored sensitivity of adrenocortical cells to apoptosis in AdKO but not in wild-type mice. In both cell lines and mouse adrenals, rapid phosphorylation of mTORC1 targets including BAD proapoptotic protein was observed in response to PKA activation. Accordingly, BAD hyperphosphorylation, which inhibits its proapoptotic activity, was increased in both AdKO mouse adrenals and human PPNAD tissues. In conclusion, mTORC1 pathway is activated by PKA signaling in human and mouse adrenocortical cells, leading to increased cell survival, which is correlated with BAD hyperphosphorylation. These alterations could be causative of tumor formation.
INTRODUCTION
Primary pigmented nodular adrenal disease (PPNAD) is a benign tumor leading to pituitary-independent hypercortisolism and Cushing's syndrome (1). It is mostly detected as part of Carney complex (CNC), a rare familial disease, in which development of multiple endocrine tumors induces endocrine overactivity and significant morbidity (2). Either isolated or as part of CNC, this bilateral micronodular adrenocortical hyperplasia is genetically associated with deregulations in cAMP/PKA pathway. Indeed, inactivating mutations in the PRKAR1A gene that encodes the type 1α regulatory subunit of the PKA (R1α) (3,4) and/or mutations in the PDE-11A and -8B genes that encode phosphodiesterases (5,6) are found in most cases of PPNAD. In the adrenal, these mutations induce constitutive activation of the cAMP/PKA pathway, resulting in hypercortisolism and adrenal tumor formation in human as well as in mice bearing Prkar1a down-regulation (4,7). Although glucocorticoid synthesis mostly depends on a direct control by the cAMP/PKA pathway (8), PKA-dependent mechanisms involved in the deregulation of cell proliferation and/or survival leading to PPNAD remain poorly identified. A better knowledge of these events could provide new targets to inhibit the tumorigenic process and/or the endocrine overactivity.
We have previously observed that both human H295R cells in culture (9) and mouse adrenocortical cells in vivo (AdKO mice) (7), showed increased resistance to apoptosis in response to R1α deficiency. In AdKO mice (for Adrenal cortex-specific Prkar1a knockout mice), this resistance coincided with the resurgence and/or incomplete clearance of the fetal X-zone that normally regresses by apoptosis. These cells with fetal characteristics that progressively invaded the entire adult cortex were proposed to be the cause of the tumorigenic process induced by R1α loss. We thus concluded that resistance to apoptosis was a major mechanism for the development of adrenal tumors in response to constitutive PKA activation.
Resistance to apoptosis in adrenocortical cells has already been associated with repression of the transforming growth factor β (TGFβ) pathway, which in vivo is implicated in the clearance of the X-zone in mouse and of the fetal zone in human (10,11). Indeed, down-regulation of PRKAR1A in H295R cells induces repression of SMAD3 that encodes a key element of the TGFβ pathway, leading to repression of this pathway and to increased cellular resistance to apoptosis (9). Moreover, inhibin-α, a negative regulator of the TGFβ pathway is overexpressed specifically in the nodular tumors of PPNAD tissues and in the adrenals of the AdKO mouse model (7). In adrenal tissues, this correlates with TGFβ pathway inhibition and subsequent tumorigenesis.
However, other pathways could also be involved in resistance to apoptosis. Recently, PKA was shown to directly activate the mTOR pathway in thyroid and ovarian cell cultures in vitro (12,13). In human PPNAD tissues, depletion of R1α protein coincides with higher mTOR activation. Furthermore, tumoral nodules are formed of hypertrophic cells that accumulate lipofuscin, indicating a decrease in autophagy typical of mTOR activation (14). Consistent with an increase in mTOR activity the fetal-like tumor cells in AdKO mice are also hypertrophic (7).
Considering the role of mTOR pathway in both cell growth and survival (15), we hypothesized that mTOR activation by PKA could play a major role in PKA-dependent micronodular adrenocortical hyperplasia. Therefore, we have investigated the impact of increased PKA signaling on mTOR pathway activity in both adrenocortical cell lines and AdKO mice. We have also tested whether mTORC1 pathway inhibition could restore sensitivity to apoptosis in the AdKO PPNAD mouse model. Finally, we have correlated the crosstalk between PKA and mTOR to hyperphosphorylation of the BAD protein in adrenocortical cells, in mice and in patients. Our results suggest that regulation of the proapoptotic activity of BAD plays a key role in the pathogenic mechanisms resulting in PPNAD tumor formation.
RESULTS
Analysis of mTOR pathway activity in AdKO mice
We previously showed that 12–14 month-old AdKO mice exhibited increased PKA activity and adrenocorticotropic hormone (ACTH)-independent Cushing's syndrome (7). At that age, we assessed the activity of three downstream targets of mTOR. Western blot analyses showed that p70 ribosomal protein S6 kinase 1 (p70S6K1) (2-fold), ribosomal protein S6 (rpS6) (1.5-fold) and eIF4E-binding protein 1 (4E-BP1) (3.1-fold) were all hyperphosphorylated in AdKO adrenal extracts when compared with wild-type (WT) (P < 0.05, Fig. 1A and B). In contrast, AKT phosphorylation was not significantly affected in AdKO adrenals. This suggested that activity of the upstream members of the phosphoinositide 3-kinase/AKT/mTOR (PI3-K/AKT/mTOR) pathway was not altered in response to Prkar1a ablation. We then examined the effect of specific inhibition of mTORC1 by rapamycin treatment on the phosphorylation of rpS6 in situ (P-rpS6). In the control conditions (vehicle), WT and AdKO adrenal sections showed P-rpS6 staining throughout medulla and cortex (Fig. 1Ca). In AdKO mice, a slightly stronger staining was specifically detected in the hypertrophic cells of the tumor lesion when compared with the adjacent normal cortex (Fig. 1Cb). Rapamycin treatment resulted in a complete abolition of P-rpS6 cortical staining in WT mice (Fig. 1Cc). In contrast, P-rpS6 staining was preserved within tumoral cells of the inner cortex in AdKO mice (Fig. 1Cd). Altogether these experiments showed that mTORC1 signaling was activated downstream of PI3-K in AdKO adrenal tumors, which resulted in rapamycin-resistant rpS6 phosphorylation.
Figure 1.
