

Abstract
Femtosecond photodynamics of the Pfr form of the red/far-red phytochrome N-terminal PAS-GAF-PHY photosensory core module of the cyanobacterial phytochrome Cph1 (termed Cph1Δ) from Synechocystis were resolved with visible broadband transient absorption spectroscopy. Multiphasic generation dynamics via global target analysis revealed parallel evolution of two pathways with distinct excited- and ground-state kinetics. These measurements resolved two subpopulations: a majority subpopulation with fast excited-state decay and slower ground-state dynamics, corresponding to previous descriptions of Pfr dynamics, and a minority subpopulation with slower excited-state decay and faster ground-state primary dynamics. Both excited-state subpopulations generated the isomerized, red-shifted Lumi-Ff photoproduct (715 nm); subsequent ground-state evolution to a blue-shifted Meta-Fr population (635 nm) proceeded on 3 ps and 1.5 ns time scales for the two subpopulations. Meta-Fr was spectrally similar to a recently described photoinactive fluorescent subpopulation of Pr (FluorPr). Thus, the reverse Pfr to Pr photoconversion of Cph1Δ involves minor structural deformation of Meta-Fr to generate the fluorescent, photochemically refractory form of Pr, with slower subsequent equilibration with the photoactive Pr subpopulation (PhotoPr).
Phytochromes are photoswitching proteins found in plants, fungi, and bacteria.1−3 In plants, phytochromes sense the ratio of red to far-red light to modulate light-induced responses such as seed germination, seedling establishment, flowering, and senescence.4−6 Phytochromes utilize heme-derived linear tetrapyrrole (bilin) chromophores for light sensing. Photoexcitation initiates a rapid photoisomerization reaction around the C15,16 double bond of the bilin chromophore followed by a series of chromophore–protein relaxation events on the ground-state surface leading to changes in biological signaling activity. Phytochromes from cyanobacteria utilize phycocyanobilin [PCB (Figure 1A)] as a chromophore, while other phytochromes utilize phytochromobilin (PΦB) and biliverdin (BV) chromophores.3,7
Figure 1.
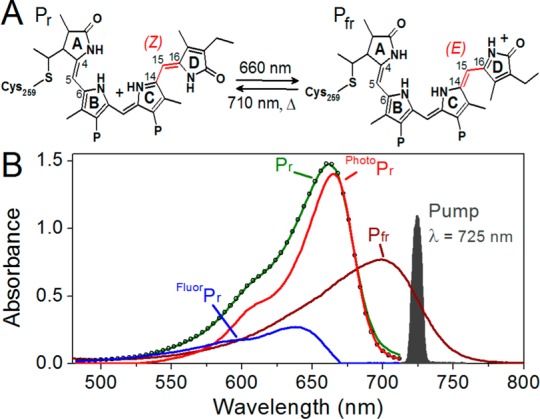
(A) Phycocyanobilin (PCB) chromophore in 15Z and 15E states. (B) Pfr pump–probe experiment with the 725 nm pump pulse spectrum (gray area) and Pfr (brown curve) spectrum compared. The spectrum of Pfr was computed to account for the residual absorbance of Pfr at photoequilibrium, and both Pr and Pfr spectra represent equimolar concentrations of each state. Also, the decomposition of the Pr spectrum (green) into fluorescent and photoactive Pr populations (blue and red, respectively) based on SVD analysis of temperature-dependent Pr absorbance bands is shown.21 The empty circles show the simulated Pr spectrum with fluorescent and photoactive Pr populations as bases.
The N-terminal PAS-GAF-PHY photosensory core module of the full length Cph1 protein (amino acids 1–514, here termed Cph1Δ) from Synechocystis sp. PCC6803 has served as an excellent model system for plant phytochromes because of its robust recombinant expression and known crystal structure.8−10 The full length Cph1 protein consists of Cph1Δ coupled to a C-terminal histidine kinase domain, and both proteins exhibit nearly identical photodynamics.11 Red illumination of the dark-adapted 15ZPr state of Cph1Δ (Figure 1B, red curve) initiates forward photoconversion (Pr to Pfr), generating the primary isomerized Lumi-Rf intermediate.a 12−17 Lumi-Rf thermally evolves via several intermediates to generate the 15EPfr photoproduct (Figure 1B, dark red curve) on a >100 ms timescale.18,19 The dark-stable 15ZPr state can be regenerated from 15EPfr either rapidly by far-red light (∼700 nm) or via spontaneous dark reversion on a very slow (>24 h) timescale.7,20
Despite numerous reports that address the primary forward photodynamics of Cph1Δ,13,15,16,21 including multipulse transient absorption studies,14,17,22 the reverse primary photodynamics (15EPfr to 15ZPr) are less well characterized. The reverse excited-state photodynamics of Cph1Δ are appreciably faster than those of the forward photoreaction,12,23 as has been reported for other phytochromes.24−27 Diller and co-workers resolved the biphasic decay of the Pfr excited state of Cph1Δ with lifetimes of 540 fs and 3.2 ps.12 More recently, we resolved 260 fs and 2.8 ps decay lifetimes using broadband transient absorption spectroscopy.23 On the basis of the evidence that the Pfr ground state of Cph1Δ is homogeneous,28,29 we attributed these components of Pfr excited state decay to the formation and decay of a photochemically nonproductive vibrationally hot ground state relaxing back to the Pfr ground state.23 Subsequent intermediates in reverse photoconversion have been characterized via cryo-trapping methods,30,31 but an equivalent study using time-resolved spectroscopy at physiological temperature has not been reported for Cph1Δ.
More recently, the circular dichroism (CD) spectra of phytochrome Pfr states have been interpreted as arising from a heterogeneous ground state.32 There is growing evidence that observed multiphasic excited-state dynamics in phytochromes and related PCB-incorporating GAF domains cyanobacteriochrome (CBCR) photosensors arises due to ground-state heterogeneity.21,33,34 In recent temperature- and excitation wavelength-dependent studies of forward photoconversion of Cph1Δ,21 we resolved five Pr* excited-state decay pathways arising from photoactive (PhotoPr) and blue-shifted fluorescent (FluorPr) subpopulations (Figure 1B, red and blue curves, respectively). PhotoPr productively formed the primary Lumi-RF photoproduct on a 20 ps timescale, whereas FluorPr did not yield Lumi-RF and exhibited longer excited-state lifetimes. The spectral properties, low quantum yield, and long-lived excited states of FluorPr were all remarkably similar to the properties of Y176H variant Cph1Δ.35,36 The ground-state bleach bands observed in transient absorption studies of the CBCRs NpR6012g4 from Nostoc punctiforme and RcaE from Fremyella diplosiphon exhibited pronounced excitation wavelength dependence.34,37 Ground-state heterogeneity may thus be more widespread in the phytochrome and CBCR family of photosensors than previously appreciated.38,39
We here extend our recent study of the ultrafast dynamics of the reverse reaction of Cph1Δ23 by using a narrowband excitation pump system (Figure 1B and Figure S1 of the Supporting Information) with an improved signal-to-noise ratio and greater temporal range (7 ns vs 100 ps). The new narrowband excitation data resolve clear multiphasic decay of the Pfr excited-state population, demonstrating heterogeneity of the Pfr ground state. Measurement up to 6 ns resolves multiphasic secondary photoproduct formation, not previously observed. We interpret the Cph1Δ reverse reaction as arising from the parallel evolution of two productive, kinetically distinct Pfr subpopulations arising via ground-state heterogeneity. Our studies also suggest that the Pfr photoreaction initially produces the fluorescent FluorPr subpopulation, which then slowly equilibrates with the photoactive PhotoPr population. This study thus illustrates the utility of transient absorption techniques to elucidate the underlying ground-state heterogeneity in photoreceptors.
Experimental Procedures
Protein Purification
Cph1Δ protein was purified after recombinant expression in Escherichia coli cells engineered to produce phycocyanobilin (PCB) as described previously.10,40
Ultrafast Experimental Setup
The ultrafast laser source consisted of an amplified Ti:sapphire laser system (Spectra Physics Spitfire Pro) that delivered 800 nm pulses with a 2.3 mJ pulse energy at a 1 kHz repetition rate and a 40 fs full width at half-maximum (fwhm) pulse duration.41 The laser output was split into two separate pathways for generating pump and probe pulses. Broadband white light probe pulses were generated by focusing the 800 nm pulses into a slowly translating 2 mm CaF2 crystal. The resulting probe light was then focused onto the sample and dispersed by a commercial spectrograph (Oriel MS125) to be detected with a linear 256-pixel photodiode array (Hamamatsu S3901 and C7884).
Excitation pulses (Figure 1B, gray shape) were generated by a two-stage home-built noncollinear optical parametric amplifier (NOPA) that produced tunable visible excitation pulses.42 The first NOPA stage generated a seed beam that was spectrally filtered (λcenter = 725 nm; 10 nm bandwidth) and amplified by a second NOPA. This amplified light was filtered again by a second interference filter of identical specifications. An instrument response function (IRF) of 120 fs was estimated by the rise time of the excited-state absorption (ESA) band of the pump–probe signals of the IR-140 laser dye. The energy of the pump pulses was 900 nJ/pulse at the sample.
