

Abstract
Cardiomyocytes derived from human pluripotent stem cells show tremendous promise for the replacement of myocardium and contractile function lost to infarction. However, until recently, no methods were available to directly determine whether these stem cell-derived grafts actually couple with host myocardium and fire synchronously following transplantation in either intact or injured hearts. To resolve this uncertainty, our group has developed techniques for the intravital imaging of hearts engrafted with stem cell-derived cardiomyocytes that have been modified to express the genetically encoded protein calcium sensor, GCaMP. When combined with the simultaneously recorded electrocardiogram, this protocol allows one to make quantitative assessments as to the presence and extent of host–graft electrical coupling as well as the timing and pattern of graft activation. As described here, this system has been employed to investigate the electromechanical integration of human embryonic stem cell-derived cardiomyocytes in a guinea pig model of cardiac injury, but analogous approaches should be applicable to other human graft cell types and animal models.
Keywords: Human pluripotent stem cells, Zinc finger nuclease, GCaMP3, Cardiac repair, Intravital imaging, Electromechanical coupling
1 Introduction
In recent years, there has been tremendous interest in stem cell- and tissue engineering-based approaches to replace the muscle that is lost following myocardial infarction. For example, our group and others have shown that human embryonic stem cell-derived cardiomyocytes (hESC-CMs) can partially remuscularize the infarct scar and improve contractile function in rodent models of myocardial infarction [1–4]. However, to contribute functionally meaningful new force-generating units and avoid creating electrically excitable but imperfectly coupled tissue that might promote arrhythmias, graft cells must undergo appropriate electromechanical integration and contract synchronously with host myocardium during systole.
A number of strategies have been employed to assess the electromechanical integration of various graft cell types following transplantation into either intact or injured hearts. Historically, the most common approach has been to transplant graft cells tagged with a static fluorescent label, such as green fluorescent protein (GFP) or DiO. With this approach, the engrafted heart is later harvested and stained with a spectrally distinct fluorescent voltage- or calcium-sensitive dye that reports cardiomyocyte activation [5–10]. One then looks for evidence of activation (e.g., optical action potentials or calcium transients) arising from the tagged graft tissue. However, a major challenge with this approach is the need to reliably distinguish between graft and host, since both will have been labeled with the dye. Graft implants commonly overlie surviving host myocardium, so distinguishing between the two becomes particularly problematic when imaging the heart from the epicardial surface. Illustrating this problem, our group recently used an independent technique to identify hearts with uncoupled grafts, and we found that deep surviving host tissue labeled with the voltage dye RH237 emitted optical action potentials [3]. Unlike the graft, these optical action potentials were perfectly synchronized with the host ECG, and we would have otherwise erroneously interpreted them as graft derived. It may therefore be cause for concern that nearly all studies using this sort of an approach have reported apparently “seamless” graft integration; that is, both graft and host tissues showed similar optical action potential amplitudes, patterns of graft activation, and conduction velocities.
One partial solution to this problem is to use higher resolution techniques such as two-photon microscopy to distinguish between host and graft cardiomyocytes at the single-cell level [8], but the latter does not allow one to assess graft behavior at the macroscopic level or acquire critical tissue-level parameters (e.g., graft conduction velocity). An arguably better solution—and the one that forms the basis for the protocol described here—is to use a graft-autonomous reporter in which the activation signal arises solely from the graft. The genetically encoded fluorescent calcium sensor GCaMP serves as a convenient reporter for this purpose, since cardiomyocytes exhibit cytosolic calcium transients with each depolarization and contractile cycle. A large family of increasingly bright GCaMP variants has now been created, each of which incorporates moieties including circularly permuted GFP (cpGFP), calmodulin (CaM), and a CaM-interacting M13 domain from myosin light chain kinase [8, 11–13]. Cardiomyocytes expressing GCaMP show robust fluorescent transients in vitro with each contractile cycle, and they form intracardiac grafts that emit fluorescence transients that can then be correlated with the ECG of recipient hearts [3, 14].
Below, we describe how to use zinc finger nuclease (ZFN)-mediated transgenesis [15, 16] to generate a stable hESC line expressing a GCaMP variant called GCaMP3 as well as how to generate large quantities of enriched GCaMP3+ hESC-CMs. We then provide detailed methods for creating a guinea pig model of cardiac injury, transplanting GCaMP3+ hESC-CMs in this model, and ultimately the intravital imaging of GCaMP3+ hESC-CM grafts. Our lab recently used these same methods to demonstrate that, while hESC-CMs exhibit reliable 1:1 host–graft coupling in uninjured hearts, their electromechanical integration is imperfect following transplantation in injured hearts (presumably due to intervening scar tissue) [3]. While this protocol is written for this specific cell type and animal model, the same general approach should be useful in assessing the electromechanical integration of other graft cell types as well as engineered tissue constructs. In addition to overcoming the aforementioned concerns about factitious graft-derived signals, the ZFN-mediated targeting strategy used to insert the GCaMP3 expression cassette can be directly applied to any human cell type.
2 Materials
2.1 Generation of the GCaMP3 Reporter hESC Line
H7/WA07 hESC line (Wicell Research Institute, Madison, WI) (see Note 1).
Electroporator.
Human stem cell Nucleofector kit 1 (Lonza).
LY294002 stock solution: 5 mg LY294002 is dissolved in 1.6 mL DMSO to make 10 mM stock solution. Aliquot, and store at −20 °C.
50 mg/mL G418.
Accutase solution.
Mouse embryonic fibroblast conditioned medium (MEF-CM) and Matrigel-coated plates (please see ref. 17, 18).
Human basic fibroblast growth factor (hbFGF): Dissolve into PBS (with 0.2 % BSA) to make 10 μg/mL stock solution. Store aliquots at −20 °C.
2.2 Generation of GCaMP3+ hESC-Derived Cardiomyocytes
Versene.
RPMI-B27 medium: 98 % (v/v) RPMI 1640, 2 % B27 serum supplement minus insulin, and 2 mM L-glutamine.
Activin A: Make 10 μg/mL stock solution in PBS (with 0.2 % BSA). Store aliquots at −20 °C.
