

Abstract
One of the goals of systems biology is the identification of regulatory mechanisms that govern an organism’s response to external stimuli. Transcription factors have been hypothesized as a major contributor to an organism’s response to various outside stimuli, and a great deal of work has been done to predict the set of transcription factors which regulate a given gene. Most of the current methods seek to identify possible binding sites from genomic sequence. Initial attempts at predicting transcription factors from genomic sequences suffered from the problem of false positives. Making the problem more difficult, it has also been shown that while predicted binding sites might be false positives, they can be shown to bind to their corresponding sequences in vitro. One method for rectifying this is through the use of phylogenetic analysis in which only regions which show high evolutionary conservation are analyzed. However such an approach may be too stringent because of the level of degeneracy shown in transcription factor binding site position weight matrices. Due to the degeneracy, there may be only a few bases that need to be conserved across species. Therefore, while a sequence may not show a high level of evolutionary conservation, these sequences may still show high affinity for the same transcription factor. In predicting transcription factor binding we explore the notion that “Co-expression implies co-regulation” [Allocco et al. BMC Bioinformatics 5:18, 2004]. With multiple genes requiring similar transcription factors binding sites, there exists a basis for eliminating false positives. This method allows for the selection of transcription factors binding sites that are active under a given experimental paradigm, thereby allowing us to indirectly incorporate the effects of chromosome and recognition site presentation upon transcription factor binding prediction. Rather than having to rationalize that a few transcription factors binding sites are over-represented in a cluster of genes, one can show that a few transcription factors are active in the cluster of genes that have been grouped together. Although the method focuses on predicting experiment-specific transcription factor binding sites, it is possible that if such a methodology were used in an iterative process where different experiments were analyzed, one could obtain a comprehensive set of transcription factors binding sites which regulate the various dynamic responses shown by biological systems under a variety of conditions hence building a more comprehensive model of transcriptional regulation.
Keywords: Corticosteroids, Gene expression, Transcription factor binding site, Phylogenetics
INTRODUCTION
In the wake of the completion of various genome projects, it was remarked that given the length of the genome, it was surprising that so much of it was not devoted to coding for protein products.31 However, taking a more holistic approach in the analysis, one realizes that given the ability of complex systems to respond to a wide range of external stimuli, the ratio of nucleotides devoted to the non-coding region vs. the coding region should not be surprising. Treating the DNA sequence as the master control program for an organism, it would follow that the majority of the sequence should be devoted to the dynamic aspect of the response or program logic, rather than mere storage for protein sequences. Researchers have begun to view the non-coding “junk” DNA as equal in importance to the coding regions due to their role in the regulation in mRNA levels and hence protein production. Without precise control of protein production via the non-coding regions, an organism would be nothing more than a static bag of different molecules and be unable to respond to changes in the environment.27
Transcription factors work by binding to specific sequences upstream of the coding region and either increase or decrease the affinity of RNA polymerase for the sequence, thereby altering the rate of mRNA production.1 The binding of these transcription factors has been determined to be sequence specific through various binding experiments.33 Previous work by Wasserman et al., have shown that this fact can be used to predict the existence of regulatory motifs within the DNA sequence. However, given the relatively short lengths of these recognition sites raging from 6 to 14 bases26,39 as well as the degeneracy possible with each given transcription factor binding site, the probability of a random hit is quite high. More problematic in this evaluation is that the transcription factors can be shown to bind in vitro even if they show no in vivo activity. This suggests that there exist other conformational factors that regulate whether a given sequence in the DNA is available for binding.
Most researchers have tackled the problem of false positives via the method of phylogenetic footprinting. 2,5,7–11,16,21,30 The core assumption in phylogenetic footprinting is that significant control mechanisms in an organism are evolutionarily conserved. Therefore, by utilizing the genomes of multiple related organisms, one should be able to identify conserved regulatory regions within the DNA. The primary benefit of this technique is that it limits the search space for which possible transcription factors binding sites can be found. This technique is exemplified by tools such as CONSITE,36 and FOOTER,11 which look for sequence homologies between two different species. CONSITE represents the basic phylogenetic analysis technique presented by Wasserman et al.39 in which only sequences which show high homology between two species such as Rat and Human would be analyzed via Position Weight Matrices (PWM) in order to determine which transcription factors binding sites are present. The primary difference between these and other tools concerns the different ways in which homologous sequences are identified.
An important point of concern with phylogenetic analysis lies in the relative degeneracy of transcription factor binding matrices.19 In many cases such as the transcription factor RE-1, a regulator of neuronal development, it was found that the transcription factor binding site had regions of high degeneracy, specifically that only 12 out of the 21 positions are highly conserved.43 Due to this fact, it is conceivable that a transcription factor can bind across multiple species with significantly different recognition sequences.28 Therefore, if sequence conservation is the primary driving force for the phylogenetic analysis of the promoter region, many important regions could be discarded. The consequence of this is that in many cases, the transcription factor binding sites predicted from the homologous sequences will be unable to satisfy the notion that co-expression implies co-regulation since it will be hard to detect a consistent set of transcription factor binding sites.
In this paper, we will show that provided that there is a high level of correlation within a set of clustered genes, there is sufficient information to extract a small set of transcription factor binding sites that can be hypothesized to co-regulate the genes in question. This is similar to the use of transcription factor enrichment to rationalize clustering results,29 or the prediction of regulatory models from a series of experiments.37 However, while studies have shown the predilection of transcription factors within groups of co-expressed genes, we will show that if the co-expressed genes have a correlation coefficient above a certain threshold, then a great majority of genes (>90%) will contain a small subset of transcription factor binding sites in common. This will eliminate many of the false positives and yield a set experimentally consistent transcription factors. Additionally, we will show that promoter regions that have been preprocessed via phylogenetic footprinting does not show an increased probability of containing transcription factor binding sites over that of the baseline sequence, suggesting that either phylogenetic footprinting is unable to preferentially select for regulatory regions, or that there are non-evolutionarily conserved regulatory sites in the sequence.
METHODS
Data Collection and Gene Expression Measurement
The microarray data was obtained from an experiment that was conducted to examine the behavior of a bolus injection of corticosteroids upon temporal gene expression profile of living cells. This dataset was specifically chosen due to the a priori knowledge that corticosteroids have powerful transcriptionally mediated effects upon the rat experimental model. The data collection and preliminary analysis were previously presented in.4 The data is available in the GEO database under the accession number GDS253.