The pump beam was chopped at 500 Hz to generate difference spectra with respect to the nonpumped probe spectrum. The probe beam was optically delayed with respect to the pump pulse with a computer-controlled linear motor stage (Newport IMS600LM), which allowed up to 6 ns temporal separation. Pump pulses were linearly polarized and set to 54.7° (magic angle) with respect to probe pulse polarization. Pump pulse spot diameters of 250–360 μm were estimated using a micrometer stage and a razor blade; the broadband probe pulses were focused to ∼50 μm. The appreciably greater pump pulse volume minimizes artifactual contributions to the signals due to varying spatial overlap between pump and probe beams. This minimization was confirmed by monitoring the signal amplitude and spectral shape while dithering the pump beam with respect to the probe beam.
The sample was passed continuously in a closed circuit to ensure fresh sample for each excitation pulse. The sample was continuously illuminated with a red light-emitting diode (Epitex Inc., L650-66-60; λcenter = 648 nm) through a quartz window to shift the Pr ⇌ Pfr equilibrium to favor the Pfr state in the sample cell. Because the 725 nm excitation light is not resonant with the Pr spectrum (Figure 1), no interfering Pr signals were observed in the data. The path length of the quartz cuvette was 2 mm, and the optical density at the red absorbance band was 0.4–0.5 at that path length. All experiments were performed at room temperature.
Results
Biphasic Photodynamics of Pfr
Pfr to Pr primary transient absorption (TA) spectra at selected times are contrasted in Figure 2. These spectra can be decomposed into four overlapping contributions: (1) negative ground-state bleach originating from the loss of ground-state population due to the excitation pulse, (2) negative stimulated emission (SE) signals arising from the probe pulse, (3) positive excited-state absorption (ESA) signals arising from S1 → Sn transitions, and (4) positive photoproduct absorption that arises from the S0 → S1 transition of photoproduct or ground-state intermediates (GSI). The ground-state bleach contribution is spectrally identical to the inverted ground-state absorption in a homogeneous population. However, in a situation with multiple ground-state subpopulations, the bleach is the sum of the inverted ground-state absorptions from the fraction of each subpopulation actually excited by the pump pulse. While both SE and ESA bands are markers of excited-state Pfr* populations, interpretation of the ESA is a simpler way of characterizing excited-state kinetics due to less complicated overlap with photoproduct, bleach, and SE bands.
Figure 2.
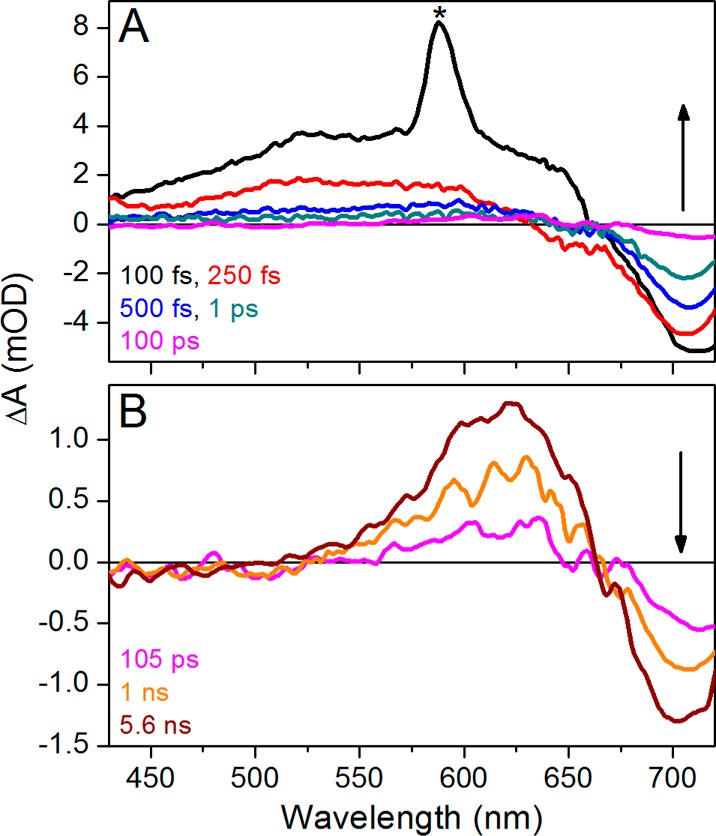
Transient absorption spectra of the Pfr to Pr photoreaction in the <100 ps time range (A) and >100 ps to 6 ns time range (B). The asterisk in panel A refers to the water Raman peak. The 100 ps transient spectrum (magenta) is shown in both panels.
At times of <100 ps, transient spectra (Figure 2A) are nearly identical to the previously reported broadband pump data23 (Figures S2 and S3 of the Supporting Information), with a slight difference in bleach amplitude attributed to differences in excitation overlap.34,37 The 100 fs transient spectrum (black curve) exhibits a broad ESA band from 430 to 675 nm, a negative band arising from bleach and/or SE beyond 675 nm, and a sharp water Raman peak at 580 nm. This spectrum rapidly decays within 100 fs (Figure 2A), reflecting the evolution of populations outside the Franck–Condon region.23 The 250 fs spectrum (Figure 2A, red curve) exhibits a broad positive band from 430 to 640 nm and a negative bleach and/or SE at >640 nm. The 520 nm signal (Figure 3A) exclusively tracks ESA and hence reports the decay of the excited-state population with biphasic relaxation kinetics with a subpicosecond component and a slower ∼2 ps component. The 520 nm kinetics are fit to both single- and double-exponential decay functions convolved over a 120 fs IRF in Figure 4. The single-exponential fit extracts a 220 fs lifetime for the best fit of the data, but the residual exhibits a systematic deviation from the data. Such a deviation is not seen with a biexponential fit, consisting of two lifetimes at 170 fs (94%) and 2 ps (6%). This may indicate either a complex bifurcating excited-state potential energy surface or the two Pfr* populations coexisting upon photoexcitation with a 94%/6% occupation.
Figure 3.
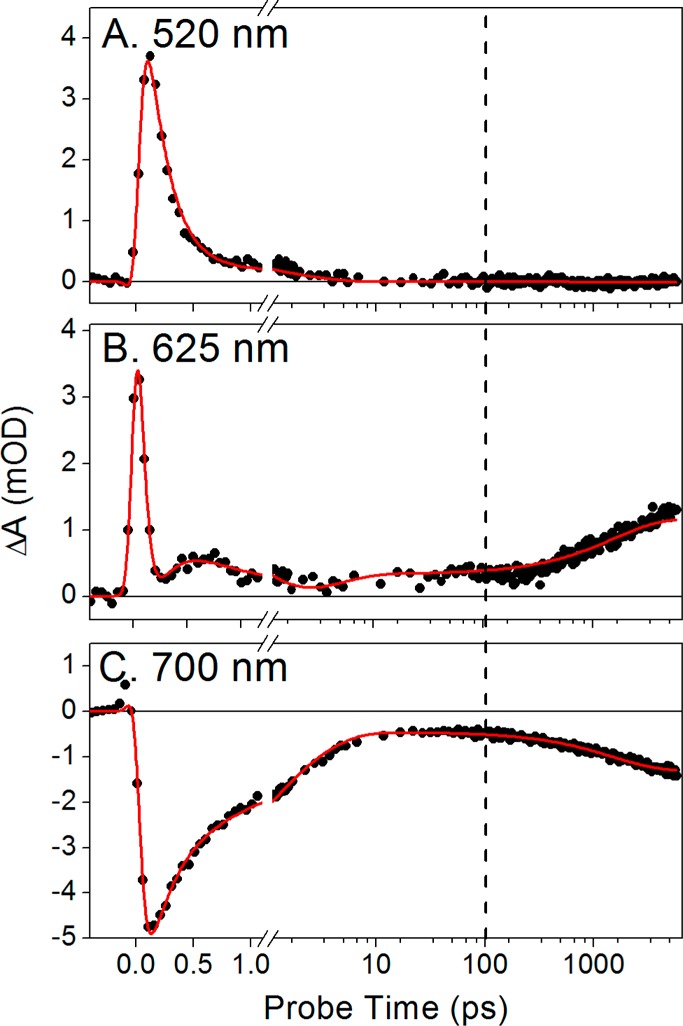
Kinetics traces of the Pfr to Pr reaction at selected probe wavelengths as indicated. The vertical dashed line marks the 100 ps probe time, which is the maximal probe time from the previous experiment.23 The traces are fit (red) with the target model in Figure 8A.
Figure 4.
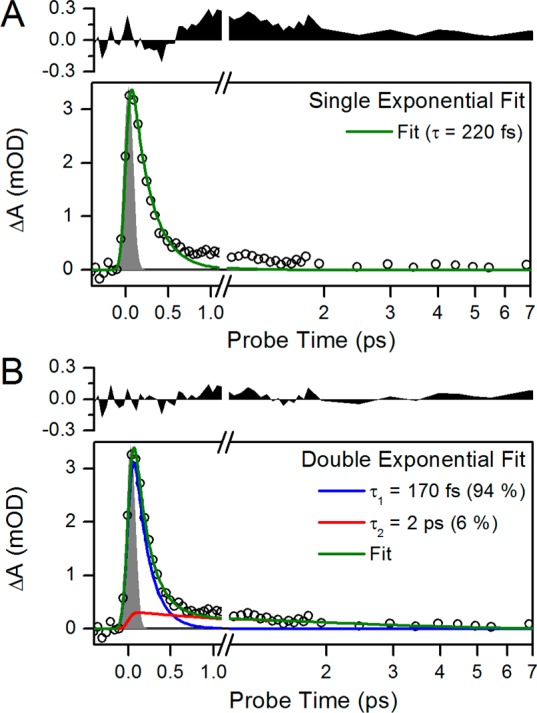
(A and B) Single- and double-exponential fitting of the 520 nm ESA kinetic traces, respectively. The fit is convoluted with a 120 fs IRF (gray area). The residual of each fit is plotted above with the same y-axes for comparison of the single- and double-exponential fits.