BMP-4: Make 1 μg/mL stock solution in PBS (with 0.2 % BSA). Store aliquots at −20°C.
Y27632 dihydrochloride (ROCK inhibitor): Dissolve in sterile water to make 10 mM stock solution. Store aliquots at −20 °C.
2.3 Cryopreservation of GCaMP3+ hESC-CMs
Trypsin–EDTA 0.05 % solution and defined trypsin inhibitor.
Insulin-like growth factor-1 (IGF-1).
Cyclosporine A (CsA).
DNase I: Dissolved to 400 U/μL with PBS. Sterile filter, and store at −20 °C.
Cryostor CS10 (BioLife Solutions).
Controlled-rate freezer.
2.4 Preparation of GCaMP3+ hESC-CMs for Intracardiac Transplantation
Matrigel-based pro-survival cocktail (PSC): Thaw 1 mL undiluted Matrigel on ice. Mix CsA (200 nM), ZVAD-FMK (10 μM), BCL-XL (50 nM), IGF-1 (0.1 μg/mL), and pinacidil (50 μM) into Matrigel to make PSC.
RPMI based PSC: Add all the preceding factors into RPMI medium instead of Matrigel.
Insulin syringes with 27G needle.
2.5 Cardiac Cryoinjury and Transplantation of GCaMP3+ hESC-CMs into Guinea Pig Hearts
Male guinea pigs (650–750 g).
A general surgical instrument set, including needle holder, tweezers, forceps, scissors, eye scissors, retractor, blades, and scalpel handle.
Sutures: 4-0 Vicryl coated, 4-0 silk, 5-0 nylon.
8 mm diameter aluminum cryoprobe, liquid nitrogen, and thermal container.
Pediatric laryngoscope and 14G shielded intravenous catheter.
Heating pad.
Skin cleaning agents: 70 % ethanol, chlorhexidine gluconate, Betadine.
Lubrifresh opthalmic ointment.
Rodent ventilator and anesthesia vaporizer with isoflurane.
Anesthetics: Ketamine (50 mg/kg), xylazine (2 mg/kg), isoflurane (1.5 %).
Analgesics: 0.025 % (v/v) Bupivacaine, buprenorphine (0.05 mg/kg).
Immunosuppressive drugs: CsA and methylprednisolone.
Antibiotic: 0.3 mL/kg Baytril® (2.27 % enrofloxacin injectable solution).
2.6 Intravital Imaging of GCaMP3+ hESC-CM Grafts in Guinea Pig Hearts
Epifluorescence stereomicroscope.
Fluorescence illumination system.
High-speed CCD camera (e.g. Andor iXon).
Data acquisition system.
Bioamplifier.
Langendorff heart system.
Langendorff perfusion solution (mM): 25.0 NaHCO3, 1.2 MgSO4, 4.7 KCl, 118.0 NaCl, 1.2 KH2PO4, 11.0 glucose, 1.0 sodium pyruvate, 1.8 CaCl2. Oxygenate with a submerged bubbler with 95 % O2/5 % CO2 gas for about 10 min before adding CaCl2 to stabilize pH, then 0.2 μm filtered.
Blebbistatin (10 μM): 10 mM Stock solution in DMSO and store at −20 °C. Dilute in Langendorff perfusion solution (above) immediately before use.
2.7 Analysis of GCaMP3 Fluorescence Signals
Matlab software (Mathworks) and a PC.
3 Methods
3.1 Generation of the GCaMP3 Reporter hESC Line (See Notes 1–4)
Transgenic hESCs stably expressing the GCaMP3 fluorescent reporter are generated by ZFN-mediated insertion of the GCaMP3 expression cassette into the AAVS1 locus using methods adapted from Hockemeyer et al. [15] (see also Fig. 1). In brief, this protocol involves transient transfection with two plasmids (the AAVS1 ZFN and pZDonor-GCaMP3 vectors), both of which are available from our group upon request or can be generated de novo as described in Note 2. Prior to transfection, undifferentiated wild-type hESCs are cultured under feeder-free conditions, using Matrigel-coated plates and daily feeding with MEF-CM plus bFGF (4 ng/mL). Methods for the feeder-free growth of hESCs have been detailed elsewhere by our group and others [17, 18].
Fig. 1.
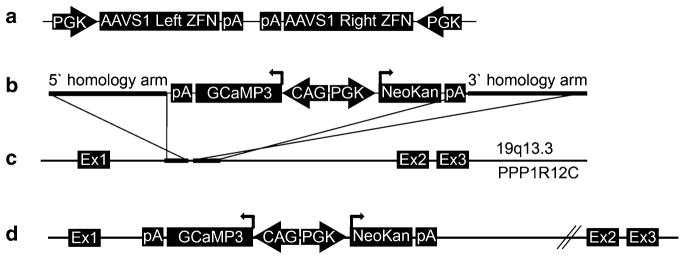
Zinc finger nuclease (ZFN)-mediated targeting of the GCaMP3 expression cassette to the AAVS1 locus. (a) The AAVS1 ZFN plasmid includes two expression cassettes, in which separate human PGK promoters drive the expression of the AAVS1 site-specific ZFN pair. (b) The pZDonor-GCaMP3 plasmid includes GCaMP3 and neomycin selection cassettes that are flanked by AAVS1 site homology arms (each ~800 bp). GCaMP3 expression is driven by the CAG promoter, while the neomycin resistance gene is driven by the human PGK promoter. (c ) The AAVS locus is located between exons 1 and 2 of the PPR1R12C gene on chromosome 19. (d) Co-transfection of AAVS1 ZFN and ZDonor-GCaMP3 plasmids in human cells results in targeting of the latter vector to the AAVS1 locus by homologous recombination (see Subheading 3.1 for details). pA bovine growth hormone polyadenylation signal, Ex exon, NeoKan neomycin/kanamycin resistance
For at least 4 h prior to transfection, treat hESCs with MEF-CM supplemented with bFGF (4 ng/mL) and LY294002 (10 μmol/L).