Identification and Classification of Relevant Gene Expression Profiles
After the data has been obtained, it is important for the expression profiles of relevant genes to be extracted. This step essentially seeks to extract genes whose expression profiles are actively being mediated by transcription factors as part of a transcriptional regulation pathway. By doing this, it ensures that the genes that were selected and grouped are part of the same transcriptional response mechanism and therefore should show clear trends when conducting transcription factor analysis.
Preliminary examination of the data lead to the observation that different clustering algorithms yielded inconsistent results which were different in the number of optimal clusters, or the genes which were grouped together.34 Further analysis suggested that the data itself was antagonistic to data clustering due primarily to the fact that no clear boundaries existed. Common data selection techniques such as various filters build around data quality checks like Affymetrix’s Absent, Present or Marginal flags or selecting genes that showed expression levels which changed by greater than 2x up or down yielded a subset of data which still was not clearly partitionable. SLINGSHOTS was an attempt at combining both clustering and selection in order to obtain a subset of genes in which boundaries could be seen.
We recently proposed a novel algorithm for the identification and classification of relevant gene expression profiles called SLINGSHOTS (SeLection of INformative Genes via Symbolic Hashing Of Time Series).42 The key motivating argument for this method is the realization that in the presence of noise and uncertainties associated with measuring mRNA abundance, looking for exact correlations or distance metrics between gene pairs may not necessarily yield the most informative interpretation. On the contrary, robust, coherent and dominating qualitative features and similarities could be a more informative proxy for the information content of the expression experiment. With our approach, the raw data is transformed into sequences of events, or symbols, and these are further analyzed for consistencies. Our algorithm is based on the assumption that genes that are relevant to the underlying dynamics of the system have two essential characteristics. The first is that they are part of a concerted mechanism and should possess expression profiles which are temporally consistent with the expression profiles of other genes involved in related molecular mechanisms. The second assumption is that the dynamics of the set of informative genes out to show significant deviations in their aggregate activity from their initial baseline activity distribution. Therefore, our algorithm performs a fine-grained clustering which results in hundreds of clusters. We then evaluate the ability of a subset of these individual clusters to satisfy these two constraints, thereby linking the selection process with the clustering result. The advantage of this technique is that we are able to perform data selection with clustering quality in mind and parse the contribution of each cluster to the overall dynamics of the system.
SLINGSHOTS uses the notion that genes which are part of large highly correlated set of genes are more likely to be significant based on the assumption that an organism responds to outside challenges to homeostasis through the utilization of a set of genes which are highly controlled in both their expression levels and temporal evolution. It has already been shown that genes which show a high degree of correlation in their expression profiles tend to be involved in related functions.3 There is an additional qualifier, that given significant perturbations to the experimental system, that a large number of genes with coordinated responses need to be brought online to deal with the challenge to homeostasis.
SLINGSHOTS deterministically clusters expression profiles into a large set of putative clusters via a hashing process. Hashing is utilized to decompose an expression profile into a single integer. Expression profiles with the same integer have very similar expression profiles. The hashing methodology used is the one proposed by Lin et al.24 What hashing accomplishes for our purposes is the grouping of expression profile into a large number of punitive clusters all with a similar range of correlation coefficients. The procedure for going from an expression profile to a hash value is given in Appendix 1.
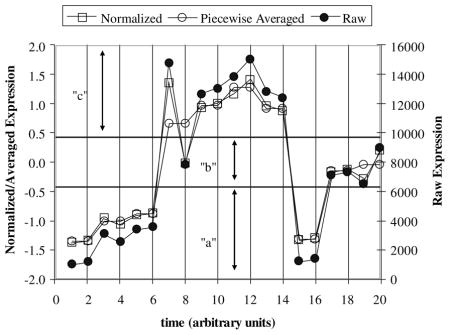
After the genes have been put into their respective clusters, the next task is to identify which of these gene clusters are actively participating in the experimental response. Given that these experiments attempt to perturb the homeostatic balance forcing the organism into a different transcriptional state, the algorithm selected clusters that when combined yield a significant deviation in the distribution of expression level values from that of baseline. Therefore, one should be looking for genes which alter the distribution of up-regulated and down-regulated expression levels during the course of the experiment, thereby pointing to their active role in changing the transcriptional state of the organism. Given that there are hundreds of clusters generated via the hashing step, a greedy selection algorithm was implemented in which the peaks are added in the order of their population. The overall algorithm is given in Appendix 2. The results of SLINGSHOTS is given in Fig. 1 indicating the 12 clusters that were identified as informative and will be further discussed in the Results section.
FIGURE 1.

A sample cluster obtained from SLINGSHOTS. All of the clusters show a reasonably correlation to the average normalized profile.
Identification of Possible Transcription Factor Binding Sites
The identification of possible transcription factor binding sites is broken down into two steps: (i) the identification of the promoter region, (ii) the identification of putative transcription factor binding sites. CORG14 was used for the identification of promoter regions as well the identification of relevant transcription factor binding sites. CORG was selected primarily for its ability to extract the 5′ upstream region up to the next gene rather than to a set number of upstream base pairs. This was important to us due to the nebulous concept of how far upstream a promoter region lies. It has been shown that the GRE (Glucocorticoid Response Element) could be found thousands of base pairs upstream of the start codon.5 Other such as TRED44 on the other hand require as a parameter the number of upstream base pairs to consider. Additionally by using CORG, one is able to utilize its built it facilities to both extract homologous sequences as well as transcription factor binding sites.
One complication which needed to be addressed was the fact that CORG returned homologous sequences between two species and is unable to return just the entire promoter region for a single species. In order to compensate for this drawback, the evaluation was conducted in the following manner. To evaluate the difference between phylogenetic footprinting and our proposed approach of looking at the promoter regions of a set of clustered genes in aggregate, a CORG search was conducted upon human/rat and mouse/rat. The human/rat case is the baseline example of phylogenetic footprinting in which ideally there will be a small set of regulators which give rise to the similar responses to corticosteroids in humans and rats. The mouse/rat case was used to give a proxy for the context specific case in which the analysis is performed only on the rat promoter region and to determine the transcription factors which are present in all of the genes in the cluster. The rationale for running this case is that the rat/mouse promoter regions have about an 85% conservation rate among homologous sequences,41 and are therefore genetically very similar. Given this high level of conservation between the two different species as well as the fact that CORG keeps sequences that show a homology of greater than 70% over 100 base pairs,13 it provides a reasonable facsimile for the rat promoter region.