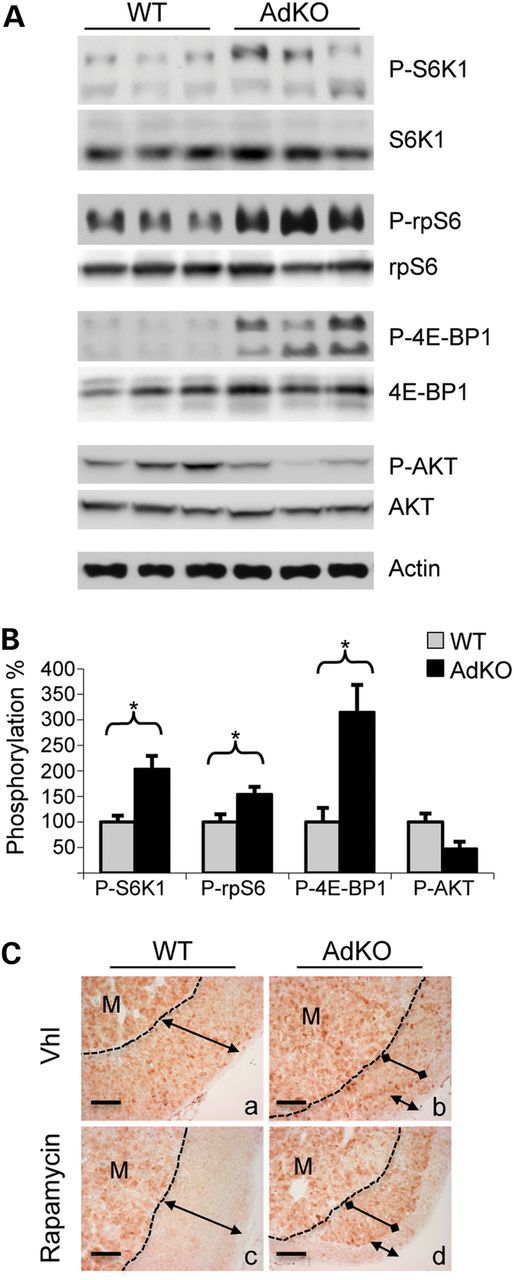
Phosphorylation of AKT/mTOR pathway proteins in adrenals of WT and AdKO mice. (A) Phosphorylated S6K1, rpS6, 4E-BP1 and AKT were detected in adrenal tissues from 12 to 14 month-old WT and AdKO mice. (B) Phosphorylation signal illustrated in A was quantified in five to six samples (values ± SEM). Values represent relative band density of the phosphorylated forms over total signal of the corresponding proteins, expressed as a percentage of the mean of WT. *P < 0.05. (C) Immunostaining for P-rpS6 in adrenal sections of vehicle-treated (Vhl) WT (a) and AdKO (b) compared with rapamycin-treated WT (c) and AdKO (d) mice. Dotted line delineates medullary (M) and cortical compartments, normal cortex (double headed arrow) and tumoral inner cortex (double dotted arrow) are indicated. Scale bars, 100 µm.
Effect of rapamycin treatment on apoptosis in WT and AdKO mice
We then treated WT and AdKO mice with dexamethasone to induce apoptosis specifically in adrenocortical cells (16). Apoptotic figures were quantified in WT and AdKO adrenal sections after TUNEL (Fig. 2A and B) and cleaved-caspase 3 stainings (data not shown and Fig. 2C). As previously described (7), the number of apoptotic cells was significantly higher in WT than in AdKO cortex, for both TUNEL (109.8 ± 9.2 versus 20.2 ± 2.3, P = 0.002) and cleaved-caspase 3 stainings (135.4 ± 19.6 versus 32.1 ± 4.9, P = 0.004). Rapamycin treatment alone had no significant effect on basal apoptosis in both genotypes (Fig. 2C), or on dexamethasone-induced apoptosis in WT mice (Fig. 2B, 109.8 ± 9.2 versus 122.4 ± 16.6, P = 0.56 and Fig. 2C). In contrast, the number of apoptotic cells per adrenal section was significantly increased in AdKO mice receiving rapamycin and dexamethasone simultaneously (Fig. 2B, 20.2 ± 2.3 versus 83.8 ± 16.6, P = 0.026 and Fig. 2C). Moreover, under these conditions, differences in cell apoptosis sensitivity between WT and AdKO genotypes were abolished (Fig. 2B, 122.4 ± 16.6 versus 83.8 ± 16.6, P = 0.17 and Fig. 2C). We concluded that rapamycin treatment specifically sensitized AdKO adrenocortical cells to dexamethasone-induced apoptosis.
Figure 2.
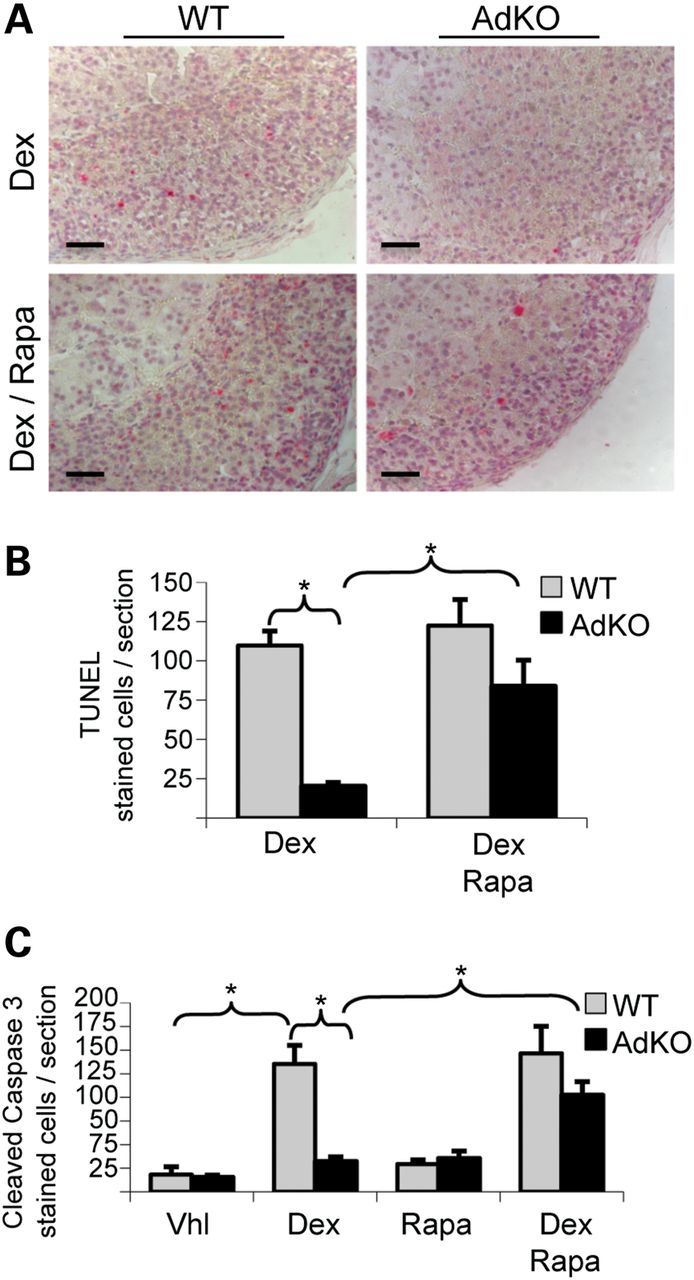
Dexamethasone-induced apoptosis in adrenals of vehicle or rapamycin-treated WT and AdKO mice. (A) TUNEL staining of adrenal sections from dexamethasone/vehicle-treated WT and AdKO (Dex) compared with dexamethasone/rapamycin-treated WT and AdKO mice (Dex/Rapa). (B) The number of TUNEL-stained cells illustrated in A was quantified in four to seven individuals per condition (values ± SEM). Values represent the number of stained cells per adrenal section expressed as a percentage of the mean of WT. *P < 0.05. (C) Cleaved-caspase 3 staining in adrenal sections was also performed in four different treatment groups (vehicle, dexamethasone, rapamycin, dexamethasone/rapamycin) in both genotypes (WT and AdKO): the number of stained cells was quantified in four to eight individuals per condition (values ± SEM). Values represent the number of cleaved-caspase 3 stained cells per adrenal section expressed as a percentage of the mean of WT. *P < 0.05. Vhl, vehicle; Dex, dexamethasone; Rapa, Rapamycin. Scale bars, 100 µm.