Prepare plasmid DNA cocktail containing 5 μg of AAVS1 ZFN and 20 μg of pZDonor GCaMP3 donor vectors. Immediately prior to transfection, mix the DNA cocktail with Amaxa Human Stem Cell Nucleofector Solution 1 (see Notes 3 and 5).
Rinse hESCs with PBS and then treat with Accutase for approximately 5 min. Remove the Accutase, rinse with PBS, and then triturate hESCs into a single-cell suspension in MEF-CM.
Count hESCs using a hemacytometer. Aliquot 4 × 105 cells into a microcentrifuge tube and spin down at 300 × g for 5 min. Resuspend the pellet in Nucleofector Solution 1 with the DNA cocktail, and place it in a nucleofector cuvette. Proceed with nucleofection as per vendor instructions using program setting B-16.
After transfection, gently resuspend hESCs in MEF-CM and then replate onto Matrigel-coated 10 cm plates in 10 mL volume of MEF-CM supplemented with bFGF (4 ng/mL) and LY294002 (10 μmol/L). Thereafter, re-feed the cells with daily exchanges of this same medium.
Begin antibiotic selection of successfully targeted hESCs 4 days after nucleofection, a time point by which typical hESC colonies should have reformed. For this, treat cells with MEF-CM supplemented with bFGF (4 ng/mL) and G418 (75 μg/mL), but no LY294002.
Continue antibiotic selection by daily re-feeding of cells with MEF-CM supplemented with bFGF (4 ng/mL) and 75 μg/mL G418 for a total of 4 days. Thereafter, reduce the G418 to a maintenance concentration of 20 μg/mL.
After obtaining stable GCaMP3+ hESC cultures, verify their karyotype and confirm proper targeting of the AAVS1 locus by Southern blotting (see Note 6).
3.2 Generation of GCaMP3+ hESC-Derived Cardiomyocytes
GCaMP3+ hESCs are differentiated into cardiomyocytes using methods we have previously reported for wild-type cultures [17, 18]. Please see Fig. 2a for an overview of the cardiac differentiation protocol. Prior to the onset of differentiation, transgenic GCaMP3+ hESCs are maintained in the undifferentiated state by culture on Matrigel-coated plates in the presence of MEF-CM plus bFGF (4 ng/mL). Importantly, because there should be no residual feeder cells present, cultures should be maintained under feeder-free conditions for at least 2–3 passages prior to attempting cardiac differentiation. When the undifferentiated hESC colonies occupy approximately two-thirds of the surface area, they are typically ready for cardiac differentiation.
Fig. 2.
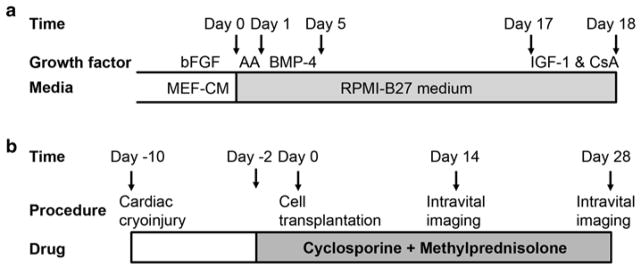
Timelines for the cardiac differentiation of GCaMP+ hESCs and the intravital imaging of GCaMP3+ hESC-CM grafts in injured hearts. (a) GCaMP3+ hESC-CMs are generated using our directed cardiac differentiation protocol, previously described for wild-type hESCs [17] (see Subheading 3.2 for details). In brief, undifferentiated GCaMP3+ hESCs are expanded under standard feeder-free conditions using mouse embryonic fibroblast-conditioned medium (MEF-CM) supplemented with human basic fibroblast growth factor (bFGF). Undifferentiated cultures are then replated to form a dense monolayer in MEF-CM plus bFGF. After a compact monolayer is formed, hESCs are switched to RPMI-B27 medium and serially pulsed with activin A (AA) on day 0 and bone morphogenetic protein-4 (BMP-4) from days 1 to 5. Thereafter, the differentiating cultures are grown in RPMI-B27 medium without any exogenous factors. One day before harvest, cells are heat-shocked and treated with insulin-like growth factor-1 (IGF-1) and cyclosporine (CsA) to improve graft survival. Cells are typically harvested on ~day 18 for cryopreservation or immediate transplantation. (b) To evaluate the electromechanical integration of GCaMP3+ hESC-CM grafts in a subacute model of cardiac injury, guinea pigs undergo a thoracotomy and direct cryoinjury of the left ventricle. 10 days later, a repeat thoracotomy is performed, and GCaMP3+ hESC-CMs are injected into the injured heart. Intravital imaging is typically performed either 14 or 28 days post-transplantation. To avoid graft rejection, animals are treated from day −2 to day 28 post-transplantation with methylprednisolone and CsA
Treat GCaMP3+ hESCs with 0.2 mL/cm2 Versene at 37 °C until all of the cells are rounded up but not yet detached. This typically requires 3–7 min, but cultures should be checked at least every 1–2 min during this step.
Gently aspirate the Versene, and replace it with 0.1 mL/cm2 MEF-CM supplemented with bFGF (4 ng/mL) and the ROCK inhibitor Y-27632 dihydrochloride (10 μmol/L). Dislodge the cells by pipetting the aforementioned medium over them, collect the cells into a 50 ml tube, and gently triturate them into a single-cell suspension (see Note 7).
Plate hESCs at 1.8–2.5 × 105 cells/cm2 onto Matrigel-coated plates. Thereafter, feed the cultures daily with 0.3 mL/cm2 MEF-CM plus bFGF (4 ng/mL).
When the resultant hESC monolayer becomes fully confluent with cells that are tightly packed but still have discernible borders, the cultures are ready for the switch to differentiating conditions At this point, replace the medium with 0.25 mL/cm2 RPMI-B27 medium supplemented with 100 ng/mL activin A (see Note 8).
24 h after treatment with activin A, gently replace the latter medium with 0.5 mL/cm2 RPMI-B27 medium supplemented with 10 ng/mL BMP-4. Culture the cells for an additional 4 days without medium changes.