Other regions of the spectrum do not exhibit time scales comparable to that seen for the excited state at 520 nm (Figure 3B,C). At 625 nm, the amplitudes of the positive absorption signals begin to increase at approximately 200 ps (Figure 3B), clearly indicating the appearance of a species with absorption at shorter wavelengths on this time scale. The amplitude of the ground-state bleach band peaking at 700 nm steadily decreases until approximately 50 ps (Figure 3C). At later times (>200 ps), the amplitude of the bleach apparently increases even in the absence of repeated photoexcitation. An isosbestic point is observed at ∼660 nm (Figure 2B). The 625 nm (Figure 3B) and 700 nm (Figure 3C) kinetic traces thus reveal secondary dynamics at >100 ps that were not resolved in the small time windows of previous studies.12,23
As a first step in interpreting the apparent increasing ground-state depletion at later times, we compared the evolution at 700 and 625 nm in more detail (Figure 5). After ∼10 ps, both traces plateau to preterminal values that remain unchanged to 200 ps (Figure 5A). The preterminal signal amplitudes are 0.36 and −0.47 mOD at 625 and 700 nm, respectively, after which the signals increase their respective amplitudes and mirror each other, again reaching stable terminal plateau values with amplitudes of 1.25 and −1.34 mOD, respectively. The ratios of the late to early signal amplitudes are 3.47 and 2.85 at 625 and 700 nm, respectively. Were these wavelengths reporting evolution of a homogeneous population between two species, these ratios would be the same. The observed discrepancy in these ratios is likely due to the presence of multiple spectral species contributing to the negative band around 700 nm. We therefore compared earlier and later probe time spectra (Figure 5B). Comparison of the experimental 5.6 ns spectrum to the mean spectrum of the plateau region (10–100 ps) shows good agreement in the positive absorption band peaking at 625 nm (Figure 5B, magenta and teal curves, respectively). However, the bleach region peaking at 710 nm differs in amplitude, also shown by the difference spectrum between the two curves (Figure 5B, black curve), which has a negative band peaking around 720 nm. This difference spectrum is attributed to that of the primary photoproduct Lumi-Ff (see below).a
Figure 5.
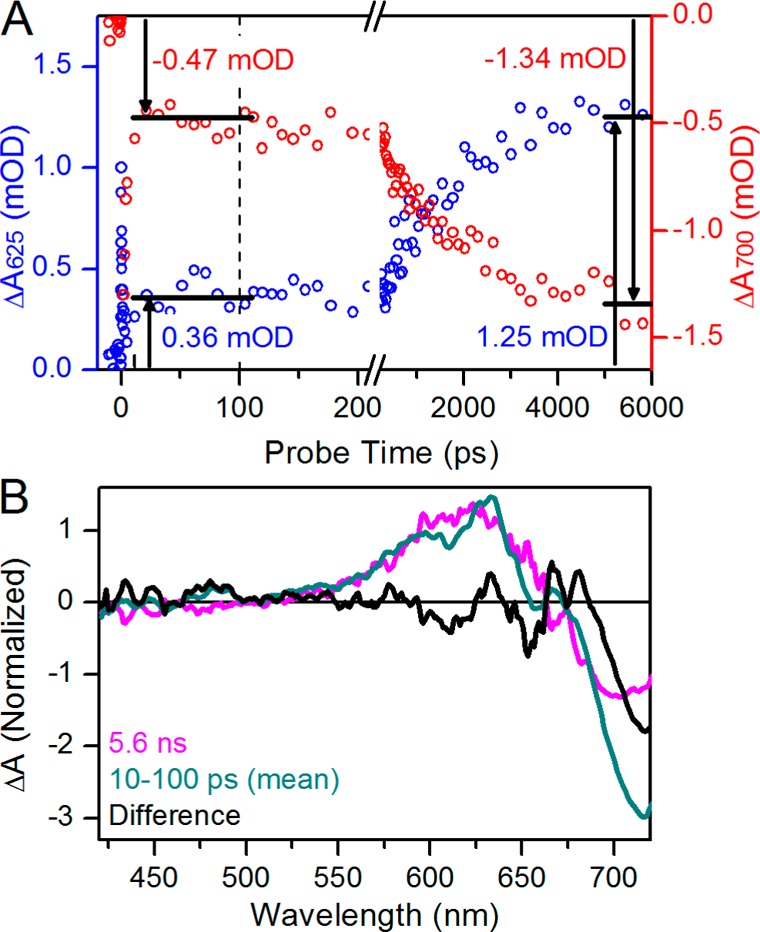
(A) Comparison of amplitudes between earlier and later probe time ranges at 625 and 700 nm probe wavelengths (blue and red and left and right y-axes, respectively). Horizontal bars indicate plateaus; the earlier time was averaged from 10 to 100 ps, and the later time was averaged from 4 to 6 ns. (B) Comparison between 5.6 ns and 10–100 ps spectra (magenta and dark cyan, respectively). The 10–100 ps spectra are averaged and scaled 5-fold for review. The difference between them is obtained (black).
The increasing magnitude of the ground-state bleach band at later times (Figure 5A, red circles) is not physically possible in a photoinitiated system under continuous flow, as the increased amplitude of the bleach signal indicates an increasing photoreaction quantum yield at later times, as if the initially photoexcited but nonproductive population becomes photoexcited again. Thus, this phenomenon suggests the presence of an intermediate population with positive absorption spectrally overlapping the Pfr ground-state bleach. The bleach band is a primary contributor to the negative signal in sub-10 ps dynamics (Figure 3C), and the preterminal amplitude at 700 nm is a permanent bleach that cannot increase its amplitude. Thus, as the spectrally overlapping population decays, the permanent bleach is restored to give the mistaken impression of a growing bleach. The rise of the 625 nm kinetics and an observed isosbestic point at 660 nm (Figure 2B) thus are consistent with evolution of Lumi-Ff into a blue-shifted secondary photoproduct on a subnanosecond time scale.
Spectral Simulation of Primary and Secondary Photoproducts
The spectral similarity between the plateau region spectrum (10–100 ps) and the terminal spectrum (5.6 ns) on the 625 nm absorption band (Figure 5B) and the presence of two photoproduct generation phases (<10 and >100 ps) suggest parallel reactions (i.e., two Lumi-Ff → the blue-shifted intermediate), and reflects a heterogeneous Pfr* excited state with different time scales. It appears that the plateau region spectrum contains both Lumi-Ff and the blue-shifted intermediate, whereas the terminal spectrum contains just the blue-shifted intermediate (Figure 5B). To test this interpretation further and facilitate global analysis of the data (see below), we conducted a spectral decomposition of the data (Figure 6). Spectra of the two photoproducts were estimated from the decomposition of both narrowband and broadband excitation spectra. The preterminal plateau spectrum is simulated from the 100 ps transient spectrum of the broadband excitation data, which exhibits positive absorption in both far-red and red regions (Figure 6A, magenta curve).23 This spectrum was previously assigned to the “Lumi-F” (distinct from Lumi-Ff) photoproduct under the assumption that Lumi-F has a broadened spectrum with absorption to both the blue and the red of Pfr.23 A similar absorption band in the far-red spectral region was also resolved in plant phytochrome reverse primary dynamics.24,25 However, absorption at 625 nm after 5.6 ns is similar to this band (Figure 5B and Figure S5F of the Supporting Information), and the red-shifted component has decayed by this point as shown by recovery of the bleach (Figure S4 of the Supporting Information). Furthermore, the rise of the blue-shifted component occurs with distinct kinetics, after decay of the excited state (Figure 3B). These data all suggest that the 100 ps spectrum (or Lumi-F) consists of two photoproducts with a shared Pfr bleach.
Figure 6.

Model simulations of >100 ps transient spectra. (A and B) Spectral decomposition of 100 ps and 5.6 ns transient absorption difference spectra by Lumi-Ff, Lumi-Fr, and Pfr bleach spectra. Both Lumi-Ff and Lumi-Fr are estimated as Gaussians with a λcenter values of 715 and 635 nm and Δλ values of 150 and 100 nm, respectively. The Pfr bleach is the inverted Pfr spectra from Figure 1B. (C) Species-associated spectra (SAS) of Pfr (black), Lumi-Ff (red), and Lumi-Fr (blue). (D) Simulation of >100 ps difference spectra with three basis sets in panel C. The Pfr bleach is fixed.
We assumed that the Pfr bleach is an inverted Pfr spectrum (Figure 1B, dark red curve), implying Pfr subpopulations exhibiting nearly identical absorption spectra but different kinetics. The spectrum of the blue-shifted intermediate was estimated from the inverted Pfr spectrum and the 5.6 ns spectrum recorded after narrowband excitation (Figure 6B) because the red-shifted intermediate Lumi-Ff has largely decayed by 5.6 ns (Figure 5B) as shown by depletion of the negative band peaking at 720 nm. Combination of a simple Gaussian with a center wavelength of 635 nm and a 100 nm bandwidth (fwhm) and the fixed Pfr bleach (Figure 6C, blue and black curves, respectively) simulates the 5.6 ns transient spectrum well (Figure 6B, cyan and magenta). Following the nomenclature protocols recently proposed for CBCRs (and also presented here),37 we designate the red-absorbing species at 635 nm as Meta-Fr, a secondary intermediate arising from far-red illumination and absorbing red light.a The combination of Meta-Fr and Pfr bleach spectra allowed extraction of the red-shifted primary photoproduct Lumi-Ff from the broadband excitation data; for simulation, Lumi-Ff was deduced by a simple Gaussian spectral shape (Figure 6A) with a center wavelength of 715 nm and a 150 nm bandwidth (fwhm).