Verification of the results was initially going to be conducted by comparing our selected transcription factors with known transcriptional regulators via RnPD.41 However, an initial evaluation of the selected and clustered genes revealed that there was insufficient data on known binding sites in order to make any sort of meaningful assessment.
Data Analysis
The primary metric which to be analyzed is the number of times a transcription factor binding site is found in the 5′ region of genes that comprise up of a highly correlated cluster. This is necessary in order to determine whether or not there are any transcription factor binding sites which were present in a sufficient percentage of genes where it would be a reasonable candidate for the co-regulation of the genes within the cluster. Secondly, once the metric is quantified, it will be possible to ascertain the overall distribution of transcription factors throughout the cluster of genes, allowing one to determine whether or not the highly conserved transcription factor was present due to a statistically significant event, or whether it was highly conserved due to chance.
The process of finding a hit for a specific sequence in the promoter region can be modeled by an exponential distribution whose PDF is given in Eq. (1). In Fig. 2, a random set of genes was selected and a distribution that relates the number of transcription factors to the number of genes that a given transcription factor is predicted to bind to is given. From this distribution, it appears that the initial assumption that one can model transcription factor occurrence rate on a cluster of gene as an exponential distribution. This also functions as a negative control. If the genes were randomly selected, then the distribution of transcription factors/cluster ought to match the exponential distribution. If there are deviations from this exponential graph near the tail end representing conservation of a significant number of transcription factors at levels higher than would be expected, then it would suggest the presence of a significant co-regulation mechanism.
FIGURE 2.

The Occurrence rate of transcription factor binding sites when random genes are grouped together. (Top) The exponential distribution. (Bottom) A Log linearized version of the plot. (Note: the tail end agrees well with the overall exponential distribution).
| (1) |
To obtain the parameters for the PDF, the mean number of times a transcription factor-binding site is present amongst the genes in a cluster as well as the standard deviation this distribution is calculated. Given the slight discrepancy between the two values, the average of the mean and the standard deviation is used as the parameter with which to model the distributions. The fits of the distributions for the 12 clusters are shown in red on Figs. 3 and 4. The exponential distribution will then allow us to obtain the probability that a single transcription factor will be conserved over x% of the time. This probability will be used below to calculate the expected number of highly conserved transcription factors.
FIGURE 3.

Distribution for the rat/mouse case that shows the number of transcription factors that are found in a given number of genes for the different clusters. Upon the initial observation, we find that the distribution can be modeled as an exponential distribution. The red curve was obtained via parameter estimation from the distribution mean and standard deviations.
FIGURE 4.

Distribution for the rat/human case that shows the number of transcription factors that are found in a given number of genes for the different clusters. Upon the initial observation, we find that the distribution can be modeled as an exponential distribution. The red curve was obtained via parameter estimation from the distribution mean and standard deviations.
After the exponential distribution had been fitted, it then becomes possible to calculate the expected number of transcription factors that ought to be highly conserved given the exponential distribution. If one has been able to filter out false positives, then one should find that the number of transcription factors that are actually conserved should be less than the expected value. The statistical significance of the number of transcription factors which are actually conserved will be calculated via the binomial distribution Eq. (2) under the assumption that the presence of a given transcription factor above any given conservation rate can be modeled as a random process.
| (2) |
RESULTS
The classification and selection step yielded 12 clusters with a total of 529 probe set, of which the clustering results are given in Fig. 1. The 529 probe sets correspond to 454 genes of which 339 genes had entries in the CORG database. The most important property of these clusters is the high level of correlation between all of the genes in the cluster. Data has presented that suggests that for genes to have a greater than baseline chance of having transcription factors in common, the correlation coefficient should be greater than 0.75.3 Our clusters show an average correlation coefficient of 0.85, comfortably over the limit. In the transcription factor dataset which they based these conclusions off of,22 they found that only 37% of the genes actually showed significant experimental binding to transcription factors. So while with a .85 correlation in the signal suggests only a 50% commonality between two genes, we believe that biologically the percentage in mammalian systems is quite higher due to the relatively sparse nature of the isolated yeast transcription factors. Additionally, we believe that if a transcription factor can be shown to be over-represented the less than perfect correlation, it has a greater chance of being significant compared to those which are not over-represented within a cluster.
Figures 3 and 4 show the distribution of transcription factor binding sites that are conserved over a certain number of genes in a cluster. The values on the x-axis are dependent on the overall number of genes in a cluster, and the values on the y-axis denote the number of transcription factor binding sites that were present in a given number of genes. The results of this plot seem to suggest that the distribution of transcription factor binding sites amongst the genes in a given cluster can be modeled via an exponential distribution. Given that the exponential distribution given in Eq. 1, is primarily defined by the parameter λ, which is the mean and the standard deviation of an exponential distribution, the means/standard deviations for the number of times a transcription factor binding site was present in a gene of that cluster is shown in Table 1. It is notable that these values are reasonably close reinforcing our assumption that the exponential distribution is a good fit for the data. To obtain the exponential fits given in Figures. 3 and 4, the means and the standard deviations were averaged for each cluster to obtain a single consistent value half way between the means and the standard deviations. Looking at the parameters, there was a direct correlation between the parameters themselves and the number of genes in a cluster. This linear relationship is illustrated in Fig. 5, where the parameters are plotted against the number of genes in a cluster. This fact will be revisited during the discussion.
TABLE 1.
THE STATISTICS WHICH DESCRIBE THE DISTRIBUTION OF TRANSCRIPTION FACTORS PER CLUSTER.
| Transcription factor occurrence statistic
| ||||
|---|---|---|---|---|
| Mouse/Rat
|
Human/Rat
|
|||
| Cluster | Mean number of occurrences | Standard deviation | Mean number of occurrences | Standard deviation |
| 1 | 7.26 | 6.44 | 3.44 | 3.80 |
| 2 | 10.95 | 10.80 | 5.59 | 5.74 |
| 3 | 6.78 | 6.02 | 3.90 | 3.15 |
| 4 | 9.65 | 9.57 | 5.68 | 5.89 |
| 5 | 5.51 | 4.60 | 3.21 | 2.97 |
| 6 | 11.01 | 11.68 | 5.66 | 6.69 |
| 7 | 5.81 | 4.48 | 3.51 | 2.99 |
| 8 | 7.54 | 6.69 | 3.41 | 3.44 |
| 9 | 8.41 | 6.58 | 4.33 | 4.07 |
| 10 | 5.96 | 5.00 | 3.11 | 3.15 |
| 11 | 7.56 | 6.64 | 3.42 | 3.45 |
| 12 | 10.40 | 10.10 | 4.63 | 5.78 |
The similarity between the means and standard deviations suggest that the distribution can be modeled via an exponential distribution. The results of this chart suggest that the primary driving force in the number of times a transcription factor is found within a gene is the length of the promoter region being analyzed.