Four days after treatment with BMP-4, gently replace the latter medium with 0.5 mL/cm2 RPMI-B27 medium without exogenous growth factors.
Feed cells every other day with 0.5 mL/cm2 RPMI-B27 medium without exogenous growth factors. Spontaneous contractile activity typically commences around 9–12 days after the initial induction with activin A. One can expect total cell yields of 3–5 × 105 cells/cm2 and ~60–90 % cardiomyocytes (based on the flow cytometry for cardiac troponin T or α-actinin) [3].
3.3 Cryopreservation of GCaMP3+ hESC-CMs
For logistical reasons and to minimize batch-to-batch variation in cell yields and purity, it is generally advisable to cryopreserve and “bank” hESC-CMs prior to use in intracardiac cell transplantation. Cells can be harvested for cryopreservation anytime 16–25 days after the initial induction with activin A. Particularly if cryopreservation is to be employed, we strongly recommend heat-shocking and treating the cells with IGF-1 and CsA prior to freezing them, as these interventions have been previously shown to significantly enhance graft survival [2, 19].
24 h prior to harvesting for cryopreservation, heat-shock cultures with 30-min exposure to RPMI-B27 medium pre-warmed to 42 °C. After 30 min, re-feed with 37 °C RPMI-B27 medium supplemented with IGF-1 (0.1 μg/ml) and CsA (0.25 μg/ml), and return the cultures to the incubator.
On the day of harvest, turn on the controlled-rate freezer and pre-chill it to 0 °C. Prepare 0.05 % trypsin–EDTA supplemented with 200 U/mL DNAse I and pre-warm to 37 °C. Chill Cryostor CS-10 solution on ice.
To begin harvesting GCaMP3+ hESC-CMs, briefly rinse the cultures twice with PBS supplemented with 200 U/mL DNase I. Next, incubate the cells with 0.2 mL/cm2 of 0.05 % trypsin–EDTA plus 200 U/mL DNase I at 37 °C for 5 min. Cells should then readily detach from the surface with brisk tapping of the flask. If necessary, cells can be further dislodged by gentle agitation and/or flowing the trypsin solution over the cell surface.
Stop the enzymatic reaction by adding an equivalent volume of RPMI-B27 medium supplemented with 10 % FBS. Transfer the cells to a 50 ml tube and gently triturate to obtain single-cell suspension. Centrifuge the cells down at 300 × g for 5 min to remove the trypsin and then resuspend in RPMI-B27 medium. This is a convenient time point to take small aliquots for cell counting (e.g., by hemacytometer) and/or determination of cardiomyocyte purity (e.g., by flow cytometry).
Centrifuge the cells down again at 300 × g for 5 min, and then resuspend them in chilled Cryostor CS-10 solution at 1 ml per 1 × 108 total cells. Aliquot into pre-labeled 2 ml cryovials on ice.
Transfer the cryovials to a controlled-rate freezer already at 0 °C. Set controlled-rate freezer to cool at 1 °C per minute until it reaches −80 °C. Transfer the vials to a −80 °C freezer and leave overnight.
The next day, transfer the vials to a liquid nitrogen tank for storage until transplantation. hESC-CMs have been successfully transplanted after >1 year in cryopreservation.
3.4 Preparation of GCaMP3+ hESC-CMs for Intracardiac Transplantation
The exact procedure for preparing GCaMP3+ hESC-CMs for transplantation will depend on whether one is using “live” cultures or cryopreserved aliquots (prepared as described in Subheading 3.3).
3.4.1 Preparation of “Live” (i.e., Non-cryopreserved) Cells for Implantation
Heat-shock and detach cells as described in steps 1–4 of Subheading 3.3.
Before commencing cell harvesting, prepare the Matrigel-based (see Note 9) and RPMI based PSC. Pretreatment and delivery in PSC have been previously shown to enhance hESC-CM graft survival [2, 19].
Resuspend the cell pellet in the RPMI based PSC, and then transfer the cell suspension to a single Eppendorf tube.
Centrifuge the latter at 300 × g for 5 min, and then resuspend the cells in the Matrigel-based PSC. Add Matrigel-based PSC to a final volume of up to 5 × 105 cells per μl. (Our general practice is to prepare aliquots of 1 × 108 cells in 200 μl, each of which is injected into a single guinea pig heart.) Gently draw the cell suspension into a precooled insulin syringe with 27G needle. Keep the syringe at 4 °C for use within 1 h.
3.4.2 Thawing and Preparation of Cryopreserved Cells for Implantation
Prepare PSC as described in step 2 of Subheading 3.4.1. Preheat RPMI-B27 medium supplemented with 200 U/mL DNase I to 37 °C.
Transfer cryovial(s) containing the cryopreserved GCaMP3+ hESC-CMs to a bucket of dry ice until ready for use.
Thaw the vial in a 37 °C water bath with gentle agitation until the pellet is almost completely melted (this generally requires <1.5 min).
Add 1 mL of preheated RPMI-B-27 medium with 200 U/mL DNase I to each cryovial, and mix by gentle shaking. Slowly transfer the resultant cell suspension in a dropwise fashion to a 50 mL tube with 25 mL of 37 °C RPMI-B27 medium. Add RPMI-B27 medium to a final volume of 50 mL.
Wash away the cryopreservative by centrifugation at 300 × g for 5 min and resuspend the cell pellet in RPMI-B27 medium with 200U/ml DNase I. Proceed with steps3–4 of Subheading 3.4.1 to prepare syringes of cells.
3.5 Cardiac Cryoinjury and Transplantation of GCaMP3+ hESC-CMs into Guinea Pig Hearts
All animal procedures should be performed in compliance with institutional, local, and federal regulations. Please see Fig. 2b for the typical sequence of events involved in cardiac cryoinjury and cell transplantation. Because the guinea pig is relatively challenging to intubate, we recommend that anesthesia be induced with injectable ketamine/xylazine, followed by maintenance with inhalational isoflurane. Note that exposure to isoflurane anesthesia for long periods of time (>2 h) or at high concentrations (≥2 %) can adversely suppress cardiac and respiratory functions in the guinea pig. It is also essential that the animal’s body temperature be maintained at 37.0 ± 1.0 °C during all procedures using a heating pad regulated by a rectal thermistor probe (see Note 9).