PKA stimulation and mTOR activation in a human adrenocortical cell line
We then activated PKA pathway in human adrenocortical cell line H295R with growing concentrations of cAMP analog 8-Br-cAMP, for 15′ (Fig. 3). ATF1, mTOR and rpS6 phosphorylation levels were all significantly increased by treatment with 1 mm 8-Br-cAMP (Fig. 3B, P < 0.05). A lower, yet not significant response was observed for rpS6 phosphorylation (P = 0.055) upon 0.1 mm 8-Br-cAMP treatment. Co-treatments with specific PKA inhibitor H89 and/or specific mTORC1 inhibitor rapamycin were performed (Fig. 3A and C). As expected, 8-Br-cAMP-induced ATF1 phosphorylation was strongly inhibited by H89 treatment (491.2 ± 95.5 versus 109.5 ± 25.1, P = 0.0058), but was unaltered in response to rapamycin (491.2 ± 95.5 versus 519.9 ± 104.4, P = 0.68). Simultaneous treatment with H89 and rapamycin induced very similar response to treatment with H89 alone (491.2 ± 95.5 versus 85.8 ± 37, P = 0.005). mTOR and rpS6 phosphorylation levels were significantly inhibited by rapamycin or H89 treatment (P < 0.05) and a maximal inhibition occurred when rapamycin and H89 were simultaneously applied (mTOR: 179.6 ± 35.5 versus 42.8 ± 13.8, P = 0.0054; rpS6: 132.1 ± 15.9 versus 57.5 ± 10.3, P = 0.0044). We concluded that PKA stimulation induced activation of mTOR pathway in human adrenocortical cells.
Figure 3.

Phosphorylation of protein targets of PKA and AKT/mTOR signaling pathways in H295R human adrenocortical cells. (A) Phosphorylated mTOR, rpS6 and ATF1 were detected in cells treated for 15′ with various combinations of PKA activator 8-Br-cAMP, PKA inhibitor H89 and mTORC1 inhibitor rapamycin. (B) Phosphorylation signals of mTOR, rpS6 and ATF1 were quantified (values ± SEM). (C) Phosphorylation signals of mTOR, rpS6 and ATF1 in the presence of 0.1 mm of PKA activator 8-Br-cAMP were quantified after treatment by either PKA inhibitor H89 (5 µm) and/or mTORC1 inhibitor rapamycin (50 nm). (values ± SEM). Values in B and C represent relative band density over tubulin expressed as a percentage of the mean of the vehicle condition. Total mTOR, rpS6 and ATF1 protein signals were unaffected by these short-time treatments (not shown). n = 4; *P < 0.05. Vhl, vehicle; Rapa, rapamycin.
PKA stimulation and mTOR activation in a mouse adrenocortical cell line
Similar experiments were then performed in the adrenocortical mouse cell line ATC7 (17) (Fig. 4). CREB, mTOR, S6K1 and rpS6 were all phosphorylated in a dose-dependent manner in response to ACTH, the natural hormonal inducer of PKA signaling in the adrenal cortex (Fig. 4A). The specific PKA activator 8-Br-cAMP had a comparable effect, indicating that the phosphorylation of all these proteins in response to ACTH was mediated by PKA activation (Fig. 4B). Kinetics experiments (2′ to 1 h, Fig. 4B) showed that phosphorylation of CREB, mTOR and rpS6 occurred as early as 2′ after ACTH treatment, with a maximum was achieved after 15′. Interestingly, AKT phosphorylation was not enhanced by ACTH and even tended to decrease over time (Fig. 4B). However, as expected, activation of PI3K/AKT/mTOR pathway by insulin induced a strong phosphorylation of AKT, showing that it was able to respond to specific activation in these cells (Fig. 4B). Finally, mechanisms of ACTH activation were tested in the presence of PKA inhibitor H89 and/or mTORC1 inhibitor rapamycin (Fig. 4C). ACTH-dependent PKA activation induced a significant increase in CREB, mTOR and rpS6 phosphorylation (P < 0.01). As in the H295R cell line, ACTH-dependent CREB phosphorylation was significantly inhibited by H89 treatment (417.6 ± 91.5 versus 284.8 ± 50.3, P = 0.0073) but was unaltered by rapamycin treatment (417.6 ± 91.5 versus 440.3 ± 118.1, P = 0.86). The combined treatment with H89 and rapamycin had a similar effect to H89 alone (417.6 ± 91.5 versus 246.3 ± 83.9, P = 0.03). mTOR phosphorylation was insensitive to treatments with rapamycin and/or H89. However, as in H295R cells, rpS6 phosphorylation was significantly inhibited by rapamycin or H89 treatment (P < 0.05) and a maximal inhibition was observed in response to simultaneous exposure to rapamycin and H89 (199.5 ± 24.7 versus 74.6 ± 14.9, P = 0.0085). We concluded that PKA stimulation induced activation of mTOR pathway in murine adrenocortical cells.
Figure 4.

Phosphorylation of protein targets of PKA and AKT/mTOR signalling pathways in murine ATC7 adrenocortical cells. (A) Phosphorylated mTOR, S6K1, rpS6 and CREB were detected in cells treated for 15′ with increasing concentrations of ACTH. (B) Phosphorylated AKT, mTOR, rpS6 and CREB were detected in cells treated for 2′ to 1 h with PKA hormonal activator ACTH or for 15′ with either the specific PKA activator 8-Br-cAMP or the PI3K/AKT/mTOR pathway hormonal activator insulin. (C) Phosphorylated mTOR, rpS6 and CREB were detected in cells treated in various combinations with ACTH, PKA inhibitor H89 and mTORC1 inhibitor rapamycin. (D) Corresponding phosphorylation signals of mTOR, rpS6 and CREB were quantified (values ± SEM). Values represent relative band density over tubulin expressed as a percentage of the mean of the control. Total mTOR, S6K1, rpS6 and CREB protein signals were unaffected by these short-time treatments (not shown). n = 4; *P < 0.05. Vhl, vehicle; 8Br, 8-Br-cAMP; Ins, insulin; Rapa, rapamycin.