The Gaussians used to simulate the Lumi-Ff and Meta-Fr intermediates are contrasted with the Pfr ground-state absorption spectra in Figure 6C. Combining these two Gaussians allowed us to simulate difference spectra at varying probe times for comparison to experiment (Figure 6D). A slight deviation from experiment was observed at approximately 100 ps, but excellent agreement was obtained at later probe times. The simulated spectra also predicted the experimentally observed 660 nm isosbestic wavelength observed for the underlying two-population kinetics (Figures 2 and 6D). The difference at 100 ps is likely to indicate that Lumi-Ff does not have a Gaussian line shape and hence could be more similar to Pfr or Pr with a higher-energy vibronic tail (Figure 1B).
Evolution between 100 ps and 1 ms
Figure 7 contrasts 5.6 ns and 1 ms transient spectra (orange and gray curves, respectively). The previous broadband excitation (Figure S1 of the Supporting Information) data,23 collected with a different probe window (450–740 nm), exhibit a positive photoproduct absorption band peaking around 730 nm at 100 ps that has decayed by the 1 ms spectrum (Figure S4B of the Supporting Information, arrow). This region of the spectrum is not detected in this study but is satisfactorily modeled in the spectral decomposition (see above) and global analysis (see below).
Figure 7.
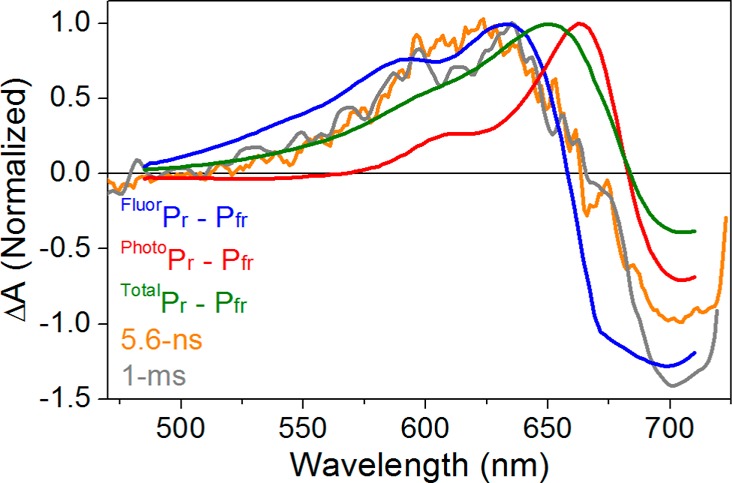
Comparison between the 6 ns and 1 ms difference spectra (orange and gray curves, respectively) in Pfr to Pr dynamics and the Pr – Pfr difference spectra. Three kinds of Pr – Pfr difference spectra are constructed on the basis of fluorescent and photoactive Pr (blue and red, respectively) and complete Pr spectra (green) represented in Figure 1B. The FluorPr – Pfr spectrum is scaled to match the 5.6 ns spectrum. The PhotoPr – Pfr and TotalPr – Pfr difference spectra used the same Pfr amplitude, and PhotoPr and TotalPr have the same respective ratio to the FluorPr amplitude in Figure 1B. The 1 ms spectrum is scaled to the 5.6 ns spectrum.
Discrepancies among the Pr – Pfr difference spectrum, the transient difference spectrum after 100 ps, and the transient difference spectrum after 1 ms were previously interpreted as evidence that evolution occurs after 100 ps for the Meta-F intermediate to generate Pr.23 This interpretation rests on the assumption that the Pr subpopulations are spectrally similar, such that photoconversion of an ensemble of Pr subpopulations in the static difference spectrum will provide an accurate comparison for transient difference spectra. However, we have recently resolved spectrally distinct FluorPr and PhotoPr (Figure 1B) subpopulations of Pr in Cph1Δ.21 We therefore compared 5.6 ns and 1 ms spectra with narrowband excitation to three Pr – Pfr difference spectra (Figure 7): FluorPr – Pfr (blue curve; λpeak = 633 nm), PhotoPr – Pfr (red curve; λpeak = 663 nm), and TotalPr – Pfr (green curves; λpeak = 650 nm). Neither the TotalPr – Pfr spectrum nor the PhotoPr – Pfr difference spectrum is in good agreement with the observed transient spectra. The FluorPr – Pfr difference spectrum is in better agreement with both transient spectra, suggesting that the secondary photoproduct Meta-Fr formed upon photoexcitation of Pfr either is very similar to the FluorPr subpopulation or is the same species within the resolution of our data.
Global Analysis of Cph1Δ Reverse Photoconversion
We analyzed the narrowband transient signals using a global analysis formalism to decompose the transient signals into the constituent evolution of a finite number of species.43,44 Within this framework, transient difference spectra are described by eq 1:
| 1 |
where the change in absorption ΔA at probe time t is the summation of n species with distinct time-dependent concentrations, cl(t), and fixed difference spectra, Δεl(λ). The time-dependent concentration of each species is given by solution of the first-order linear differential equation shown in eq 2:
| 2 |
where c(t) is the 1 × n vector describing the time-dependent concentration of the respective lth species, K is the matrix describing the connectivity scheme among species dictated by the chosen target model, Ipump(t) is the IRF of the pump pulse, and xl is the initial fractional occupation of lth species by the excitation pulse. This analysis extracts the number of species, their connectivity scheme, and the extracted spectra. If the model successfully describes the underlying sample dynamics, then the extracted spectra, Δεl(λ), represent the true difference spectra of the constituent transient populations and are called species-associated difference spectra (SADS). If the analysis unsuccessfully models the data, then the estimated spectra are linear combinations of the SADS.
In constructing a target model, we first used a simpler scheme that describes the transient signals as a sequential flow of spectral species with single-exponential kinetics (i.e., species 1 → species 2 → ... → species n). The extracted difference spectra from this analysis are termed sequential evolution-associated difference spectra (EADS).45 This procedure estimates experimentally observed time scales, the number of species (n), and the general spectral evolution of the signals. Sequential analysis also provides valuable model-independent constraints for target model construction.34,45
Both narrowband and broadband excitation data sets were initially subjected to a sequential analysis (Figure S5 of the Supporting Information), which give an excellent fit to the respective signals (Figures S6 and S7 of the Supporting Information, respectively). The broadband excitation signals have one less EADS (n = 5 vs n = 6) because these data were only collected to 100 ps and do not resolve the second phase of Meta-Fr growth observed in the narrowband excitation data. The estimated EADS lifetimes are consistent between the two data sets at 60 fs, 180 fs, 770 fs, 2.8 ps, and 1.5 ns for the first five EADS. EADS5 for the broadband excitation data is constrained to a 1.5 ns lifetime to be consistent with the lifetime extracted from narrowband excitation signals, while the narrowband EADS6 are stable to the end of the experiment (τ = ∞). The EADS5 to EADS6 transition in the narrowband excitation signals corresponds to the experimentally observed dynamics observed after 100 ps (Figure 5A).
Side-by-side comparison of the sequential EADS for broadband and narrowband excitation data shows good agreement (Figure S8 of the Supporting Information), with a greater bleach magnitude observed for broadband excitation data. This difference is tentatively attributed to excitation wavelength-dependent dynamics, which have been reported for the Cph1Δ Pr state21 and in CBCR systems.34,37 EADS6 estimated from the narrowband excitation signals is nearly identical to the 1 ms spectrum, demonstrating negligible spectral evolution from 6 ns to 1 ms (Figure S5E of the Supporting Information). EADS6 of the narrowband excitation signals is also comparable to EADS5 of the broadband signals, with good spectral overlap at the 625 nm positive band (Figure S5F of the Supporting Information).
The biphasic decay of ESA, the sequential analysis described above, and the simulation of Pfr CD spectra32 argue for the adoption of a parallel evolving target model for interpreting the narrowband transient data (Figure 8A). By analogy to other phytochrome and CBCR systems,21,22,33,34,45 we modeled this as arising because of an inhomogeneous ground state rather than bifurcation on a homogeneous population on a excited-state surface. In the absence of evidence to the contrary, we assumed that the two subpopulations possess identical ground-state and excited-state spectra. Kinetic analysis of the ESA band at 520 nm (Figure 4) extracted fractional occupancies for the 1.90 ps and 180 fs components of 6 and 94%, respectively. These subpopulations are termed Pfr I and Pfr II, respectively, using the nomenclature adopted for CBCR systems.37 The overall quantum yield was set at ∼15% [15.4% for the target model (Table 1)], based on the previously reported quantum yield for reverse photoconversion of Cph1Δ.8 The low quantum yield indicates that nonproductive regeneration of the Pfr ground state is the dominant route for de-excitation of Pfr*. Branching between productive and nonproductive Pfr* is posited to occur at progression through the conical intersection and not during subsequent evolution of the ground-state photoproducts (discussed below).
Figure 8.