FIGURE 5.

The parameters used to fit the exponential distribution vs. the cluster population. This linear trend further reinforces our contention in our belief that the CDF representing the number of times a transcription factor is present amongst a set of genes is governed only by the length of sequence analyzed. Note that in both the cases, the parameters show a good linear fit. This suggests that phylogenetic footprinting in the Rat/human case has not selected for sequences in the promoter region that show a greater number of correct promoter regions.
A cutoff of 95% was set to determine which transcription factors ought to be examined. In the case where the mouse/rat promoter region was analyzed, it was found that there were one or more transcription factors that was present on average 99.7% of the time in each of the clusters. In the case where the human/rat promoter region was analyzed, the most conserved transcription factor was present in only 85.2% of the genes of a given cluster. From this immediate result, it would seem that there is a sizable gap in terms of the ability of the phylogenetic analysis conducted via CORG in the rat/human case to obtain transcription factors that are likely candidates for the co-regulation of the genes in the cluster. The transcription factors which were highly conserved in both cases are given in Table 2, 3 with Table 2 utilizing a lower cutoff of 80% of the genes for rat/human and Table 3 utilizing a cutoff of 95% for rat/mouse. Different cutoffs were set given the fact that in the rat/mouse case, there was a transcription factor present in 99.7% of the genes/cluster whilst in the rat/human case, the most conserved transcription factors were only present at only 85.2% of the time. Further investigation of the parameters that were used for the exponential fits suggested that the means and the standard deviations in the human/rat case were roughly half that in the mouse/rat case. What makes this association more interesting is the fact that after phylogenetic footprinting through CORG, the sequence being analyzed by position weight matrices has decreased roughly by half. This suggests that the hit rate of the transcription factors is sequence independent, and that the two results despite having very different cutoffs have the same overall characteristic.
TABLE 2.
TRANSCRIPTION FACTORS CONSERVED MORE THAN 80% OF THE TIME BETWEEN HUMAN AND RAT.
| Cluster | Transcription factors | |
|---|---|---|
| 1 | STAT 6 | |
| 3 | STAT 6 | |
| 4 | STAT 6 | |
| 5 | STAT 6 | |
| 7 | STAT 6 | STAT5 |
| 9 | STAT 6 | TEF-1 |
| 10 | TEF-1 | |
| 12 | STAT 6 | |
Note that 4 of the clusters (2,6,8,11) do not contain highly conserved transcription factors and that all of the transcription factors are those that are highly represented in the genome.
TABLE 3.
TRANSCRIPTION FACTORS CONSERVED MORE THAN 95% OF THE TIME BETWEEN MOUSE AND RAT.
| Cluster | Transcription factors | ||||||
|---|---|---|---|---|---|---|---|
| 1 | STAT 5 | STAT 6 | TEF-1 | ||||
| 2 | STAT 5 | STAT 6 | TEF-1 | CDX | |||
| 3 | STAT 6 | TEF-1 | AP2 ALPHA | ||||
| 4 | STAT 5 | ||||||
| 5 | STAT 5 | STAT 6 | |||||
| 6 | STAT 5 | STAT 6 | TEF-1 | ||||
| 7 | STAT 5 | STAT 6 | USF1 | ||||
| 8 | STAT 5 | STAT 6 | TEF-1 | GE II | CDX | GATA6 | |
| 9 | STAT 5 | STAT 6 | TEF-1 | GE II | CDXA | AP2 ALPHA | PAX4 |
| GATA6 | CIZ | SRY | USF1 | ||||
| 10 | STAT 5 | STAT 6 | GATA6 | ||||
| 11 | STAT 5 | TEF-1 | STAT 6 | CDX | AP2 ALPHA | ||
| 12 | STAT 5 | STAT 6 | TEF-1 | ||||
In contrast to the human/rat case, all of the clusters show transcription factors conserved more than 95% of the time as well as transcription factors which are highly conserved and not found in a random sampling of genes.
Random analysis was conducted to ascertain the significance of these transcription factors. Thirty random genes were grouped from the microarray data and the same procedure was conducted upon this synthetic cluster. What was found was that 3 of the transcription factors that were highly conserved in both the rat/human case and the rat/mouse case were also found in a random sampling of the data. These transcription factors are TEF, and STAT5, and STAT6. Removing these transcription factors from consideration, it was observed that the rat/human homologous promoter case has no transcription factor that is conserved in more than 80% of the genes. In fact, there are no transcription factors that are conserved in more than 75% of the genes in any given cluster. In contrast to this, when TEF1, STAT5 and STAT6 where removed, 8 out of the 12 clusters still had transcription factors that were conserved in more than 95% of the case, with the remaining four clusters containing transcription factors that were conserved more than 90% of the time. The transcription factors that are conserved more than 95% of the time which are not STAT6, STAT5, and TEF1 are highlighted in red in Table 3. This suggests that aside from the global non-specific activation of transcription, in our specific experimental data, phylogenetic analysis in the human/rat case has been unable to find a reasonable candidate for co-regulation.
Given the following facts, the distribution of transcription factors amongst genes in a cluster, the parameters that fit the distribution, and the fact that there are 457 possible transcription factors, one can begin to calculate the probability of a the number of transcription being highly conserved within a cluster in the rat/mouse case. This evaluation was not conducted in the rat/human case due to the fact that did not exist a set of transcription factors which can be hypothesized to co-regulate the set of genes. Excluding the transcription factors STAT5, STAT6 and TEF1 and assuming a conservation rate of greater than 95% one has a 4% chance of finding a transcription factor. This is consistent due to the linear relationship between the cluster size and the mean values. Given 457 possible transcription factors, this would lead to an expected value of 18. Therefore in a random case one would expect 18 transcription factor to be conserved over 95% of the time. However, what is found that there are between 1 and 8 transcription factors being highly conserved. This result suggests that solely by looking at the genes that are clustered with a very high correlation it is possible to throw out a significant number of transcription factors which may be indicative of false positives. The associated p-value assuming a binomial distribution in this case ranges from 1.58 × 10−7 to 5.27 × 10−3.