PKA stimulation and resistance to apoptosis in a mouse adrenocortical cell line
ATC7 cells were treated with HA14-1, a molecule designed to trap anti-apoptotic BCL-2/BCL-XL proteins and to induce apoptosis as a result (Fig. 5) (18). Immunodetection of the cleaved forms of PARP and Caspase-3 showed an induction of apoptosis upon HA14-1 treatment (Fig. 5A). Interestingly, forskolin co-treatment, which induced PKA activation as shown by AKR1B7 induction, had a significant inhibitory effect on the cleavage of apoptotic markers PARP (100 ± 10.2 versus 42.8 ± 4.1, P = 0.005) and Caspase-3 (100 ± 3.3 versus 26.6 ± 10.6, P = 0.02) (Fig. 5A and B). This inhibitory effect of PKA activation on HA14-1-induced apoptosis was further confirmed by the decreased percentage of sub-G0/G1 cells in flow cytometric analyses (vehicle-treated, 47.0 ± 3.5% versus Fsk-treated, 26.2 ± 4.1%, P = 0.017 and not shown). We then evaluated the phosphorylation, i.e. inactivation, of the proapoptotic protein BAD in ACTH-treated ATC7 cells. Two residues were studied, serine 112, which is known to be phosphorylated by PKA, and serine 136, whose phosphorylation depends on AKT and p70S6K1. Both residues were phosphorylated in a dose-dependent manner after 15′ of ACTH treatment (Fig. 5C). A similar result was also observed in response to the specific PKA activator 8-Br-cAMP (Fig. 5D). Interestingly, during induction kinetics with ACTH, BAD phosphorylation at both positions paralleled CREB phosphorylation, but seemed cumulative over the time of experiment (Fig. 5D), exceeding the phosphorylation levels induced by 15′ treatment with either 8-Br-cAMP or insulin (Fig. 5D).
Figure 5.
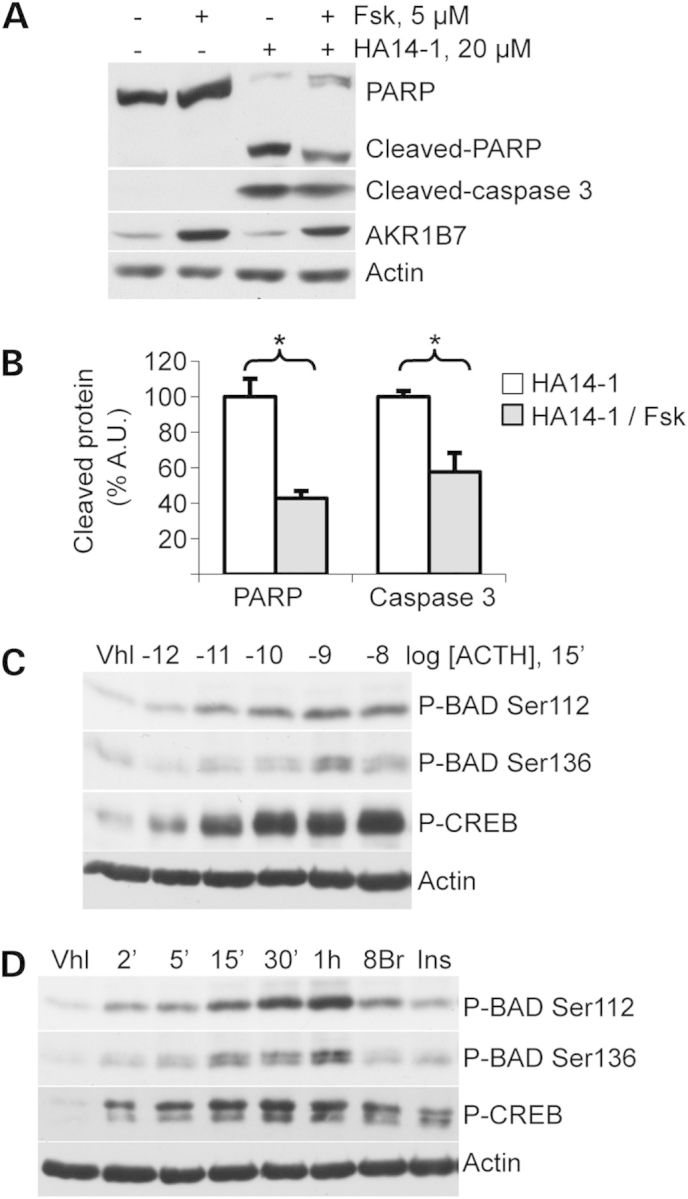
Apoptosis induction and BAD phosphorylation in murine ATC7 adrenocortical cells. (A) PARP and Caspase 3 cleavage and AKR1B7 levels were detected in ATC7 cells treated with PKA pathway activator forskolin (Fsk) and/or apoptosis inductor HA14-1 for 4 h. (B) PARP and Caspase 3 cleavage was quantified in response to HA14-1 in the presence or absence of forskolin (values ± SEM). Values represent relative band density over actin for cleaved-caspase 3 and relative cleaved protein over total protein for cleaved-PARP, expressed as a percentage of the mean of the control. n = 5; *P < 0.05. Vhl, vehicle. (C) BAD phosphorylation at Ser112 or Ser136 positions was detected in cells treated for 15′ with increasing concentrations of ACTH. (D) BAD phosphorylation at Ser112 or Ser136 positions was detected in cells treated for various amounts of time with the PKA pathway activator ACTH or for 15′ with either the specific PKA activator 8-Br-cAMP (8Br) or PI3K/AKT/mTOR pathway activator insulin (Ins). Total BAD and CREB protein signals were unaffected by these short-time treatments (not shown).
PKA stimulation and BAD phosphorylation in mouse and human PPNAD
Next, we looked for the same kind of correlation in vivo. WT mice were injected with ACTH and culled 2 h later. Strong PKA activation resulted in an increase in plasma corticosterone levels (vehicle-treated, 15.2 ± 2.3 versus ACTH-treated, 310 ± 24, in ng/ml, P < 0.001). Immunodetection of total and phosphorylated forms of BAD and rpS6 was performed on adrenal extracts (Fig. 6A and B). As expected, rpS6 was more phosphorylated in ACTH-treated than in vehicle-treated mice (2-fold, P < 0.001). Total (2.6-fold, P < 0.001) and phosphorylated BAD levels were significantly increased by ACTH treatment. When normalized according to total BAD levels, BAD phosphorylation was increased at Ser112 (3.8-fold, P < 0.001) but was unaltered at Ser136 (1.08-fold, P = 0.64). We then evaluated phosphorylation and global expression of BAD in AdKO mice (Fig. 6C and D). Total BAD levels were not significantly affected in these mice, although they tended to increase by 26% (P = 0.07). Interestingly, an increased phosphorylation level of BAD was found at Ser112 as well as at Ser136 (2.6-fold, P = 0.0028 and 1.5-fold, P = 0.025, respectively). Levels of the anti-apoptotic BCL-XL protein were found unchanged between WT and AdKO adrenal extracts (Fig. 6C and D). Finally, immunohistological stainings were performed on human PPNAD sections. Both Ser112 and Ser136 residues of BAD were found specifically hyperphosphorylated in the cells of the tumor nodules (Fig. 6E and F). Importantly, the expected hyperphosphorylation of rpS6 was detected in the nodules while no hyperphosphorylation of AKT was revealed in the same tissues (Fig. 6G). Therefore, we concluded that a positive correlation existed between PKA activation and BAD phosphorylation both in a mouse model of Prkar1a inactivation and in patients with PPNAD.