Global analysis of Pfr to Pr dynamics. (A) Proposed target model with two Pfr subpopulations (Pfr I and Pfr II). Each spectral species is represented in a box, and its apparent time constant is given in parentheses. (B) Estimated SADS of the target model. FCPfr*, Pfr*, GSI1, GSI2, Lumi-Ff, and Meta-Fr from populations I and II have identical respective SADS. (C) Comparison between Lumi-Ff SADS and the difference spectra in Figure 5B. (D) Comparison between Meta-Fr SADS (green) and FluorPr – Pfr and PhotoPr – Pfr difference spectra (blue and red, respectively). (E) Concentration profile of each constituent population. The color scheme is the same as that in panel A, with population I represented by the dashed curves.
Table 1. Kinetic Parameters and Branching Ratios of the Pfr* State Based on the Target Model from Figure 8Aa.
| initial occupancy (%) | apparent τ | % (Pfr* to GSI) | % (Pfr* to Lumi-Fr) | Φ (Lumi-Fr) (%) | |
|---|---|---|---|---|---|
| Pfr* I | 6 | 180 fs | 27 | 73 | 4.4 |
| Pfr* II | 94 | 1.9 ps | 88 | 12 | 11 |
The apparent τ is the observed lifetime of the spectral species. % (Pfr* to GSI) is the branching ratio of the respective Pfr* population to GSI1, and % (Pfr* to Lumi-Fr) is the branching ratio from Pfr* to Lumi-Fr. The addition of both parameters makes up 100% for respective subpopulations. Φ (Lumi-Fr) is the final quantum yield of Lumi-Fr in reference to the total (Pfr I + Pfr II) excited Pfr molecules.
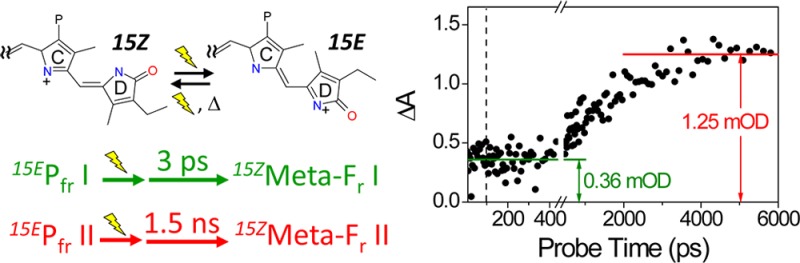
The target model includes branching between productive and nonproductive components in each Pfr* population (Table 1), with only the overall quantum yield constrained. The raw data at 625 nm provide an independent estimate of the branching ratios. Two phases of Meta-Fr photoproduct formation are clear in the 625 nm kinetics: a fast formation phase completed by approximately 10 ps and a slower phase proceeding on a 1.5 ns timescale (Figure 5A). The difference in time scales results in steady plateaus at 625 nm, allowing the ratio of absorption intensities for the early and later plateau regions to be 0.36:1.25. Assuming that Meta-Fr does not decay on the experimental timescale, the ratio of Meta-Fr yield between the earlier and later formation would thus be 0.36:0.89, which translates to absolute Φ values of 4.3 and 10.7%, respectively, if total yield is 15%.8 Because the initial excited-state occupancy of Pfr I is only 6%, it cannot account for the slowly arising component of the Meta-Fr yield, which requires at least 10.7% occupancy at 100% quantum yield. Given the initial occupancies and absolute Φ, we can set the branching ratio of the Pfr ground state to photoproduct at ∼28:72 and ∼89:11 for Pfr I and Pfr II, respectively. The calculated values derived from the target model (Table 1) are 27:73 and 88:11 to be consistent with the experimental estimate.
In the target model, each of the two Pfr* populations generates one component of Lumi-Ff that then decays into Meta-Fr (Figure 8A). The extracted Lumi-Ff SADS compares well with the difference spectrum calculated from the 5.6 ns transient spectrum and the mean transient spectrum from 10 to 100 ps (Figures 5B and 8C). The decay of Lumi-Ff after 100 ps is thus sufficient to explain the spectral evolution observed in the bleach region (Figure 6). The Meta-Fr SADS is in reasonable agreement with the FluorPr – Pfr difference spectrum (Figure 8D) and is consistent with the final 5.6 ns and 1 ms spectrum (Figure 7). The concentration of each species is plotted in Figure 8E (Pfr I in dashed lines and Pfr II in solid lines). The final Lumi-Fr concentration associated with Pfr I is 4.4% and with Pfr II is 11% (Table 1), in good agreement with the independent estimate derived from analysis of spectral evolution at 625 nm (see above).
The sequential EADS analysis estimated lifetimes of 60 fs, 180 fs, 770 fs, and 2.8 ps (Figure S7 of the Supporting Information). The 60 fs component is assigned to Franck–Condon relaxation as reported previously.23 The 180 fs EADS exhibits a clear ESA band peaking around 520 nm, indicating the presence of Pfr*. Pfr* decay is biphasic as discussed above, so the 2.8 ps component is likely to be a superposition of Pfr* decay and subsequent dynamics. The 770 fs component was not resolved in the previous broadband excitation data because of the lower signal-to-noise ratio in this data set (compare Figures S6 and S7 of the Supporting Information); however, a spectrally similar component can be extracted from these data using the current analysis (Figure S5D of the Supporting Information). In the narrowband data, this component is necessary to describe the <1 ps dynamics (Figure 3B). The 770 fs component (EADS3) is spectrally distinct from the Pfr* and HotPfr spectra resolved in the previous study, which are also resolved here as EADS2 and EADS4 (Figure S5 of the Supporting Information). In the current target model, the 770 fs component is assigned as the first GSI population [GSI1 (Figure 8A)], with spectral features qualitatively similar to those of the Meta-Fr photoproduct (Figure 8B, magenta and green curves, respectively). Because this component appears before the appearance of the primary Lumi-Ff photoproduct, we assign the 770 fs component to a nonisomerized GSI (GSI1) that adapts a structurally twisted, blue-shifted conformation.46−48 The 2.8 ps component (EADS4) is spectrally and kinetically analogous to the HotPfr GSI extracted from the previous broadband data, with red-shifted product absorption (Figure S5C,D of the Supporting Information). We therefore interpret this species as a second GSI species (GSI2) arising from the earlier GSI1 via vibrational relaxation before decaying back to the Pfr ground state.23,46
We tested this interpretation by fitting the narrowband data to a target model with just one GSI having a lifetime of 2.8 ps (Figure S9 of the Supporting Information). This model does not accurately describe the <1 ps dynamics (Figure S10B of the Supporting Information). By contrast, including two GSI populations in the final target model (Figure 8A) allows a good fit to the 625 nm kinetic trace (Figure 3B). The decay of nonproductive Pfr* back to the Pfr ground state is therefore postulated to occur via two resolved ground-state intermediate (GSI) populations.46−49 The final target model (Figure 8A) gives good agreement with the narrowband data set on all time scales examined (Figure 3).
We next applied the heterogeneous target model to the broadband excitation signals (Figure S11 of the Supporting Information), with excellent results (Figures S11 and S12 of the Supporting Information). Because the broadband excitation signals were collected only up to 100 ps, the 1.5 ns Lumi-Ff II to Meta-Fr II evolution was not modeled. The side-by-side comparison with respective SADS between the narrowband and broadband excitation signals (Figure S11 of the Supporting Information) shows consistencies in Pfr* and two GSI populations, albeit with variation in the bleach-band amplitude (∼700 nm). The initial Franck–Condon relaxation FCPfr* SADS are expected to vary between these data sets, because the location of the initially excited wavepacket is strongly affected by the excitation energy. The Lumi-Ff and Meta-Fr SADS show the most notable discrepancy (panels E and F, respectively, of Figure S11 of the Supporting Information). Unfortunately, analysis of the broadband excitation signals could not extract Lumi-Ff as a separate component because of the limited time window, and the Meta-Fr SADS was the mixture of Lumi-Ff and Meta-Fr populations. The concentration of Meta-Fr at 100 ps (Figure 8E) is too low for separation of Lumi-Ff and Meta-Fr by global analysis.
Discussion
Our previous interpretation of Cph1Δ primary reverse reaction dynamics was based on a homogeneous model that attributed a “broadened” spectrum to the Lumi-F primary photoproduct in comparison with the Pfr ground state.23 That study neither considered nor excluded the possibility that the Lumi-F’s broadened spectrum reflects simultaneous photogeneration of blue- and red-absorbing photoproducts.23 The main limitation of that study was the short temporal window examined. In this study, we have extended the analysis to longer times and have used a narrower excitation pulse, with a higher signal-to-noise ratio in the transient spectra. This allowed us to observe spectral evolution from the red-shifted Lumi-Ff primary photoproduct to a blue-shifted Meta-Fr species, a process described well by a target model with a heterogeneous Pfr ground state. Resonance Raman intensity analysis of the Cph1Δ Pfr state28 supported a homogeneous Pfr ground state, consistent with characterization of Pfr by solid-state NMR29 but inconsistent with a recent re-evaluation of Pfr CD spectra32 and with the transient absorption signals we report here. In comparable analyses of the Cph1Δ Pr state, solid-state NMR29 and transient absorption21 both revealed considerable heterogeneity, consistent with an earlier analysis of temperature and excitation wavelength effects50 but at odds with resonance Raman intensity analysis.51 For Pfr, the two kinetically distinct phases of Meta-Fr generation reported here strongly support the presence of at least two subpopulations. Other transient absorption studies of phytochromes and CBCRs have also reported multiphasic excited-state relaxation, typically interpreted as arising from coevolving subpopulations.33,34,45,47,52 This is consistent with characterization of other photoreceptor systems such as PYP and phototropin, which also exhibit multiexponential kinetics attributed to inhomogeneity.46,53
An inverse correlation between excited-state lifetime and quantum yield for primary photoproduct formation of red/green CBCR has been proposed,54 under the assumption that rapidly decaying excited states will be less prone to competing reactions. Two bacteriophytochromes from Rhodopseudomonas palustris also demonstrated an inverse correlation between excited-state lifetime and isomerization efficiency.55 However, Cph1Δ provides a counterexample to this trend, because Pfr* decays much more rapidly than Pr* despite comparable quantum yields.8,22 Interestingly, the Cph1Δ Pfr excited-state dynamics for the two subpopulations described here demonstrate such a counterexample within a single reaction: Pfr* I exhibits a slower lifetime but a higher branching ratio to the isomerized photoproduct when compared to Pfr* II (Table 1). It is not yet clear whether there is a general physical interpretation for such correlations.