3.5.1 Induction of Cardiac Cryoinjury
Induce anesthesia with an intraperitoneal injection of ketamine (50 mg/kg)/xylazine (2 mg/kg).
After confirming anesthesia by toe-pinch, carefully shave the animal’s chest and left axillary region. Clean the skin site by serial wiping with ethanol (70 %) and chlorhexidine gluconate. Coat the animal’s eyes with Lubrifresh ointment to prevent dehydration.
Intubate the animal using a pediatric laryngoscope to visualize the vocal cords. A tracheal tube can be formed using a 14G shielded intravenous catheter bent into a curve. One can confirm proper intubation by placing a small amount of water in the catheter ahead of the procedure. Thus, if the endotracheal tube is properly positioned, this water should be expelled with the animal’s first breath. After confirming proper intubation, connect the catheter to a mechanical ventilator set at a rate of 60 breaths per minute and a tidal volume of 3 mL/kg. At this point, the animal can be switched to inhalational anesthesia with 1.5 % isoflurane in oxygen.
Place the animal on the heating pad, and position it to expose the surgical site. Sterilize the site by gentle wiping with Betadine three times.
Using a surgical blade, make a 3–4 cm long skin incision from the left axilla to the area of the xiphoid process using blade and scissors. Use blunt forceps to gently separate the connective tissues between the skin and underlying muscle. Separate the first layer of muscle with blunt forceps, and then cut the second layer of muscle with scissors to expose the chest wall.
After it has been adequately exposed, open the chest wall using blunt forceps. Take care to avoid injury to the adjacent viscera. Separate the second and third ribs with a retractor.
After the beating heart is clearly visible, gently remove the pericardium with blunt forceps.
Induce cardiac cryoinjury by firmly contacting the anterior left ventricle with the aluminum cryoprobe that has been pre-cooled in liquid nitrogen. The cryoprobe should be applied four times for 30-s duration each, with re-cooling for at least 30 s in liquid nitrogen between each application.
After inspecting the heart and lungs for bleeding or other unexpected injuries, carefully close the chest wall and deep tissues using 4-0 silk sutures. Suture the skin wound closed using nylon 5-0 sutures.
For analgesia, infiltrate the wound site during closure with 0.025 % (v/v) topical bupivacaine. Inject buprenorphine (0.05 mg/kg subcutaneous in a 1 mL volume) every 8–12 h for up to 48 h or as directed by veterinary staff (see Note 10).
To avoid perioperative dehydration, inject 4 mL of sterile saline subcutaneously (divided between at least two injection sites).
3.5.2 Cell Transplantation
To ensure survival of the human xenografts in the guinea pig hearts, we recommend a regimen combining cyclosporine (15 mg/kg/day subcutaneous for 7 days, later reduced to 7.5 mg/kg/day) and methylprednisolone (2 mg/kg/day intraperitoneal). To ensure adequate levels of immunosuppression at the time of cell transplantation, administration of the cyclosporine and methylprednisolone should commence 2 days prior to transplantation. We have also found that a one-time perioperative administration of Baytril (enrofloxacin) greatly reduces the incidence of infections associated with this second round of survival surgery in chronically immunosuppressed animals.
Prepare syringes loaded with GCaMP3+ hESC-CMs as described above in Subheading 3.4. Bend the needle tip to facilitate injection into the myocardium.
Immediately before surgery, inject the animal with 0.3 mL/kg intraperitoneal Baytril.
-
Repeat steps 1–7 of Subheading 3.5.1 for anesthesia, intubation, and repeat thoracotomy.
Note that exposure of the chest wall and heart can sometimes be more complicated due to scarring and adhesion related to the earlier procedure.
After the heart is exposed, inject cells into three locations (i.e., the center of the scar and each of the flanking border zones) with each site receiving approximately one-third of the total injection. Proper cell injection is indicated by blanching and the appearance of a white halo surrounding the needle entry site.
Close the chest wall and deep tissues using 4-0 vicryl sutures. Administer analgesia (i.e., bupivacaine and buprenorphine), and rehydrate as detailed in steps 10–11 of Subheading 3.5.1.
3.6 Intravital Imaging of GCaMP3+ hESC-CM Grafts in Guinea Pig Hearts
Our laboratory has successfully imaged normal and cryoinjured guinea pig hearts with GCaMP3+ hESC-CM grafts at both 2 and 4 weeks post-transplantation [3]. In this work, we have employed two different experimental preparations, each of which provides slightly different information and has its own advantages and disadvantages. The first approach employs an open chest preparation, performed as a terminal procedure in an already anesthetized animal. This prep has the advantage that all (or nearly all) of the neurohormonal signaling of the intact animal will be in place, but image acquisition is somewhat limited by motion artifact. The second approach involves an ex vivo preparation in which the heart is harvested and mounted on a Langendorff apparatus. While the latter preparation obviously cannot model all of the signaling and other factors that would apply in the intact animal, it does allow the use of mechanical uncouplers (e.g., blebbistatin [20]) to remove motion artifacts. It also allows one to image hearts with GCaMP3+ grafts after treatment with drugs and/or other fluorescent indicators (e.g., voltage-sensitive dyes). Below, we describe the methods for both experimental preparations separately.
3.6.1 Imaging Using an Open Chest Preparation
Follow steps 1–4 of Subheading 3.5.1 to prepare the animal for surgery as well for anesthesia, intubation, and mechanical ventilation.
Place the anesthetized animal in a supine position on a heated surface, and outfit it for standard surface ECG recordings.
Perform a wide thoracotomy by incising the full width of the epicardium and reflecting chest wall away anteriorly.
Use an appropriate optical system to visualize the GCaMP3 grafts. In brief, our own system employs a high-speed EM-CCD camera that is mounted on an epifluorescence stereomicroscope (see Fig. 3a). GCaMP3 is excited at 470 ± 20 nm, and emitted light is collected. Image acquisition is typically performed at 80–140 frames per second, and signals from the CCD camera and the surface ECG are fed through a computer for digital storage and off-line analysis as described below (see Note 11).