Figure 6.
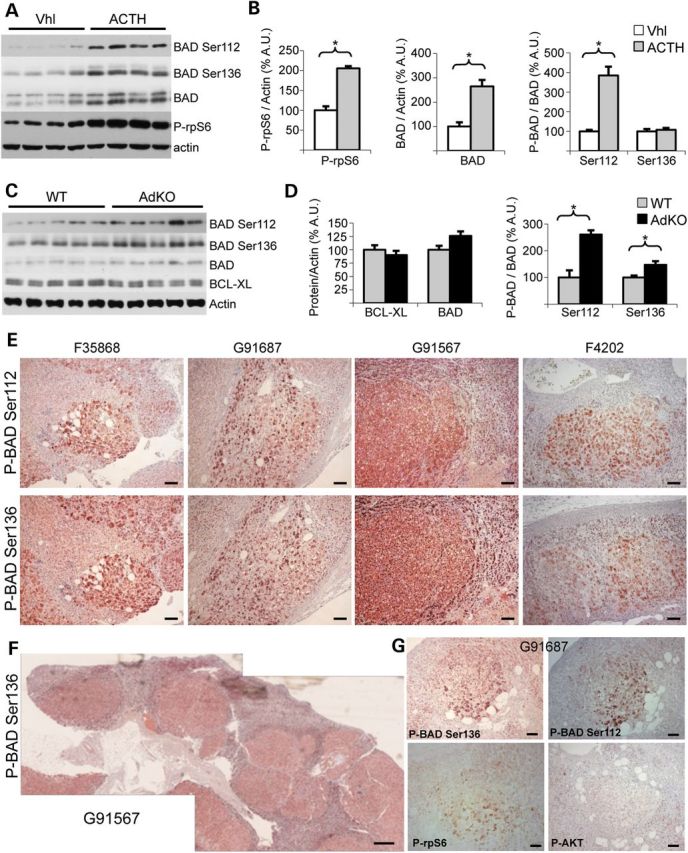
Ser112 and Ser136 phosphorylation of BAD in adrenals from WT and AdKO mice. (A) P-rpS6, BAD, P-BAD Ser112 and P-BAD Ser136 were detected in adrenal tissues from ACTH-treated or from vehicle-treated WT mice (Vhl). (B) Results illustrated in A were quantified (values ± SEM). Values represent relative band density over actin expressed as a percentage of the mean of vehicle-treated mice for P-rpS6 and BAD, and relative band density over BAD expressed as a percentage of the mean of vehicle-treated mice for the two forms of P-BAD. n = 7. (C) BCL-XL, BAD, P-BAD Ser112 and P-BAD Ser136 were detected in adrenal tissues from 12 to 14 month-old WT and AdKO mice. (D) Results illustrated in C were quantified on five WT and six AdKO mice (values ± SEM). Values represent relative band density over actin expressed as a percentage of the mean of WT mice for BCL-XL and BAD, and relative band density over BAD expressed as a percentage of the mean of WT mice for the two forms of P-BAD. (E–G) P-BAD Ser112, P-BAD Ser136, P-rpS6 and P-AKT were detected in PPNAD human adrenal samples from patients carrying germline PRKAR1A-inactivating mutation (19) and counterstained with hematoxylin. *P < 0.05. Scale bars, 100 µm in magnification and 500 µm in entire adrenal.
DISCUSSION
Although PKA plays a central role in the development of PPNAD, the molecular underpinnings of its tumorigenic activity remain elusive. On the basis of in vitro data, it had been proposed that PKA may act at least in part, through stimulation of mTOR signaling (14). Here we show that mTORC1 pathway activity is partially dependent on PKA pathway activity in adrenocortical cells in culture and in vivo both in a mouse model of PPNAD and in patients.
Interaction between PKA and mTOR pathways
Interestingly, at least three different mechanisms were described for this crosstalk. In PPNAD, it has been proposed that R1α was able to bind to and regulate mTOR kinase activity through a mechanism reminiscent of its regulation of the catalytic subunits of PKA (14,20). In thyroid cells, PKA phosphorylated Thr246 residue of the negative mTOR regulator PRAS40 and consequently induced its dissociation from mTOR (12). In pancreatic β-cells, rpS6 protein was described as a direct target of the PKA catalytic subunit (21). Our observations in both ATC7 and H295R cell lines indicate a short-term interaction between PKA activation and mTOR pathway activation (Fig. 3 and 4). In human adrenocortical H295R cells, cAMP-induced mTOR phosphorylation was decreased by H89 (Fig. 3A and C), showing that PKA catalytic activity was required to achieve this phosphorylation. In murine ATC7 cells, mTOR phosphorylation was only mildly induced by PKA stimulation and was insensitive to H89 or rapamycin treatment (Fig. 4C and D). However, in these cells, Thr389 phosphorylation of S6K1 was increased by ACTH, serum or insulin treatment but completely abolished in the presence of rapamycin (Supplementary Material, Fig. S1). This suggested that although there was no obvious change in mTOR phosphorylation, mTOR kinase activity was obviously affected. In ATC7 cells, rpS6 was still phosphorylated in response to ACTH treatment in the presence of rapamycin (Fig. 4C, compare lanes 2 and 6 : 52.6 ± 14.3 versus 127.3 ± 19.7, P = 0.025). Considering the dramatic effect of rapamycin on S6K1, this rapamycin-resistant rpS6 phosphorylation may be due to a direct action of PKA kinase activity on rpS6 (21). Accordingly, in our PPNAD mouse model, rapamycin treatment sensitized adrenal cortical cells to apoptosis (Fig. 2), in the absence of effect on rpS6 phosphorylation in the tumor (Fig. 1C). Taken together, these results suggested that in the adrenal cortex, PKA–mTOR crosstalk is ensured by at least two non-exclusive PKA-dependent mechanisms: the phosphorylation of mTOR and the phosphorylation of mTOR downstream targets (Supplementary Material, Fig. S2).
Role of AKT/PKB in the interaction between PKA and mTOR pathways
mTOR is a downstream target of AKT/PKB (22) and AKT/PKB kinase activity has also been implicated in tumorigenesis (23) and could participate to PPNAD. We thus analysed a potential involvement of AKT/PKB in the crosstalk between PKA and mTOR signaling pathways. The phosphorylation of Ser473 reflects the activity of the AKT/PKB kinase (24). In ATC7 cells, insulin treatment induced a robust phosphorylation of AKT, mTOR, rpS6 (Fig. 4B) and S6K1 (Supplementary Material, Fig. S1). However, Ser473 phosphorylation of AKT was insensitive to ACTH treatment, and even tended to decrease over 1 h (Fig. 4B). In AdKO mice, mTOR pathway activation was observed in the absence of increased AKT Ser473 phosphorylation. These results are consistent with human data. Indeed, no hyperphosphorylation of AKT/PKB was detected in PPNAD nodules (25), a result we confirmed in the PPNAD samples that we processed in this study (Fig. 6G). Taken together, these data indicate that AKT is not involved in PKA-driven mTOR pathway activation in adrenocortical cell cultures as well as in vivo in AdKO mice adrenals and in human PPNAD tissues. Thus, AKT is not essential to the tumorigenic process in the context of PPNAD.