In R. palustris bacteriophytochromes, excited-state proton transfer was proposed as a nonproductive excited-state quenching mechanism.55 For the forward reaction of the red/green CBCR NpR6012g4, pump–dump–probe experiments supported a twisted GSI population as the dominant pathway of excited-state decay.47 Such failed attempts at photoisomerization usually decay back to the original ground state,37 but the twisted GSI chromophore conformation in NpR6012g4 was shown to partition between the original ground state and the primary photoproduct.47 Such second-chance mechanisms would enhance quantum yield with no change in excited-state lifetime. In the Cph1Δ Pfr state, which has a low quantum yield, the need for two GSI populations in the target model (Figure 8A) suggests a rugged ground-state potential energy surface and raises the possibility of unproductive depopulation of the excited state to generate GSI species.
This study demonstrates that the primary photoproduct is a mixture of red-shifted Lumi-Ff species that decay to the secondary intermediate Meta-Fr on vastly different time scales. Lumi-Ff exhibits a red-shifted absorption maximum at ∼715 nm (Figure 6C), consistent with the red-shifted primary photoproducts seen in other phytochromes and CBCRs.12,24,34,45,56 The Lumi-Ff to Meta-Fr transition exhibits a significant blue shift from 715 to 635 nm, suggesting a major structural rearrangement. A combination of cryo-trapping and solid-state NMR allowed characterization of two intermediates in the Cph1Δ reverse reaction, “Lumi-F” and “Meta-F”.30 The Lumi-F to Meta-F conversion triggered more significant chemical shift changes than those observed for the Meta-F to Pr conversion, especially for atoms associated with the D-ring of the chromophore. These observations led to the conclusion that Lumi-F possesses an isomerized 15Z configuration but is structurally more constrained than Meta-F. Two intermediates were also resolved in a temperature-scan crystallographic study of Pfr to Pr photoconversion in a bacteriophytochrome from Pseudomonas aeruginosa.57 The first intermediate exhibited D-ring isomerization, whereas the second intermediate exhibited a more twisted chromophore conformation but a more relaxed protein environment. These structural changes are consistent with the blue shift observed upon the analogous transition from Lumi-Ff to Meta-Fr in Cph1Δ.
In our previous study of the Cph1Δ reverse reaction photodynamics,23 the TotalPr spectrum was used to calculate the static Pr – Pfr difference spectrum used as a comparison (Figure 7, green curve). Since then we resolved two spectrally distinct subpopulations with the Cph1Δ Pr state, FluorPr and PhotoPr.21 We therefore compared the individual FluorPr – Pfr and PhotoPr – Pfr difference spectra both to the transient spectra taken at 5.6 ns and 1 ms (Figure 7) and to the Meta-Fr SADS (Figure 8D). Remarkably, the FluorPr – Pfr difference spectrum (blue curve) is the best match to the experimental transient spectra. This is consistent with the lack of major structural changes of the chromophore during the formation of Pr from Meta-F in Cph1Δ,30,57 so the Meta-Fr species resolved in this study seems to be consistent with the Meta-F intermediate resolved by cryo-trapping. The strong spectral similarity between Meta-Fr and FluorPr also suggests that reverse photoconversion initially forms FluorPr and the regeneration of the red-shifted, photoactive PhotoPr subpopulation occurs on a slower timescale via thermal equilibration of the FluorPr and PhotoPr subpopulations. This observation may also imply the existence of a short refractory period within the Cph1Δ photocycle, during which newly regenerated Pr cannot be photoconverted to Pfr because it is in the less active FluorPr substate.
The schematic of the photocycle of Cph1Δ is demonstrated in Figure 9, including both ultrafast and secondary dynamics of the Pr and Pfr states. The dark-adapted Pr state consists of two coexisting populations in thermal equilibrium: FluorPr and PhotoPr. Upon red light excitation, only PhotoPr generates Lumi-R photoproduct, which then proceeds through Meta-Ra and Meta-Rc intermediates before forming the Pfr state.58 We have now demonstrated that Pfr is also heterogeneous, with at least two subpopulations that exhibit parallel primary and secondary dynamics upon far-red light excitation. The red-shifted primary photoproduct Lumi-Ff undergoes further structural relaxation to generate the blue-shifted Meta-Fr intermediate with relaxation time scales of 3 ps and 1.5 ns for the two subpopulations. These time constants are much faster than those observed for structural relaxations in forward photoconversion of Cph1Δ and the related cyanobacterial phytochrome CphA.19,59 In contrast to the productive and nonproductive Pr subpopulations, both Pfr subpopulations can generate Meta-Fr populations that strongly resemble the FluorPr subpopulation. In Cph1Δ, further evolution may involve formation of an equilibrium with the photoactive PhotoPr subpopulation, but whether other phytochromes have similar Pr subpopulations is not yet clear. It will also be interesting to probe the molecular details underlying the dynamics observed in this study via transient CD and vibrational experiments to elucidate the molecular nature of phytochrome inhomogeneity.
Figure 9.
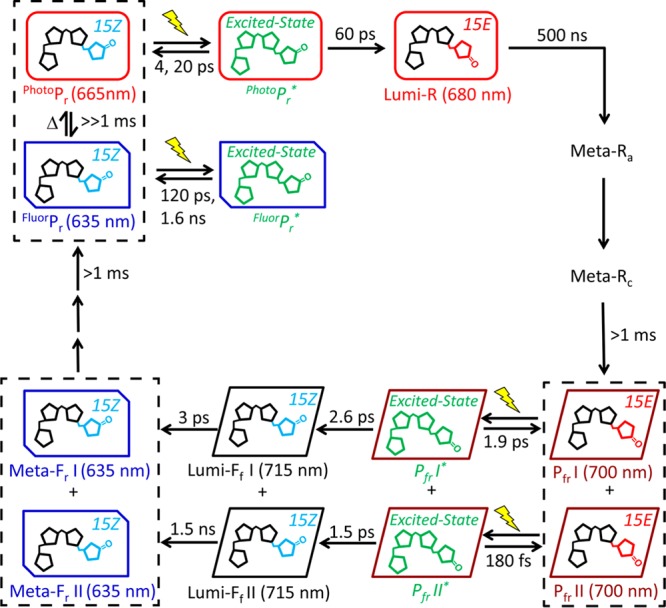
Scheme of the total Cph1Δ photocycle including both forward (Pr to Pfr) and reverse (Pfr to Pr). The spectrally and kinetically distinct fluorescent (FluorPr) and photoactive (PhotoPr) Pr subpopulations are in slow equilibrium (≫1 ms), which make up the overall Pr state. The light-activated pathways are indicated by photoexcitation (yellow light sign) and subsequent evolution via thermally activated steps (solid arrows). The evolution of Pfr to Lumi-Ff to Lumi-Fr occurs via heterogeneous pathways with different reaction kinetics and yields. Populations enclosed within black boxes indicate inhomogeneous coexisting populations.
Acknowledgments
Dr. Mikas Vengris from Light Conversion Ltd. is acknowledged for the donation of global and target analysis software package.
Glossary
Abbreviations
- Cph1Δ
PAS-GAF-PHY photosensory core module of Cph1
- BV
biliverdin
- CBCR
cyanobacteriochrome
- CD
circular dichroism
- EADS
evolution-associated difference spectrum
- GSI
ground-state intermediate
- ESA
excited-state absorption
- SE
stimulated emission
- IRF
instrument response function
- NOPA
noncollinear optical parametric amplifier
- PCB
phycocyanobilin
- PΦB
phytochromobilin
- Pr
red-absorbing dark state of red/far-red phytochromes
- Pfr
far-red-absorbing photoproduct state of red/far-red phytochromes
- FluorPfr
nonphotoactive, fluorescent Pr subpopulation
- PhotoPfr
photoactive Pr subpopulation
- Pfr*
excited-state population(s) derived from photoexcitation of Pfr
- PYP
photoactive yellow protein
- SADS
species-associated difference spectra
- SNR
signal-to-noise ratio
- Φ
photocycle quantum yield.
Supporting Information Available
Sequential EADS analysis of both narrowband and broadband excitation signals and extra target model analysis. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
This work was supported by grants from the Chemical Sciences, Geosciences, and Biosciences Division, Office of Basic Energy Sciences, Office of Science, U.S. Department of Energy (DOE DE-FG02-09ER16117). to both J.C.L. and D.S.L. and the National Institutes of Health (GM068552) to J.C.L.