Fig. 3.

Schematic diagram of our GCaMP3 imaging system and representative recordings. (a) Our intravital imaging system includes a high-speed, high-sensitivity CCD camera (Andor iXon 860 EM-CCD) mounted on an epifluorescence stereomicroscope (Nikon SMZ1000). GCaMP3 is excited with a 450–490 band-pass filter, and its emission passes through a 500–550 nm band-pass filter before detection. The host electrocardiogram (ECG) is simultaneously recorded using a bioamplifier and data acquisition board (ADInstruments PowerLab system). Fluorescence and ECG signals are digitized and fed to a computer for storage and off-line analysis. (b) Left: Representative still images of GCaMP3+ hESC-CM grafts during systole and diastole. Two graft regions of interest (ROIs) have been circled by the dotted line. Right: Representative GCaMP3 fluorescence traces for the aforementioned ROIs as well as the simultaneously acquired ECG. Note that, in this case, fluorescence transients in both graft ROIs occur in a 1:1 relationship with the host ECG, indicating reliable graft–host coupling
3.6.2 Imaging Using an Ex Vivo Langendorff Preparation
Prepare Langendorff perfusion buffer as described above. (A total of ~4 L is usually sufficient for a typical imaging experiment.) Set aside two 1 L bottles, and add blebbistatin to it for a final concentration of 10 μM.
Connect 1 L reservoirs containing the control and blebbistatin-supplemented perfusate to a non-recirculating Langendorff apparatus, and bubble each solution with 95 % O2–5 % CO2 gas mix using submerged bubblers. The Langendorff apparatus should be configured to deliver retrograde coronary perfusion at ~70 mmHg constant pressure, and it should warm the perfusate to 37 °C prior to flow into the heart, e.g., by flow through a glass heat exchange coil fed by a warm circulating water bath.
Euthanize the animal, and excise the engrafted heart for mounting onto the Langendorff apparatus. To harvest the heart, perform a wide thoracotomy, quickly identify the aorta, and cut across the aorta and other great vessels to dissect the heart from the surrounding tissues. Cannulate the aorta without introducing air bubbles, connect the heart to the Langendorff system, and perfuse the heart with the blebbistatin-free solution.
Monitor the electrical activity of the isolated heart by placing negative and positive ECG leads at the base of the right ventricle and left ventricular apex, respectively. These leads are then connected to an appropriate bioamplifier system for recording and off-line storage.
Once the preparation is electrically stable, switch to the blebbistatin-containing perfusate. Motion arrest typically occurs within 10 min.
After mechanical arrest, maneuver the heart into focus on an appropriate imaging system (see Fig. 3 and Subheading 3.6.1). Record the epicardial GCaMP3 signals as described above (see Note 11).
3.7 Analysis of GCaMP3 Intravital Imaging Data
Our lab has created a number of custom Matlab scripts for analyzing the experiments described in the preceding section, and these tools are available upon request. Among other functions, they allow one to examine the relative intensity of GCaMP3 fluorescence transients, the relationship between the activation of GCaMP3+ graft(s) and host myocardium, and, in some circumstances, even the timing and pattern of graft activation. (See Fig. 3b for representative images of GCaMP3+ hESC-CM grafts during systole and diastole as well as representative fluorescence intensity traces.) The methods below assume that the reader will apply these tools, although we do describe alternative approaches for those who prefer to work outside of the Matlab software environment (see Note 11).
Arguably the most common reason for intravital imaging of GCaMP3+ grafts is to determine the temporal relationship between host and graft activation. For this, one obviously needs to precisely correlate the graft-derived GCaMP3 fluorescence transients and the host ECG. In our case, we have found that one convenient way of accomplishing this is to use the shutter fire signal emitted by the Andor EM-CCD to “time-sync” the imaging and ECG data recordings. ADInstruments’ PowerLab multichannel data acquisition system can simultaneously receive both this TTL signal from the camera shutter and the digitized ECG. This information can be used later to align the GCaMP3 and ECG traces, either in a spreadsheet or using our custom Matlab scripts.
In either custom Matlab scripts or in the camera controller software package (in our case Andor Solis), play the recorded data and identify graft regions of interest (ROIs).
Calculate the mean pixel intensity of each ROI, and plot this against the time-synced ECG. Assess the extent of graft coupling by visually identifying which ROIs activate in synchrony with the QRS complex of the ECG. We recommend recording at least ten contractile cycles to ensure appropriate image smoothing and statistical analyses.
Our custom Matlab scripts can also be used to assess the activation times for graft regions that are reliable coupled to the host ECG. For this, apply available spatial and temporal filters to the imaging data and background subtract the imaging data. Next, identify the peaks and troughs in the mean pixel intensity traces for each coupled ROI as well as the corresponding R wave of the host ECG. Select a time window that spans several graft activations, and then determine (on a per pixel basis) the time difference between the beginning of the rise in the GCaMP3 signal and the corresponding R wave of the host ECG. These time intervals can then be stored in a separate matrix for each cycle of activation. Average these matrices to generate an ensemble-averaged map of activation times. Our scripts greatly automate this analysis and generate two-dimensional color maps to help visualize the pattern and timing of graft activation (see Note 12).
Acknowledgments
This work was supported by funding from the National Institutes of Health (grants R01-HL064387, P01-HL094374, U01-HL100405, and RO1-HL117991). The authors would also like to thank Mr. Benjamin Van Biber for helpful comments on this manuscript.
Footnotes
While this protocol is written for the H7/WA07 hESC line, we have had good success using an equivalent ZFN-mediated targeting strategy to introduce the GCaMP3 expression cassette into multiple human pluripotent stem cell lines, including RUES2 hESCs and IMR90 human induced pluripotent stem cells. Greater transfection efficiency may be possible by using line-optimized electroporation settings and solutions in the nucleofection system.