DISCUSSION
The main point of phylogenetic analysis has been the reduction of false positives in transcription factor binding predictions. However, it is our hypothesis that one cannot perform such reduction if the result of the operation cannot satisfy the notion that co-expression implies co-regulation. We believe that by performing phylogenetic analysis between human and rat as well as utilizing mouse and rat to extract a homologue for the rat promoter region, it has been shown that phylogenetic footprinting does a poor job in keeping the necessary transcription factors that would co-regulate clusters of co-expressed genes. Therefore, it is our contention that due to this fact, phylogenetic footprinting utilizing sequence information only may not be the best way to tackle the issue of false positives. One may argue that the notion of requiring that all of the genes in the highly correlated clusters must have a set of common active regulators is a naïve approach. In spite of the simplicity of this approach, the proposed method was still able to find a small subset of transcription factors that were highly conserved across all of the genes in a given cluster.
Our second contention is that performing phylogenetic footprinting does not yield results that were characteristically different than in the case where phylogenetic footprinting was not performed. In both cases, there was an observed exponential distribution with parameters that vary by the total amount of base pairs analyzed. We had expected that while there were numerous false positives generated via standard transcription factor binding site prediction that transcription factor binding sites were more prevalent in “true” regulatory regions that were conserved through evolution than over the baseline rate. However, we did not find a greater affinity for transcription factor binding sites to be localized to regions of evolutionary conservation than over that of non-evolutionary conserved segments of the 5′ region. So while there was a difference in the parameters for the rat/human case vs. rat/mouse case, it was not due specifically to the presence of certain conserved regions that were present in the different species, but rather due only to the length of the sequence being analyzed. Had there been a true species dependent conservation of phylogenetic footprinting, then the correlation between the parameters which fit the curves in Figs. 3 and 4, ought not to be accurate correlated with the length of the promoter sequence to be analyzed.
This leads to the hypothesis that the primary driving force in the number of times a given transcription factor occurs within a gene cluster is driven by the length of the promoter region analyzed. Furthermore, the general fit of the exponential distribution in both cases suggests that the phylogenetic footprinting does not add information to the system. If phylogenetic footprinting in its current formulation is correct, then it should be able to extract a set of regulatory hotspots in which the presence of transcription factors were over-represented. If this were the case, then there wouldn’t be a correlation between the parameters for the exponential distribution and the length of the promoter regions being analyzed. However, this was found not to be the case. There was no greater probability for transcription factors in the regions conserved via phylogenetic footprinting.
While it has been shown that the probability of a transcription factor “hit” is dependent upon the length of the sequence analyzed, the number of transcription factors that are actually conserved over a high number of genes cannot be. If the probability for a transcription factor to be highly conserved in >95% of the genes per cluster is around 4% one would expect around 18 transcription factors to show similar conservation. However, we find that this is not the case, The number of transcription factors that are highly conserved in the rat/mouse case range from 1 to 8 after the highly non-specific transcription factors have been eliminated. This is due primarily to the fact that while the exponential distribution is a reasonable fit for the data, the tail end of the distribution, i.e. the highly conserved transcription factors, deviate from the exponential distribution.
What is evident in Fig. 6 is that the graph is bimodal with a linear regime that describes the random occurrences of transcription factors, and a nonlinear regime in which the transcription factors show a non-random occurrence rate. This suggests that by lumping the genes by their expression profile together, it allows one to isolate a set of transcription factors that have a high probability of being active under the experimental regime. Taking into account the random trial, one can further cut down on the number of isolated transcription factors by removing the non-specific initiators of transcription, i.e. those that are part of widespread signaling cascades.
FIGURE 6.

Log Normalized version of Fig. 3. The lines are fits obtained by fitting those transcription factors that are not highly conserved within a cluster. What is evident is that there are a number of transcription factors at the tail region that cannot be adequately modeled by the exponential distribution suggesting a non-random preference for a given cluster of genes.
In Fig. 7, we illustrate what we term the “parameter gap”. In all of the cases shown in Fig. 6, we were able to get a better fit in terms of the R2 value if we fitted the genes that were conserved over a few genes. This “parameter gap” allows for the determination of both the limits of the expected number of transcription factor binding sites in a given cluster and the limits of the conservation rate. In this case, the bounds for the expected number of transcription factors are 0–18, and the bounds for the conservation rate is 80–100%. This allows us to discount human/rat phylogenetic case as isolating any transcription factor binding sites that co-regulate a cluster, and allows us to calculate the p-values for the number of transcription factor binding sites in a cluster.
FIGURE 7.

This illustrates the gap caused by fitting all of the data, and fitting only the first ten data points. The number of informative transcription factors should be less than the expected value if the exponential distribution is estimated from all of the data, and greater than the expected value of the exponential distribution accounts only for the first ten points. R2 value in with the first 10 data points is 0.74 and 0.69 when all of the data was used.
While the presence of STAT6 and STAT5 are highly non-specific, we feel that the results are still rather interesting. Given the relative promiscuity of the transcription factor for genes, we believe that presence of STAT5 and STAT6 show the relative widespread effects of the various JAK-STAT pathways that are activated by cytokines and growth factors.15 Further examination of their binding matrices on TRANSFAC shows the fact that the STAT5 and STAT6 are highly nonspecific with base specificity in 3/8 and 4/8 of the binding matrix, leading to a high rate of positive hits in the promoter region. We hypothesize that the relative promiscuity of the STAT5 and STAT6 transcription factor makes it a possible candidate as one of the primary initiators of transcription, and that it is the other cluster specific transcription factors binding sites that serve to control the relative shapes of the expression profiles.
However, for many of the other transcription factors such as CDX (Caudal-type homeodomain protein), AP2-Alpha (activating enhancer binding protein 2), USF (Upstream Stimulating Factor), GATA6 (GATA Binding Protein 6), and PAX4 (Paired Box Gene 4) their presence within the various clusters are more specific. Utilizing information from iHOP,18 it is possible to establish links between them and the effects of corticosteroid administration. For the transcription factors GE II, Sry and CIZ, there was not sufficient information about their functions in the context of corticosteroid response to make a meaningful evaluation.
For these five transcription factors, the RG-U34A microarray had expression data on four of them (CDX, USF1, AP2-Alpha and PAX4). Of these, CDX and USF were down-regulated after the administration of corticosteroids, while the rest of the transcription factors are up-regulated. The down-regulation of CDX may be evidence of the suppression of proliferation by corticosteroids. CDX has been characterized as a regulator of cancer cell proliferation and is often up-regulated in malignant tumors.17 Therefore, it would follow that the down-regulation of this transcription factor would lead to the suppression of cellular proliferation, one of the hallmarks of malignant tumors. The down-regulation of USF is characteristic of the decrease in lipid and glucose metabolism by the liver,23 leading to the increase in the levels of circulating free fatty acids and glucose in the bloodstream leading to the associated steroid induced diabetes.