Funding Statement
National Institutes of Health, United States
Footnotes
For the nomenclature of phytochrome photostates, in Lumi-Xy Lumi refers to the primary intermediate formed within 10 ns of photoexcitation, the uppercase X refers to the spectral range of the parent state (R for Pr and F for Pfr), and lowercase subscript y refers to the spectral range of the intermediate-state difference absorption peak. For the intermediates after 10 ns time windows, the intermediate is termed Meta-Xy. The color code for the spectral range is v for violet (380–450 nm), b for blue (450–495 nm), g for green (495–570 nm), y for yellow (570–590 nm), o for orange (590–620 nm), r for red (620–710 nm), and fr for far-red (710–750 nm). More details can be found in the Supporting Information of reference (60).
Supplementary Material
References
- Rockwell N. C.; Lagarias J. C. (2010) A Brief History of Phytochromes. ChemPhysChem 11, 1172–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Romero J.; Hedtke M.; Kastner C.; Muller S.; Fischer R. (2010) Fungi, Hidden in Soil or Up in the Air: Light Makes a Difference. Annu. Rev. Microbiol. 64, 585–610. [DOI] [PubMed] [Google Scholar]
- Auldridge M. E.; Forest K. T. (2011) Bacterial Phytochromes: More than Meets the Light. Crit. Rev. Biochem. Mol. Biol. 46, 67–88. [DOI] [PubMed] [Google Scholar]
- Quail P. H. (2002) Phytochrome Photosensory Signalling Networks. Nat. Rev. Mol. Cell Biol. 3, 85–93. [DOI] [PubMed] [Google Scholar]
- Franklin K. A.; Quail P. H. (2010) Phytochrome Functions in Arabidopsis Development. J. Exp. Bot. 61, 11–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M.; Chory J. (2011) Phytochrome Signaling Mechanisms and the Control of Plant Development. Trends Cell Biol. 21, 664–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockwell N. C.; Su Y. S.; Lagarias J. C. (2006) Phytochrome Structure and Signaling Mechanisms. Annu. Rev. Plant Biol. 57, 837–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamparter T.; Mittmann F.; Gartner W.; Borner T.; Hartmann E.; Hughes J. (1997) Characterization of Recombinant Phytochrome from the Cyanobacterium Synechocystis. Proc. Natl. Acad. Sci. U.S.A. 94, 11792–11797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essen L. O.; Mailliet J.; Hughes J. (2008) The Structure of a Complete Phytochrome Sensory Module in the Pr Ground State. Proc. Natl. Acad. Sci. U.S.A. 105, 14709–14714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockwell N. C.; Shang L.; Martin S. S.; Lagarias J. C. (2009) Distinct Classes of Red/Far-Red Photochemistry within the Phytochrome Superfamily. Proc. Natl. Acad. Sci. U.S.A. 106, 6123–6127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sineshchekov V.; Koppel L.; Esteban B.; Hughes J.; Lamparter T. (2002) Fluorescence Investigation of the Recombinant Cyanobacterial Phytochrome (Cph1) and its C-Terminally Truncated Monomeric Species (Cph1 Delta 2): Implication for Holoprotein Assembly, Chromophore-Apoprotein Interaction and Photochemistry. J. Photochem. Photobiol., B 67, 39–50. [DOI] [PubMed] [Google Scholar]
- Heyne K.; Herbst J.; Stehlik D.; Esteban B.; Lamparter T.; Hughes J.; Diller R. (2002) Ultrafast Dynamics of Phytochrome from the Cyanobacterium Synechocystis, Reconstituted with Phycocyanobilin and Phycoerythrobilin. Biophys. J. 82, 1004–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Thor J. J.; Ronayne K. L.; Towrie M. (2007) Formation of the Early Photoproduct Lumi-R of Cyanobacterial Phytochrome Cph1 Observed by Ultrafast Mid-Infrared Spectroscopy. J. Am. Chem. Soc. 129, 126–132. [DOI] [PubMed] [Google Scholar]
- van Wilderen L.; Clark I. P.; Towrie M.; van Thor J. J. (2009) Mid-Infrared Picosecond Pump-Dump-Probe and Pump-Repump-Probe Experiments to Resolve a Ground-State Intermediate in Cyanobacterial Phytochrome Cph1. J. Phys. Chem. B 113, 16354–16364. [DOI] [PubMed] [Google Scholar]
- Dasgupta J.; Frontiera R. R.; Taylor K. C.; Lagarias J. C.; Mathies R. A. (2009) Ultrafast Excited-State Isomerization in Phytochrome Revealed by Femtosecond Stimulated Raman Spectroscopy. Proc. Natl. Acad. Sci. U.S.A. 106, 1784–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y.; Linke M.; von Haimberger T.; Hahn J.; Matute R.; Gonzalez L.; Schmieder P.; Heyne K. (2012) Real-Time Tracking of Phytochrome’s Orientational Changes During Pr Photoisomerization. J. Am. Chem. Soc. 134, 1408–1411. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick A. E.; Lincoln C. N.; van Wilderen L. J.; van Thor J. J. (2012) Pump-Dump-Probe and Pump-Repump-Probe Ultrafast Spectroscopy Resolves Cross Section of an Early Ground State Intermediate and Stimulated Emission in the Photoreactions of the Pr Ground State of the Cyanobacterial Phytochrome Cph1. J. Phys. Chem. B 116, 1077–1088. [DOI] [PubMed] [Google Scholar]
- Rohmer T.; Lang C.; Hughes J.; Essen L. O.; Gartner W.; Matysik J. (2008) Light-Induced Chromophore Activity and Signal Transduction in Phytochromes Observed by C-13 and N-15 Magic-Angle Spinning NMR. Proc. Natl. Acad. Sci. U.S.A. 105, 15229–15234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Thor J. J.; Borucki B.; Crielaard W.; Otto H.; Lamparter T.; Hughes J.; Hellingwerf K. J.; Heyn M. P. (2001) Light-Induced Proton Release and Proton Uptake Reactions in the Cyanobacterial Phytochrome Cph1. Biochemistry 40, 11460–11471. [DOI] [PubMed] [Google Scholar]
- Yeh K. C.; Wu S. H.; Murphy J. T.; Lagarias J. C. (1997) A Cyanobacterial Phytochrome Two-Component Light Sensory System. Science 277, 1505–1508. [DOI] [PubMed] [Google Scholar]
- Kim P. W.; Rockwell N. C.; Martin S. S.; Lagarias J. C.; Larsen D. S. (2014) Dynamic Inhomogeneity in the Photodynamics of Cyanobacterial Phytochrome Cph1. Biochemistry 53, 2818–2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim P. W.; Rockwell N. C.; Freer L. H.; Chang C.-W.; Martin S. S.; Lagarias J. C.; Larsen D. S. (2013) Unraveling the Primary Isomerization Dynamics in Cyanobacterial Phytochrome Cph1 with Multipulse Manipulations. J. Phys. Chem. Lett. 4, 2605–2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim P. W.; Pan J.; Rockwell N. C.; Chang C.-W.; Taylor K. C.; Lagarias J. C.; Larsen D. S. (2012) Ultrafast E to Z Photoisomerization Dynamics of the Cph1 Phytochrome. Chem. Phys. Lett. 549, 86–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller M. G.; Lindner I.; Martin I.; Gartner W.; Holzwarth A. R. (2008) Femtosecond Kinetics of Photoconversion of the Higher Plant Photoreceptor Phytochrome Carrying Native and Modified Chromophores. Biophys. J. 94, 4370–4382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischoff M.; Hermann G.; Rentsch S.; Strehlow D. (2001) First Steps in the Phytochrome Phototransformation: A Comparative Femtosecond Study on the Forward (Pr → Pfr) and Back Reaction (Pfr → Pr). Biochemistry 40, 181–186. [DOI] [PubMed] [Google Scholar]
- Schumann C.; Gross R.; Wolf M. M. N.; Diller R.; Michael N.; Lamparter T. (2008) Subpicosecond Midinfrared Spectroscopy of the P-fr Reaction of Phytochrome Agp1 from Agrobacterium tumefaciens. Biophys. J. 94, 3189–3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumann C.; Gross R.; Michael N.; Lamparter T.; Diller R. (2007) Sub-Picosecond Mid-Infrared Spectroscopy of Phytochrome Agp1 from Agrobacterium tumefaciens. ChemPhysChem 8, 1657–1663. [DOI] [PubMed] [Google Scholar]
- Spillane K. M.; Dasgupta J.; Mathies R. A. (2012) Conformational Homogeneity and Excited-State Isomerization Dynamics of the Bilin Chromophore in Phytochrome Cph1 from Resonance Raman Intensities. Biophys. J. 102, 709–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song C.; Psakis G.; Lang C.; Mailliet J.; Gartner W.; Hughes J.; Matysik J. (2011) Two Ground State Isoforms and a Chromophore D-Ring Photoflip Triggering Extensive Intramolecular Changes in a Canonical Phytochrome. Proc. Natl. Acad. Sci. U.S.A. 108, 3842–3847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohmer T.; Lang C.; Bongards C.; Gupta K.; Neugebauer J.; Hughes J.; Gartner W.; Matysik J. (2010) Phytochrome as Molecular Machine: Revealing Chromophore Action during the Pfr → Pr Photoconversion by Magic-Angle Spinning NMR Spectroscopy. J. Am. Chem. Soc. 132, 4431–4437. [DOI] [PubMed] [Google Scholar]