Two plasmids are required for ZFN-mediated insertion of the GCaMP3 expression cassette into the AAVS1 locus: the AAVS1 ZFN and pZDonor-GCaMP3 vectors (see Fig. 1a, b). Both are readily available from our group upon request. Alternatively, these plasmids can be re-created using previously described methods [3]. To create the AAVS1 ZFN vector, we had the left and right arms of the AAVS1-specific ZFN de novo synthesized using sequences reported by Hockemeyer et al. [15]. These arms were then cloned into a pUC57 backbone plasmid to create a polycistronic vector in which the expression of each ZFN arm is driven by a separate human PGK promoter. This ZFN pair causes a double-strand break in the intron between exon 1 and 2 of the AAVS1 locus (19q13.3, see Fig. 1c). The donor vector was generated by modifying the pZDonor plasmid (Sigma Aldrich, #Z3027), which includes a multi-cloning site and ~800 bp homology arms flanking the AAVS1 ZFN cut site. Two expression cassettes were inserted between these homology arms: one in which the CAG promoter drives expression of GCaMP3 (Addgene, #22692) and another in which the PGK promoter drives neomycin resistance. Please see Fig. 1d for a schematic of the correctly targeted transgene including both expression cassettes.
Plasmid DNA should be prepared using a “midi-prep” kit (Qiagen), rather than phenol/chloroform extraction. The DNA should then be concentrated to a volume of <10 μL in water. Salts used in other DNA buffers (e.g., TE buffer) can interfere with transfection efficiency.
As an alternative to ZFN-mediated insertion of the GCaMP3 expression cassette, both TALEN- and CRISPR-mediated targeting of the human AAVS1 locus has been described [21, 22], and suitable vector systems are now available via the Addgene plasmid repository. These systems should also be compatible with the pZDonor-GCaMP3 vector described above.
Immediately before electroporation, prepare the nucleofection solution as per kit instructions. In brief, the nucleofection solution is a 4.5:1 mix of solution 1 to supplement solution (corresponding to 81.8 μL:18.2 μL for a typical 100 μL reaction).
The following primer sets can be used to generate Southern blot probes useful for confirming proper targeting of the GCaMP3 expression cassette to the AAVS1 locus: genomic probe (forward): GGAGGTGGTGCGCTTCTTGG, genomic probe (reverse): CGCATCCCCTCCCAGAAAGAC, neomycin cassette forward: GAAGAACTCGTCAAGAAGGCG, and neomycin cassette reverse: GAAGAACTCGTCAAGAAGGCG. DNA were cut with NdeI and NheI overnight then isolated by ethanol precipitation and loaded into a 1 % agarose gel. DNA transfer and membrane preparation was performed using Biorad Zeta-probe GT instruction manual section 2.1 and probe labeling was accomplished using Amersham Megaprime DNA labeling system (GE Healthcare RPN1604/5 kit). Southern blotting with the genomic probe produces a wild-type/nontargeted band at 3,968 bp, while the properly targeted allele shifts to 5,604 bp. In properly targeted cells, the neomycin probe will label a 5,604 bp band, whereas off-targeted integration events will be seen at other positions.
This seeding density works well for the cardiac differentiation of GCaMP3+ hESCs generated from the H7/WA07 hESC line. However, the optimal seeding density varies slightly from line to line, so it should be determined empirically when testing other lines. The goal should be to obtain a uniformly distributed monolayer of hESCs that occupies 80–100 % of the growth area 1 day post-plating.
The timing of this step is critical. The properly timed application of activin A should be based on the appearance of the culture (i.e., the formation of a compact monolayer without gaps), rather than a pre-specified interval of time. This will vary slightly from line to line, depending on its plate efficiency, growth kinetics, etc. We have also found that the efficiency of this cardiac differentiation protocol can be improved for some (but not all) lines by adding a variation of the Matrigel “overlay” step proposed by Kamp and colleagues [23]. The latter is performed during the activin A induction step. For this, thaw growth factor-reduced Matrigel on ice for at least 2 h. Chill RPMI-B27 medium on ice for 10 min. Mix the two reagents thoroughly to prepare a 1:60 dilution of Matrigel in medium. Warm the mixture to 37 °C, add activin A (100 ng/mL), and then gently overlay this onto cells at a volume of 0.25 mL/cm2. The remainder of the protocol is unchanged from that described above in Subheading 3.2 (i.e., continue on with steps 5–7).
The guinea pig has a large tongue, narrow oral cavity, and other anatomic features that make endotracheal intubation in this species particularly challenging. While beyond the scope of the present protocol, other authors have provided invaluable advice both on obtaining and confirming successful intubation in this model [24, 25]. One other issue with the guinea pig is that this species has excellent collaterals, so experimental myocardial infarction cannot be induced by coronary ligation as in other rodents. However, cardiac cryoinjury is a reasonable alternative and results in a highly reproducible area of injury and degree of dysfunction.
Like other rodents, guinea pigs usually recover from thoracotomy and cardiac cryoinjury procedures remarkably well. We allow animals to recover in a 37 °C warmed environment in which they can be closely monitored, and we typically find that they resume their normal activities within 3–4 h post-procedure. Animals that recover well are then returned to their cages with some hydrating gel and food placed directly onto the bedding, so that they do not become dehydrated due to incision discomfort when reaching up to the cage insert with their water bottle and food pellets. Veterinary staff should be consulted if there are concerning signs such as weight loss, altered posture, unusual vocalizations, listless behavior, and respiratory distress. Our experience suggests that a <25 % mortality rate can be expected after two rounds of survival surgery (i.e., cardiac cryoinjury and cell transplantation), with most deaths occurring shortly after cryoinjury.
Although our group has found that custom Matlab scripts provide more powerful analysis tools, there are simpler alternatives for those uncomfortable with work in the Matlab software environment. Most relevant EM-CCD systems are controlled by acquisition software that usually include limited analysis functions. For example, our imaging system includes an Andor iXon EM-CCD, which is controlled by the Andor Solis software package. Solis allows one to directly export the mean pixel intensity values for rectangular ROIs into a spreadsheet such as Microsoft Excel. Alternatively, if ROI shapes other than rectangles are desired, Solis data can be exported as a stack of *.tiff files into ImageJ. There, the freehand selection tool can be used to define a custom ROI and determine the mean pixel intensity. In either case, this pixel intensity data can then be manually aligned with the digital ECG recordings in a spreadsheet using the available camera-derived signals (e.g., shutter fire pulse) as a “time stamp.” This approach is tedious, but it works.