BAD as a potential anti-apoptotic effector of PKA-mTOR crosstalk
BAD is a proapoptotic member of the BCL-2 family, which induces apoptosis by preventing BAX from binding to BCL-2 and BCL-XL (26). BAD is phosphorylated in response to many survival pathways, its phosphorylation resulting in an inactivation through cytoplasmic retention by 14-3-3 sigma (27). The phosphorylation of Ser112 and Ser155 residues is essentially dependent on PKA activity (28,29), while Ser136 phosphorylation can be achieved by AKT or S6K1 (30,31). Our data showed that BAD phosphorylation was achieved through PKA stimulation in both adrenocortical cell cultures and in vivo. Moreover, when observed in WT mice, PKA stimulation induced a classical phosphorylation on BAD Ser112, but no response from BAD Ser136 (Fig. 6A and B). In contrast, in AdKO mice, basal phosphorylation at both Ser112 and Ser136 was increased when compared with WT mice (Fig. 6C and D). Consistent with these data, both Ser112 and Ser136 were highly phosphorylated in PPNAD nodules (Fig. 6E). Our data show that AKT activity is not increased in AdKO mice and PPNAD nodules, which suggests that BAD Ser136 phosphorylation results from increased S6K1 activity in response to PKA activation. Altogether, these observations support the hypothesis that activation of mTOR pathway could be involved in PKA-dependent resistance to apoptosis, which could sensitize adrenocortical cells to tumor development. However, at present, it remains uncertain whether BAD hyperphosphorylation is the principal effector of the anti-apoptotic effects of increased PKA signaling. Alterations in other pathways cannot be ruled out in PPNAD and might cooperate with mTOR activation. For instance, we previously showed that increased cell survival in both PPNAD and animal models appeared to be correlated to inhibition of TGFβ/activin signaling (7,9). To what extent these different signaling pathways are responsible for apoptosis resistance and tumor development remains to be explored.
mTOR as therapeutical target: the possibility of rapamycin treatment in Carney complex?
Our data show that rapamycin treatment suppresses apoptosis resistance in adrenal tumors from mice lacking R1α (AdKO) but does not trigger apoptosis in WT (Fig. 2). These results indicate that (i) mTORC1 pathway is involved in the (R1α-)PKA-dependent increased cell survival, (ii) mTORC1 pathway is likely to be involved in adrenal tumorigenesis and (iii) rapamycin treatment might be a potential tool to correct the deleterious effect of (R1α-)PKA-dependent mTORC1 overactivity in PPNAD, as previously hypothesized (14).
In summary, we demonstrated that mTOR pathway is activated through PKA overactivity in adrenocortical cells. This phenomenon occurs in vitro as well as in vivo and in PPNAD patients (14). We revealed a correlation, in vitro, in vivo and in PPNAD samples, between PKA/mTOR activation and BAD phosphorylation, which could be a mechanism accounting for resistance to apoptosis. We hypothesized that this could be important in adrenal tumorigenesis, and found that in a mouse model of PPNAD, mTORC1 pathway down-regulation by rapamycin sensitized adrenocortical cells to apoptosis, the phenomenon thought to be responsible for hyperplasia in this model. Considering that mTOR is overactivated in many tumors of different tissues (32), mTOR pathway appears as a good candidate for early steps of adrenal tumorigenesis. Consequently, rapamycin could present a therapeutic potential in the treatment of adrenal hyperplasia. Furthermore, in the context of Carney complex, PKA pathway overactivation is known for its tumorigenic potential in other endocrine tissues and in the heart and skin (2,33,34). It would be interesting to study the relevance of PKA-mTOR crosstalk in these tissues and its potential involvement in their tumorigenesis.
MATERIALS AND METHODS
Human PPNAD tissue sections
Informed signed consent for the analysis of adrenal tissue and for genetic diagnosis was obtained from the patients. The study was approved by an institutional review board (Comité Consultatif de Protection des Personnes dans la Recherche Biomédicale, Cochin Hospital, Paris). PPNAD paraffin sections were cut from adrenal samples of patients who underwent bilateral adrenalectomy for ACTH-independent Cushing's syndrome in the context of isolated PPNAD or PPNAD within Carney complex. All patients carried germline inactivating mutations of the PRKAR1A gene.
Animals and hormonal treatments
Animal studies were conducted in agreement with international standards for animal welfare and approved by Auvergne Ethics Committee. Treatments were performed on adult male mice, with procedures depending on the considered compound: dexamethasone acetate in sesame oil was injected subcutaneously for 4 days (75 µg/mouse twice a day, Sigma-Aldrich); vehicle (NaCl 0.9%) or ACTH (1.2 U/mouse, Synacthene Retard, Novartis Pharma S.A.) were given as a single intra-muscular injection 2 h before culling. Rapamycin (5 mg/kg bodyweight/day) or corresponding vehicle was provided as intraperitoneal injections for 4 days. Rapamycin (#R-5000, LC Laboratories) was diluted at a final concentration of 50 μg/ml in a vehicle solution of phosphate buffered saline/10%, tween80/5%, ethanol/5%, cremophor (#C5135, Sigma-Aldrich).
Cultured cells and treatments
Mouse adrenocortical ATC7 cells and human adrenocortical H295R cancer cells were cultured as previously described and maintained in serum-free media for 12 h before treatments (17,35). ACTH (0.25 mg/ml Synacthene Immediat, Novartis Pharma S.A) or Insulin (#I3536, Sigma-Aldrich) were diluted in DMEM/Ham's F12 medium; Rapamycin (#R-5000, LC laboratories), H89 (#EI196, Biomol Research Laboratories), 8-Br-cAMP, forskolin, HA14-1 (#B7880, #F6886, #H8787, respectively, Sigma-Aldrich) were resuspended in DMSO, which was also used as the control in all experiments. In all cases, the final concentration of DMSO was maintained <0.1%. Where required, cell cultures were pre-incubated with inhibitors (rapamycin or H89) for 1 h before adding inducers. To minimize side-effects of treatments in the short-time experiments, inductors were added to prewarmed media at a two-fold concentration. Prewarmed media were then added directly to culture media so as to be diluted two-fold. To terminate experiments, culture media were removed and cells were directly treated with cold extraction buffer on ice.
Histology and immunostaining
Preparation of adrenal samples and stainings/counterstainings were performed as previously described (7). The following primary antibodies were used according to manufacturers’ protocols: anti-cleaved-caspase 3 (Asp175) (Cell Signaling, #9661), anti-P-rpS6 ribosomal protein Ser235-236 (Cell Signaling, #2211), anti-P-BAD Ser112 (Cell Signaling, #5284); anti-P-BAD Ser136 (Abcam, #ab28824); anti-P-AKT1/PKBa Ser473 (Epitomics, #2118-1).