- Foerstendorf H.; Lamparter T.; Hughes J.; Gartner W.; Siebert F. (2000) The Photoreactions of Recombinant Phytochrome from the Cyanobacterium Synechocystis: A Low-Temperature UV-Vis and FT-IR Spectroscopic Study. Photochem. Photobiol. 71, 655–661. [DOI] [PubMed] [Google Scholar]
- Rockwell N. C.; Duanmu D.; Martin S. S.; Bachy C.; Price D. C.; Bhattacharya D.; Worden A. Z.; Lagarias J. C. (2014) Eukaryotic Algal Phytochromes Span the Visible Spectrum. Proc. Natl. Acad. Sci. U.S.A. 111, 3871–3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb S. M.; Kim P. W.; Rockwell N. C.; Hirose Y.; Ikeuchi M.; Lagarias J. C.; Larsen D. S. (2013) Primary Photodynamics of the Green/Red-Absorbing Photoswitching Regulator of the Chromatic Adaptation E Domain from Fremyella diplosiphon. Biochemistry 52, 8198–8208. [DOI] [PubMed] [Google Scholar]
- Kim P. W.; Freer L. H.; Rockwell N. C.; Martin S. S.; Lagarias J. C.; Larsen D. S. (2012) Femtosecond Photodynamics of the Red/Green Cyanobacteriochrome NpR6012g4 from Nostoc punctiforme. 2. Reverse Dynamics. Biochemistry 51, 619–630. [DOI] [PubMed] [Google Scholar]
- Fischer A. J.; Lagarias J. C. (2004) Harnessing Phytochrome’s Glowing Potential. Proc. Natl. Acad. Sci. U.S.A. 101, 17334–17339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller A. E.; Fischer A. J.; Laurence T.; Hollars C. W.; Saykally R. J.; Lagarias J. C.; Huser T. (2006) Single-Molecule Dynamics of Phytochrome-Bound Fluorophores Probed by Fluorescence Correlation Spectroscopy. Proc. Natl. Acad. Sci. U.S.A. 103, 11136–11141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C.-W.; Gottlieb S. M.; Kim P. W.; Rockwell N. C.; Lagarias J. C.; Larsen D. S. (2013) Reactive Ground-State Pathways Are Not Ubiquitous in Red/Green Cyanobacteriochromes. J. Phys. Chem. B 117, 11229–11238. [DOI] [PubMed] [Google Scholar]
- Scurlock R. D.; Evans C. H.; Braslavsky S. E.; Schaffner K. (1993) A Phytochrome Phototransformation Study Using 2-Laser 2-Color Flash-Photolysis: Analysis of the Decay Mechanism of I(700). Photochem. Photobiol. 58, 106–115. [Google Scholar]
- Holzwarth A. R.; Venuti E.; Braslavsky S. E.; Schaffner K. (1992) The Phototransformation Process in Phytochrome. 1. Ultrafast Fluorescence Component and Kinetic-Models for the Initial Pr-]Pfr Transformation Steps in Native Phytochrome. Biochim. Biophys. Acta 1140, 59–68. [Google Scholar]
- Gambetta G. A.; Lagarias J. C. (2001) Genetic Engineering of Phytochrome Biosynthesis in Bacteria. Proc. Natl. Acad. Sci. U.S.A. 98, 10566–10571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll E. C.; Compton O. C.; Madsen D.; Osterloh F. E.; Larsen D. S. (2008) Ultrafast Carrier Dynamics in Exfoliated and Functionalized Calcium Niobate Nanosheets in Water and Methanol. J. Phys. Chem. C 112, 2394–2403. [Google Scholar]
- Cerullo G.; Nisoli M.; Stagira S.; De Silvestri S. (1998) Sub-8-fs Pulses from an Ultrabroadband Optical Parametric Amplifier in the Visible. Opt. Lett. 23, 1283–1285. [DOI] [PubMed] [Google Scholar]
- Holzwarth A. R. (1996) Data Analysis of Time-Resolved Measurements. In Biophysical techniques in photosynthesis (Amesz J., and Hoff A. J., Eds.) pp 75–92, Springer, Dordrecht, The Netherlands. [Google Scholar]
- van Stokkum I. H. M.; Larsen D. S.; van Grondelle R. (2004) Global and Target Analysis of Time-Resolved Spectra. Biochim. Biophys. Acta 1657, 82–104. [DOI] [PubMed] [Google Scholar]
- Kim P. W.; Freer L. H.; Rockwell N. C.; Martin S. S.; Lagarias J. C.; Larsen D. S. (2012) Femtosecond Photodynamics of the Red/Green Cyanobacteriochrome NpR6012g4 from Nostoc punctiforme. 1. Forward Dynamics. Biochemistry 51, 608–618. [DOI] [PubMed] [Google Scholar]
- Larsen D. S.; van Stokkum I. H. M.; Vengris M.; van der Horst M. A.; de Weerd F. L.; Hellingwerf K. J.; van Grondelle R. (2004) Incoherent Manipulation of the Photoactive Yellow Protein Photocycle with Dispersed Pump-Dump-Probe Spectroscopy. Biophys. J. 87, 1858–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim P. W.; Freer L. H.; Rockwell N. C.; Martin S. S.; Lagarias J. C.; Larsen D. S. (2012) Second-Chance Forward Isomerization Dynamics of the Red/Green Cyanobacteriochrome NpR6012g4 from Nostoc punctiforme. J. Am. Chem. Soc. 134, 130–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupenyan A. B.; Vreede J.; van Stokkum I. H. M.; Hospes M.; Kennis J. T. M.; Hellingwerf K. J.; Groot M. L. (2011) Proline 68 Enhances Photoisomerization Yield in Photoactive Yellow Protein. J. Phys. Chem. B 115, 6668–6677. [DOI] [PubMed] [Google Scholar]
- Kennis J. T. M.; Larsen D. S.; van Stokkum I. H. M.; Vengris M.; van Thor J. J.; van Grondelle R. (2004) Uncovering the Hidden Ground State of Green Fluorescent Protein. Proc. Natl. Acad. Sci. U.S.A. 101, 17988–17993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sineshchekov V.; Hughes J.; Hartmann E.; Lamparter T. (1998) Fluorescence and Photochemistry of Recombinant Phytochrome from the Cyanobacterium Synechocystis. Photochem. Photobiol. 67, 263–267. [DOI] [PubMed] [Google Scholar]
- Spillane K. M.; Dasgupta J.; Lagarias J. C.; Mathies R. A. (2009) Homogeneity of Phytochrome Cph1 Vibronic Absorption Revealed by Resonance Raman Intensity Analysis. J. Am. Chem. Soc. 131, 13946–13948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freer L. H.; Kim P. W.; Corley S. C.; Rockwell N. C.; Zhao L.; Thibert A. J.; Lagarias J. C.; Larsen D. S. (2012) Chemical Inhomogeneity in the Ultrafast Dynamics of the DXCF Cyanobacteriochrome Tlr0924. J. Phys. Chem. B 116, 10571–10581. [DOI] [PubMed] [Google Scholar]
- Song S.-H.; Freddolino P. L.; Nash A. I.; Carroll E. C.; Schulten K.; Gardner K. H.; Larsen D. S. (2011) Modulating LOV Domain Photodynamics with a Residue Alteration outside the Chromophore Binding Site. Biochemistry 50, 2411–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb S. M., Kim P. W., Chang C.-W., Hanke S. J., Hayer R. J., Rockwell N. C., Lagarias J. C., and Larsen D. S. (2014) Not All Red/Green Photoswitching Cyanobacteriochromes Are Built Alike: 1. Diversity in the Primary Forward Photoreaction Dynamics. Manuscript in preparation.
- Toh K. C.; Stojkovic E. A.; van Stokkum I. H. M.; Moffat K.; Kennis J. T. M. (2010) Proton-Transfer and Hydrogen-Bond Interactions Determine Fluorescence Quantum Yield and Photochemical Efficiency of Bacteriophytochrome. Proc. Natl. Acad. Sci. U.S.A. 107, 9170–9175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischoff M.; Hermann G.; Rentsch S.; Strehlow D. (1998) Ultrashort Processes of Native Phytochrome: Femtosecond Kinetics of the Far-Red-Absorbing Form Pfr. J. Phys. Chem. A 102, 4399–4404. [Google Scholar]
- Yang X.; Ren Z.; Kuk J.; Moffat K. (2011) Temperature-Scan Cryocrystallography Reveals Reaction Intermediates in Bacteriophytochrome. Nature 479, 428–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mroginski M. A.; Murgida D. H.; Hildebrandt P. (2007) The Chromophore Structural Changes during the Photocycle of Phytochrome: A Combined Resonance Raman and Quantum Chemical Approach. Acc. Chem. Res. 40, 258–266. [DOI] [PubMed] [Google Scholar]
- Chizhov I.; Zorn B.; Manstein D. J.; Gaertner W. (2013) Kinetic and Thermodynamic Analysis of the Light-induced Processes in Plant and Cyanobacterial Phytochromes. Biophys. J. 105, 2210–2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb S. M.; Kim P. W.; Corley S. C.; Madsen D.; Hanke S. J.; Chang C.-W.; Rockwell N. C.; Martin S. S.; Lagarias C.; Larsen D. S. (2014) Primary and Secondary Photodynamics of the Violet/Orange Dual-Cysteine NpF2164g3 Cyanobacteriochrome Domain from Nostoc Punctiforme. Biochemistry 53, 1029–1040. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.