The up-regulation of AP2-Alpha could again be evidence of the suppression of cellular proliferation by corticosteroids. AP2-Alpha has been cited as a tumor suppressor,38 and combined with the down regulation of CDX, it may point to the mechanism by which corticosteroids suppress cellular proliferation. PAX-4 is normally associated with the differentiation of beta islet cells in the pancreas. This is consistent with the observation that the levels of circulating glucose are increased via administration of corticosteroids. However while its presence in the liver has not been substantiated in the literature, it is conceivable that given that it is active in one organ under administration of corticosteroids, that it could play a less visible though still important role in the liver as well. An interesting question that arises from this observation is whether or not the differentiation of beta islet cells in the pancreas is driven primarily by the levels of circulating glucose levels, or whether it is driven by the levels of corticosteroid.
We acknowledge that there is significant disagreement between the results that we have obtained and those obtained via phylogenetic analysis. However, we feel that our results are correct, given its success at identifying possible co-regulators. The fact that the algorithm has identified a very small subset of transcription factors that show significant biological roles related to the pharmacological effect of corticosteroid leads us to believe that the algorithm has been successful in predicting transcription factor/gene relations. Such data will allow us to build regulatory networks that can be used to build PK/PD models which will allow us to predict the behavior of the system under different dosing conditions.
If the disagreement between the results obtained from the presented method and phylogenetic footprinting is a paradox rather than a contradiction, an interesting possibility arises. Currently, there is a similar and perhaps related paradox in the field of transcriptional network analysis. It has been widely noted that maps of transcriptional interactions appear to have a scale-free topography in which the distribution of links between different genes follows an exponential distribution. 20,25,35 However, is has also been observed that despite the apparent scale free nature of the network, biological transcription networks illustrate a higher degree of robustness than could be normally explained via a scale free network.6 Specifically that the removal of a large number of hubs are not lethal to an organism. It has been shown that in yeast, the removal of 28 out of 33 highly connected hubs did not lead to the death of the given yeast cells6 with little correlation between the connectivity of a node and its importance to viability. Additionally, simulations which explore the evolution of metabolic networks have resulted in networks that contain the existence of hubs but do not exhibit a clear power law32 in their network connectivity suggesting there are non scale-free elements in the overall network.
Both the analysis of the random clusters of genes as well as the transcriptional networks obtained via phylogenetic analysis seem to confirm the existence of a scale-free network as evidenced by the exponential distribution of links between transcription factors and a set of genes. Such an observation can be justified by the fact that some transcription factors appear to be highly selective while others such as STAT5 and STAT6 appear to be highly promiscuous. However, as shown in Fig. 6, there also appears to be a significantly non-exponential portion to the distribution. This suggests that the genes in a cluster and their respective co-regulators may not follow a scale free network. Our hypothesis is that while the overall topography of a network is scale free, if one were to look at important response pathways, one may obtain a sub-graph with a different topography given the need to maintain a high degree of robustness. This means that there are certain co-expressed genes in which the pathway is common over multiple organisms which have a evolutionarily conserved transcription factor binding sites. However, there are also genes that augment this central pathway which contain regulatory regions that may be species specific.
We find this notion attractive given the fact that it has been shown that rats and humans often have different responses to medication or treatment regimens12 despite the fact that the same primary pathway is being targeted. Given the relative importance of the highly connected hubs in many different biological processes, these auxiliary genes would allow the system to maintain consistency within the primary response pathway in the presence of significant cross-talk between different signaling pathways as well as perturbations such as disease or injury.
If this were the case, then it would allow us to reconcile our results with those obtained through standard phylogenetic footprinting. The network corresponding to the links extracted via phylogenetic analysis may correspond to the primary response pathways, i.e. those that code for enzymatic products that the organism uses to deal with alterations to homeostasis are evolutionarily conserved. The results obtained via our algorithm includes this primary response pathway as well as the extra links that give the network a composite characteristic rather than a simple scale free architecture.
Assuming that this hypothesis is correct, then the following questions arise: What are the properties of this network; Can we find this sub-network efficiently given the properties; If we identify this network, can we show that the genes that make up the nodes of the network are co-expressed? If these questions can be answered in the affirmative, then it would give a powerful tool to molecular biologists in the identification of key pathways. Currently, we can only provide a vague notion as to what the property of this transcriptional sub-network would be, namely that it should be robust to the removal of highly connected hubs, i.e. the removal of a hub would not separate the network into two disjoint subsets thus rendering it non-functional.
CONCLUSION/FUTURE WORK
The primary goal behind the prediction of transcription factor binding sites is the creation of a global gene interaction network that can be used to predict an organism’s response to different stimuli. Therefore, it is our contention that any sort of network must be coherent with experimental results. Our initial analysis of the results of transcription factor binding sites via phylogenetic footprinting suggests that oftentimes this is not the case, and that there were many genes that were co-expressed that did not appear to be co-regulated under the experimental regime. While it is plausible and highly likely that unrelated regulatory factors can lead to co-expression, it is our belief that the bulk of co-regulated genes ought to have similar regulatory mechanisms given prior work by Wolfe et al.40 Therefore we focused whether it was possible to predict a set of transcription factors that would give rise to the observed co-expression. Our method focused primarily upon the notion that instead of eliminating false positives by comparing the predictions between different organisms, we ought to be able to eliminate false positives by comparing predictions between different genes which show the same response.
Ideally, there results between the two methods should agree to a large extent. However, the results which we obtained were different from those obtained through phylogenetic analysis, and lead to the following conclusions. Either there was a paradox and both methods gave correct answers, one of the methods is correct, or neither of the methods is correct. Out of these possibilities, we found the first consequence the most intriguing because if one assumes the correctness of both, it provides a mechanism for the possible elucidation of primary response pathways in a highly connected network structure, an explanation for the phenomenon of differing side effects in different organisms, and resolving the paradox of a highly robust scale-free network.
Additionally, we believe that we have found only the transcription factors that are active under a given condition, which is not the overall set of transcription factors. We believe that with additional experiments of the response of an organism under different conditions it would be possible for us to isolate a set of transcription factors that are active under those conditions in order to obtain a clearer picture as to the overall regulatory structure of the organism as a whole. Therefore an iterative processes in which every new experiment yields a few transcription factors can eventually lead to a more complete picture as to the network regulatory structure.