Western blot analysis
Extraction of adrenal samples or cellular samples and western blotting were done as previously described (7). The following primary antibodies, if not specified, were used as described in manufacturers' protocols: anti-CREB #9192, anti-P-CREB Ser133 #9191, anti-mTOR #2972, anti-P-mTOR Ser2448 #2971, anti-S6K1 #9202, anti-P-S6K1 Thr389 #9205, anti-rpS6 #2217, anti-P-rpS6 Ser235-236 #2211 (1/5000), anti-4EBP1 #9644, anti-P-4EBP1 Thr37-46 #2855 (1/3000), anti-cleaved-caspase 3 (Asp175) #9661, anti-BAD #9292, anti-P-BAD Ser112 #5284, anti-P-BAD Ser136 #4366 (from Cell Signaling); anti-actin #A2066, anti-tubulin #T0198 (from Sigma-Aldrich); anti-P-AKT1/PKBa Ser473 (Epitomics, #2118-1); anti-AKR1B7 antibody (36). Western blot signals were quantified with a DNR MF ChemiBis 3.2 camera and Multi Gauge Software suite (Fujifilm).
Statistical analysis
Statistical analyses were performed with Student's t test. P-values under 0.05 were considered significant.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by Université Blaise Pascal, Université d′Auvergne, Centre National de la Recherche Scientifique, Institut National de la Santé et de la Recherche Médicale, Agence National de la Recherche (grant ANR-08-GENOPAT-007).
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Sandrine Plantade, Khirredine Ouchen and Philippe Mazuel for excellent animal care. Tissue analyses were performed at Anipath Clermont core facility.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Shenoy B.V., Carpenter P.C., Carney J.A. Bilateral primary pigmented nodular adrenocortical disease. Rare cause of the Cushing syndrome. Am. J. Surg. Pathol. 1984;8:335–344. doi: 10.1097/00000478-198405000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Carney J.A., Gordon H., Carpenter P.C., Shenoy B.V., Go V.L. The complex of myxomas, spotty pigmentation, and endocrine overactivity. Medicine (Baltimore) 1985;64:270–283. doi: 10.1097/00005792-198507000-00007. [DOI] [PubMed] [Google Scholar]
- 3.Casey M., Vaughan C.J., He J., Hatcher C.J., Winter J.M., Weremowicz S., Montgomery K., Kucherlapati R., Morton C.C., Basson C.T. Mutations in the protein kinase A R1alpha regulatory subunit cause familial cardiac myxomas and Carney complex. J. Clin. Invest. 2000;106:R31–R38. doi: 10.1172/JCI10841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kirschner L.S., Carney J.A., Pack S.D., Taymans S.E., Giatzakis C., Cho Y.S., Cho-Chung Y.S., Stratakis C.A. Mutations of the gene encoding the protein kinase A type I-alpha regulatory subunit in patients with the Carney complex. Nat. Genet. 2000;26:89–92. doi: 10.1038/79238. [DOI] [PubMed] [Google Scholar]
- 5.Horvath A., Boikos S., Giatzakis C., Robinson-White A., Groussin L., Griffin K.J., Stein E., Levine E., Delimpasi G., Hsiao H.P., et al. A genome-wide scan identifies mutations in the gene encoding phosphodiesterase 11A4 (PDE11A) in individuals with adrenocortical hyperplasia. Nat. Genet. 2006;38:794–800. doi: 10.1038/ng1809. [DOI] [PubMed] [Google Scholar]
- 6.Horvath A., Mericq V., Stratakis C.A. Mutation in PDE8B, a cyclic AMP-specific phosphodiesterase in adrenal hyperplasia. N. Engl. J. Med. 2008;358:750–752. doi: 10.1056/NEJMc0706182. [DOI] [PubMed] [Google Scholar]
- 7.Sahut-Barnola I., de Joussineau C., Val P., Lambert-Langlais S., Damon C., Lefrancois-Martinez A.M., Pointud J.C., Marceau G., Sapin V., Tissier F., et al. Cushing’s syndrome and fetal features resurgence in adrenal cortex-specific Prkar1a knockout mice. PLoS Genet. 2010;6:e1000980. doi: 10.1371/journal.pgen.1000980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rae P.A., Gutmann N.S., Tsao J., Schimmer B.P. Mutations in cyclic AMP-dependent protein kinase and corticotropin (ACTH)-sensitive adenylate cyclase affect adrenal steroidogenesis. Proc. Natl. Acad. Sci. USA. 1979;76:1896–1900. doi: 10.1073/pnas.76.4.1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ragazzon B., Cazabat L., Rizk-Rabin M., Assie G., Groussin L., Fierrard H., Perlemoine K., Martinez A., Bertherat J. Inactivation of the Carney complex gene 1 (protein kinase A regulatory subunit 1A) inhibits SMAD3 expression and TGF beta-stimulated apoptosis in adrenocortical cells. Cancer Res. 2009;69:7278–7284. doi: 10.1158/0008-5472.CAN-09-1601. [DOI] [PubMed] [Google Scholar]
- 10.Beuschlein F., Looyenga B.D., Bleasdale S.E., Mutch C., Bavers D.L., Parlow A.F., Nilson J.H., Hammer G.D. Activin induces x-zone apoptosis that inhibits luteinizing hormone-dependent adrenocortical tumor formation in inhibin-deficient mice. Mol. Cell Biol. 2003;23:3951–3964. doi: 10.1128/MCB.23.11.3951-3964.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spencer S.J., Mesiano S., Lee J.Y., Jaffe R.B. Proliferation and apoptosis in the human adrenal cortex during the fetal and perinatal periods: implications for growth and remodeling. J. Clin. Endocrinol. Metab. 1999;84:1110–1115. doi: 10.1210/jcem.84.3.5513. [DOI] [PubMed] [Google Scholar]
- 12.Blancquaert S., Wang L., Paternot S., Coulonval K., Dumont J.E., Harris T.E., Roger P.P. cAMP-dependent activation of mammalian target of rapamycin (mTOR) in thyroid cells. Implication in mitogenesis and activation of CDK4. Mol. Endocrinol. 2010;24:1453–1468. doi: 10.1210/me.2010-0087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Palaniappan M., Menon K.M. Human chorionic gonadotropin stimulates theca-interstitial cell proliferation and cell cycle regulatory proteins by a cAMP-dependent activation of AKT/mTORC1 signaling pathway. Mol. Endocrinol. 2010;24:1782–1793. doi: 10.1210/me.2010-0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mavrakis M., Lippincott-Schwartz J., Stratakis C.A., Bossis I. Depletion of type IA regulatory subunit (RIalpha) of protein kinase A (PKA) in mammalian cells and tissues activates mTOR and causes autophagic deficiency. Hum. Mol. Genet. 2006;15:2962–2971. doi: 10.1093/hmg/ddl239. [DOI] [PubMed] [Google Scholar]
- 15.Watanabe R., Wei L., Huang J. mTOR signaling, function, novel inhibitors, and therapeutic targets. J. Nucl. Med. 2011;52:497–500. doi: 10.2967/jnumed.111.089623. [DOI] [PubMed] [Google Scholar]
- 16.Thomas M., Keramidas M., Monchaux E., Feige J.J. Dual hormonal regulation of endocrine tissue mass and vasculature by adrenocorticotropin in the adrenal cortex. Endocrinology. 2004;145:4320–4329. doi: 10.1210/en.2004-0179. [DOI] [PubMed] [Google Scholar]
- 17.Ragazzon B., Lefrancois-Martinez A.M., Val P., Sahut-Barnola I., Tournaire C., Chambon C., Gachancard-Bouya J.L., Begue R.J., Veyssiere G., Martinez A. Adrenocorticotropin-dependent changes in SF-1/DAX-1 ratio influence steroidogenic genes expression in a novel model of glucocorticoid-producing adrenocortical cell lines derived from targeted tumorigenesis. Endocrinology. 2006;147:1805–1818. doi: 10.1210/en.2005-1279. [DOI] [PubMed] [Google Scholar]
- 18.Wang J.L., Liu D., Zhang Z.J., Shan S., Han X., Srinivasula S.M., Croce C.M., Alnemri E.S., Huang Z. Structure-based discovery of an organic compound that binds Bcl-2 protein and induces apoptosis of tumor cells. Proc. Natl. Acad. Sci. USA. 2000;97:7124–7129. doi: 10.1073/pnas.97.13.7124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Groussin L., Horvath A., Jullian E., Boikos S., Rene-Corail F., Lefebvre H., Cephise-Velayoudom F.L., Vantyghem M.C., Chanson P., Conte-Devolx B., et al. A PRKAR1A mutation associated with primary pigmented nodular adrenocortical disease in 12 kindreds. J. Clin. Endocrinol. Metab. 2006;91:1943–1949. doi: 10.1210/jc.2005-2708. [DOI] [PubMed] [Google Scholar]
- 20.Mavrakis M., Lippincott-Schwartz J., Stratakis C.A., Bossis I. mTOR kinase and the regulatory subunit of protein kinase A (PRKAR1A) spatially and functionally interact during autophagosome maturation. Autophagy. 2007;3:151–153. doi: 10.4161/auto.3632. [DOI] [PubMed] [Google Scholar]
- 21.Moore C.E., Xie J., Gomez E., Herbert T.P. Identification of cAMP-dependent kinase as a third in vivo ribosomal protein S6 kinase in pancreatic beta-cells. J. Mol. Biol. 2009;389:480–494. doi: 10.1016/j.jmb.2009.04.020. [DOI] [PubMed] [Google Scholar]
- 22.Inoki K., Li Y., Zhu T., Wu J., Guan K.L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell. Biol. 2002;4:648–657. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- 23.Chen M.L., Xu P.Z., Peng X.D., Chen W.S., Guzman G., Yang X., Di Cristofano A., Pandolfi P.P., Hay N. The deficiency of Akt1 is sufficient to suppress tumor development in Pten+/- mice. Genes. Dev. 2006;20:1569–1574. doi: 10.1101/gad.1395006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sarbassov D.D., Guertin D.A., Ali S.M., Sabatini D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 25.Robinson-White A., Meoli E., Stergiopoulos S., Horvath A., Boikos S., Bossis I., Stratakis C.A. PRKAR1A mutations and protein kinase A interactions with other signaling pathways in the adrenal cortex. J. Clin. Endocrinol. Metab. 2006;91:2380–2388. doi: 10.1210/jc.2006-0188. [DOI] [PubMed] [Google Scholar]
- 26.Yang E., Zha J., Jockel J., Boise L.H., Thompson C.B., Korsmeyer S.J. Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell. 1995;80:285–291. doi: 10.1016/0092-8674(95)90411-5. [DOI] [PubMed] [Google Scholar]
- 27.Zha J., Harada H., Yang E., Jockel J., Korsmeyer S.J. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L) Cell. 1996;87:619–628. doi: 10.1016/s0092-8674(00)81382-3. [DOI] [PubMed] [Google Scholar]
- 28.Harada H., Becknell B., Wilm M., Mann M., Huang L.J., Taylor S.S., Scott J.D., Korsmeyer S.J. Phosphorylation and inactivation of BAD by mitochondria-anchored protein kinase A. Mol. Cell. 1999;3:413–422. doi: 10.1016/s1097-2765(00)80469-4. [DOI] [PubMed] [Google Scholar]
- 29.Lizcano J.M., Morrice N., Cohen P. Regulation of BAD by cAMP-dependent protein kinase is mediated via phosphorylation of a novel site, Ser155. Biochem. J. 2000;349:547–557. doi: 10.1042/0264-6021:3490547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Datta S.R., Dudek H., Tao X., Masters S., Fu H., Gotoh Y., Greenberg M.E. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 31.Harada H., Andersen J.S., Mann M., Terada N., Korsmeyer S.J. p70S6 kinase signals cell survival as well as growth, inactivating the pro-apoptotic molecule BAD. Proc. Natl. Acad. Sci. USA. 2001;98:9666–9670. doi: 10.1073/pnas.171301998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morgensztern D., McLeod H.L. PI3K/Akt/mTOR pathway as a target for cancer therapy. Anticancer Drugs. 2005;16:797–803. doi: 10.1097/01.cad.0000173476.67239.3b. [DOI] [PubMed] [Google Scholar]
- 33.Carney J.A., Headington J.T., Su W.P. Cutaneous myxomas. A major component of the complex of myxomas, spotty pigmentation, and endocrine overactivity. Arch. Dermatol. 1986;122:790–798. doi: 10.1001/archderm.122.7.790. [DOI] [PubMed] [Google Scholar]
- 34.Pringle D.R., Vasko V.V., Yu L., Manchanda P.K., Lee A.A., Zhang X., Kirschner J.M., Parlow A.F., Saji M., Jarjoura D., et al. Follicular thyroid cancers demonstrate dual activation of PKA and mTOR as modeled by thyroid specific deletion of Prkar1a and Pten in mice. J. Clin. Endocrinol. Metab. 2014;99:E804–E812. doi: 10.1210/jc.2013-3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Groussin L., Massias J.F., Bertagna X., Bertherat J. Loss of expression of the ubiquitous transcription factor cAMP response element-binding protein (CREB) and compensatory overexpression of the activator CREMtau in the human adrenocortical cancer cell line H295R. J. Clin. Endocrinol. Metab. 2000;85:345–354. doi: 10.1210/jcem.85.1.6307. [DOI] [PubMed] [Google Scholar]
- 36.Lefrancois-Martinez A.M., Bertherat J., Val P., Tournaire C., Gallo-Payet N., Hyndman D., Veyssiere G., Bertagna X., Jean C., Martinez A. Decreased expression of cyclic adenosine monophosphate-regulated aldose reductase (AKR1B1) is associated with malignancy in human sporadic adrenocortical tumors. J. Clin. Endocrinol. Metab. 2004;89:3010–3019. doi: 10.1210/jc.2003-031830. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.