Acknowledgments
EY, DS, and IPA would like to acknowledge the financial support from the NSF under the grant NSF-0519563 and the EPA under the grant EPA-GAD R 832721-010. RRA, DCD, and WJJ acknowledge the NIH under grants GM 24211 and GM 67650, and a grant from NASA.
APPENDIX 1
The normalization of the gene expression profile to N(0,1) via the z-score transform.
If the sequences are longer than 10 time points, piecewise averaging is conducted, i.e. averaging together sets of n time points to reduce the exponential expansion of the search space. In the case of our data, the 17 time points are interpolated to 18 time points, and the time series are broken down into sets of 2 to be piecewise averaged
-
These piecewise averaged points are then converted into symbols through the use of Gaussian breakpoints. Gaussian breakpoints are divisions in the Gaussian distribution such that the cumulative probability of each section are equivalent. These can be obtained through the use of CDF tables found in statistics text books or by solving the following equation for b:
The overall process of assigning a letter to each piecewise averaged point is illustrated in below:
-
After the symbolic transformation, the series of symbols is converted into a single integer via the formula:
Where c is the letter assigned to each piecewise averaged point, a is the size of the alphabet,27 and w is the total length of the expression profile divided by the number of points per piecewise average.31 The parameters of the alphabet were selected to so that the population distribution of motifs is non-exponential, to reflect the non-random distribution of expression profiles present in the data. w was chosen to preserve as much of the high frequency component of the signal as possible.
APPENDIX 2
|
References
- 1.Alberts B. Essential Cell Biology: An Introduction to the Molecular Biology of the Cell. New York: Garland Pub; 1998. [Google Scholar]
- 2.Allende ML, Manzanares M, Tena JJ, Feijoo CG, Gomez-Skarmeta JL. Cracking the genome’s second code: enhancer detection by combined phylogenetic footprinting and transgenic fish and frog embryos. Methods. 2006;39:212–219. doi: 10.1016/j.ymeth.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 3.Allocco DJ, Kohane IS, Butte AJ. Quantifying the relationship between co-expression, co-regulation and gene function. BMC Bioinformatics. 2004;5:18. doi: 10.1186/1471-2105-5-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Almon RR, DuBois DC, Pearson KE, Stephan DA, Jusko WJ. Gene arrays and temporal patterns of drug response: corticosteroid effects on rat liver. Funct Integr Genomics. 2003;3:171–179. doi: 10.1007/s10142-003-0090-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Almon RR, Lai W, DuBois DC, Jusko WJ. Corticosteroid-regulated genes in rat kidney: mining time series array data. Am J Physiol Endocrinol Metab. 2005;289:E870–882. doi: 10.1152/ajpendo.00196.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Balaji S, Iyer LM, Aravind L, Babu MM. Uncovering a hidden distributed architecture behind scale-free transcriptional regulatory networks. J Mol Biol. 2006;360:204–212. doi: 10.1016/j.jmb.2006.04.026. [DOI] [PubMed] [Google Scholar]
- 7.Berezikov E, Guryev V, Plasterk RH, Cuppen E. CONREAL: conserved regulatory elements anchored alignment algorithm for identification of transcription factor binding sites by phylogenetic footprinting. Genome Res. 2004;14:170–178. doi: 10.1101/gr.1642804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bigelow HR, Wenick AS, Wong A, Hobert O. CisOrtho: a program pipeline for genome-wide identification of transcription factor target genes using phylogenetic footprinting. BMC Bioinformatics. 2004;5:27. doi: 10.1186/1471-2105-5-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blanchette M, Tompa M. FootPrinter: A program designed for phylogenetic footprinting. Nucleic Acids Res. 2003;31:3840–3842. doi: 10.1093/nar/gkg606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.von Bubnoff A, Peiffer DA, Blitz IL, Hayata T, Ogata S, Zeng Q, Trunnell M, Cho KW. Phylogenetic footprinting and genome scanning identify vertebrate BMP response elements and new target genes. Dev Biol. 2005;281:210–226. doi: 10.1016/j.ydbio.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 11.Corcoran DL, Feingold E, Benos PV. FOOTER: a web tool for finding mammalian DNA regulatory regions using phylogenetic footprinting. Nucleic Acids Res. 2005;33: W442–446. doi: 10.1093/nar/gki420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Corpet DE, Pierre F. How good are rodent models of carcinogenesis in predicting efficacy in humans? Asystematic review and meta-analysis of colon chemoprevention in rats, mice and men. Eur J Cancer. 2005;41:1911–1922. doi: 10.1016/j.ejca.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 13.Dieterich C, Cusack B, Wang H, Rateitschak K, Krause A, Vingron M. Annotating regulatory DNA based on man-mouse genomic comparison. Bioinformatics. 2002;18(Suppl 2):S84–90. doi: 10.1093/bioinformatics/18.suppl_2.s84. [DOI] [PubMed] [Google Scholar]
- 14.Dieterich C, Wang H, Rateitschak K, Luz H, Vingron M. CORG: a database for COmparative Regulatory Genomics. Nucleic Acids Res. 2003;31:55–57. doi: 10.1093/nar/gkg007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ehret GB, Reichenbach P, Schindler U, Horvath CM, Fritz S, Nabholz M, Bucher P. DNA binding specificity of different STAT proteins. Comparison of in vitro specificity with natural target sites. J Biol Chem. 2001;276:6675–6688. doi: 10.1074/jbc.M001748200. [DOI] [PubMed] [Google Scholar]
- 16.Fang F, Blanchette M. FootPrinter3: phylogenetic footprinting in partially alignable sequences. Nucleic Acids Res. 2006;34:W617–620. doi: 10.1093/nar/gkl123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hinoi T, Lucas PC, Kuick R, Hanash S, Cho KR, Fearon ER. CDX2 regulates liver intestine-cadherin expression in normal and malignant colon epithelium and intestinal metaplasia. Gastroenterology. 2002;123:1565–1577. doi: 10.1053/gast.2002.36598. [DOI] [PubMed] [Google Scholar]
- 18.Hoffmann R, Valencia A. A gene network for navigating the literature. Nat Genet. 2004;36:664. doi: 10.1038/ng0704-664. [DOI] [PubMed] [Google Scholar]
- 19.Holloway DT, Kon M, DeLisi C. Integrating genomic data to predict transcription factor binding. Genome Inform. 2005;16:83–94. [PubMed] [Google Scholar]
- 20.Jeong H, Tombor B, Albert R, Oltvai ZN, Barabasi AL. The large-scale organization of metabolic networks. Nature. 2000;407:651–654. doi: 10.1038/35036627. [DOI] [PubMed] [Google Scholar]
- 21.Jiao K, Nau JJ, Cool M, Gray WM, Fassler JS, Malone RE. Phylogenetic footprinting reveals multiple regulatory elements involved in control of the meiotic recombination gene, REC102. Yeast. 2002;19:99–114. doi: 10.1002/yea.800. [DOI] [PubMed] [Google Scholar]
- 22.Lee JC, Lusis AJ, Pajukanta P. Familial combined hyperlipidemia: upstream transcription factor 1 and beyond. Curr Opin Lipidol. 2006;17:101–109. doi: 10.1097/01.mol.0000217890.54875.13. [DOI] [PubMed] [Google Scholar]
- 23.Lin J, Keogh E, Lonardi S, Chiu B. A symbolic representation of time series, with implication for streaming algorithms. ACM SIGMOD Workshop on Research Issues in Data Mining and Knowledge Discovery; San Diego, CA: ACM; 2003. [Google Scholar]
- 24.Madan Babu M, Teichmann SA, Aravind L. Evolutionary dynamics of prokaryotic transcriptional regulatory networks. J Mol Biol. 2006;358:614–633. doi: 10.1016/j.jmb.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 25.Matys V, Fricke E, Geffers R, Gossling E, Haubrock M, Hehl R, Hornischer K, Karas D, Kel AE, Kel-Margoulis OV, Kloos DU, Land S, Lewicki-Potapov B, Michael H, Munch R, Reuter I, Rotert S, Saxel H, Scheer M, Thiele S, Wingender E. TRANSFAC: transcriptional regulation, from patterns to profiles. Nucleic Acids Res. 2003;31:374–378. doi: 10.1093/nar/gkg108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moraru II, Loew LM. Intracellular signaling: spatial and temporal control. Physiology (Bethesda) 2005;20:169–179. doi: 10.1152/physiol.00052.2004. [DOI] [PubMed] [Google Scholar]
- 27.Moses AM, Chiang DY, Kellis M, Lander ES, Eisen MB. Position specific variation in the rate of evolution in transcription factor binding sites. BMC Evol Biol. 2003;3:19. doi: 10.1186/1471-2148-3-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Murphy CT, McCarroll SA, Bargmann CI, Fraser A, Kamath RS, Ahringer J, Li H, Kenyon C. Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature. 2003;424:277–283. doi: 10.1038/nature01789. [DOI] [PubMed] [Google Scholar]
- 29.Neph S, Tompa M. MicroFootPrinter: a tool for phylogenetic footprinting in prokaryotic genomes. Nucleic Acids Res. 2006;34:W366–368. doi: 10.1093/nar/gkl069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nowak R. Mining treasures from ‘junk DNA’. Science. 1994;263:608–610. doi: 10.1126/science.7508142. [DOI] [PubMed] [Google Scholar]
- 31.Pfeiffer T, Soyer OS, Bonhoeffer S. The evolution of connectivity in metabolic networks. PLoS Biol. 2005;3:e228. doi: 10.1371/journal.pbio.0030228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ren B, Robert F, Wyrick JJ, Aparicio O, Jennings EG, Simon I, Zeitlinger J, Schreiber J, Hannett N, Kanin E, Volkert TL, Wilson CJ, Bell SP, Young RA. Genome-wide location and function of DNA binding proteins. Science. 2000;290:2306–9. doi: 10.1126/science.290.5500.2306. [DOI] [PubMed] [Google Scholar]
- 33.Richard R, Almon DCD, Jin Jin Y, Yao Zhenling, Hazra Anasuya, Samtani M, Snyder G, Piel W, Jusko W. New Research on Pharmacogenetics (Nova) 2006. [Google Scholar]
- 34.Rodriguez-Caso C, Medina MA, Sole RV. Topology, tinkering and evolution of the human transcription factor network. Febs J. 2005;272:6423–6434. doi: 10.1111/j.1742-4658.2005.05041.x. [DOI] [PubMed] [Google Scholar]
- 35.Sandelin A, Wasserman WW, Lenhard B. ConSite: web-based prediction of regulatory elements using cross-species comparison. Nucleic Acids Res. 2004;32:W249–252. doi: 10.1093/nar/gkh372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schneier B. Applied Cryptography: Protocols, Algorithms, and SourceCode in C. New York: Wiley; 1996. [Google Scholar]
- 37.Segal E, Shapira M, Regev A, Pe’er D, Botstein D, Koller D, Friedman N. Module networks: identifying regulatory modules and their condition-specific regulators from gene expression data. Nat Genet. 2003;34:166–176. doi: 10.1038/ng1165. [DOI] [PubMed] [Google Scholar]
- 38.Tellez C, Bar-Eli M. Role and regulation of the thrombin receptor (PAR-1) in human melanoma. Oncogene. 2003;22:3130–3137. doi: 10.1038/sj.onc.1206453. [DOI] [PubMed] [Google Scholar]
- 39.Wasserman WW, Sandelin A. Applied bioinformatics for the identification of regulatory elements. Nat Rev Genet. 2004;5:276–287. doi: 10.1038/nrg1315. [DOI] [PubMed] [Google Scholar]
- 40.Wolfe CJ, Kohane IS, Butte AJ. Systematic survey reveals general applicability of “guilt-by-association” within gene coexpression networks. BMC Bioinformatics. 2005;6:227. doi: 10.1186/1471-2105-6-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xuan Z, Zhao F, Wang J, Chen G, Zhang MQ. Genome-wide promoter extraction and analysis in human, mouse, and rat. Genome Biol. 2005;6:R72. doi: 10.1186/gb-2005-6-8-r72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang E, Berthiamume F, Yarmush ML, Androulakis IP. Proceedings of the Joint 9th International Symposium. Processing Systems Engineering and 16th European Symposium; 2006. [Google Scholar]
- 43.Zhang C, Xuan Z, Otto S, Hover JR, McCorkle SR, Mandel G, Zhang MQ. A clustering property of highly-degenerate transcription factor binding sites in the mammalian genome. Nucleic Acids Res. 2006;34:2238–2246. doi: 10.1093/nar/gkl248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao F, Xuan Z, Liu L, Zhang MQ. TRED: a transcriptional regulatory element database and a platform for in silico gene regulation studies. Nucleic Acids Res. 2005;33:D103–107. doi: 10.1093/nar/gki004. [DOI] [PMC free article] [PubMed] [Google Scholar]