Ensemble averaging of activation time matrices can only be performed if the signal is periodic and steady. If the signal signature changes with time, then this method cannot be reliably applied.
References
- 1.Caspi O, Huber I, Kehat I, et al. Transplantation of human embryonic stem cell-derived cardiomyocytes improves myocardial performance in infarcted rat hearts. J Am Coll Cardiol. 2007;50:1884–1893. doi: 10.1016/j.jacc.2007.07.054. [DOI] [PubMed] [Google Scholar]
- 2.Laflamme MA, Chen KY, Naumova AV, et al. Cardiomyocytes derived from human embryonic stem cells in pro-survival factors enhance function of infarcted rat hearts. Nat Biotechnol. 2007;25:1015–1024. doi: 10.1038/nbt1327. [DOI] [PubMed] [Google Scholar]
- 3.Shiba Y, Fernandes S, Zhu WZ, et al. Human ES-cell-derived cardiomyocytes electrically couple and suppress arrhythmias in injured hearts. Nature. 2012;489:322–325. doi: 10.1038/nature11317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Laake LW, Passier R, Monshouwer-Kloots J, et al. Human embryonic stem cell-derived cardiomyocytes survive and mature in the mouse heart and transiently improve function after myocardial infarction. Stem Cell Res. 2007;1:9–24. doi: 10.1016/j.scr.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 5.Gepstein L, Ding C, Rahmutula D, et al. In vivo assessment of the electrophysiological integration and arrhythmogenic risk of myocardial cell transplantation strategies. Stem Cells. 2010;28:2151–2161. doi: 10.1002/stem.545. [DOI] [PubMed] [Google Scholar]
- 6.Lai PF, Panama BK, Masse S, et al. Mesenchymal stem cell transplantation mitigates electrophysiological remodeling in a rat model of myocardial infarction. J Cardiovasc Electrophysiol. 2013;24:813–821. doi: 10.1111/jce.12162. [DOI] [PubMed] [Google Scholar]
- 7.Rota M, Kajstura J, Hosoda T, et al. Bone marrow cells adopt the cardiomyogenic fate in vivo. Proc Natl Acad Sci U S A. 2007;104:17783–17788. doi: 10.1073/pnas.0706406104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rubart M, Soonpaa MH, Nakajima H, et al. Spontaneous and evoked intracellular calcium transients in donor-derived myocytes following intracardiac myoblast transplantation. J Clin Invest. 2004;114:775–783. doi: 10.1172/JCI21589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xue T, Cho HC, Akar FG, et al. Functional integration of electrically active cardiac derivatives from genetically engineered human embryonic stem cells with quiescent recipient ventricular cardiomyocytes: insights into the development of cell-based pacemakers. Circulation. 2005;111:11–20. doi: 10.1161/01.CIR.0000151313.18547.A2. [DOI] [PubMed] [Google Scholar]
- 10.Costa AR, Panda NC, Yong S, et al. Optical mapping of cryoinjured rat myocardium grafted with mesenchymal stem cells. Am J Physiol Heart Circ Physiol. 2012;302:H270–H277. doi: 10.1152/ajpheart.00019.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen TW, Wardill TJ, Sun Y, et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature. 2013;499:295–300. doi: 10.1038/nature12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tallini YN, Ohkura M, Choi BR, et al. Imaging cellular signals in the heart in vivo: cardiac expression of the high-signal Ca2+ indicator GCaMP2. Proc Natl Acad Sci U S A. 2006;103:4753–4758. doi: 10.1073/pnas.0509378103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tian L, Hires SA, Mao T, et al. Imaging neural activity in worms, flies and mice with improved GCaMP calcium indicators. Nat Methods. 2009;6:875–881. doi: 10.1038/nmeth.1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roell W, Lewalter T, Sasse P, et al. Engraftment of connexin 43-expressing cells prevents post-infarct arrhythmia. Nature. 2007;450:819–824. doi: 10.1038/nature06321. [DOI] [PubMed] [Google Scholar]
- 15.Hockemeyer D, Soldner F, Beard C, et al. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat Biotechnol. 2009;27:851–857. doi: 10.1038/nbt.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zou J, Maeder ML, Mali P, et al. Gene targeting of a disease-related gene in human induced pluripotent stem and embryonic stem cells. Cell Stem Cell. 2009;5:97–110. doi: 10.1016/j.stem.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu WZ, Van Biber B, Laflamme MA. Methods for the derivation and use of cardiomyocytes from human pluripotent stem cells. Methods Mol Biol. 2011;767:419–431. doi: 10.1007/978-1-61779-201-4_31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu C, Inokuma MS, Denham J, et al. Feeder-free growth of undifferentiated human embryonic stem cells. Nat Biotechnol. 2001;19:971–974. doi: 10.1038/nbt1001-971. [DOI] [PubMed] [Google Scholar]
- 19.Laflamme MA, Gold J, Xu C, et al. Formation of human myocardium in the rat heart from human embryonic stem cells. Am J Pathol. 2005;167:663–671. doi: 10.1016/S0002-9440(10)62041-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dou Y, Arlock P, Arner A. Blebbistatin specifically inhibits actin-myosin interaction in mouse cardiac muscle. Am J Physiol Cell Physiol. 2007;293:C1148–C1153. doi: 10.1152/ajpcell.00551.2006. [DOI] [PubMed] [Google Scholar]
- 21.Holkers M, Maggio I, Liu J, et al. Differential integrity of TALE nuclease genes following adenoviral and lentiviral vector gene transfer into human cells. Nucleic Acids Res. 2013;41:e63. doi: 10.1093/nar/gks1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mali P, Yang L, Esvelt KM, et al. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang J, Klos M, Wilson GF, et al. Extracellular matrix promotes highly efficient cardiac differentiation of human pluripotent stem cells: the matrix sandwich method. Circ Res. 2012;111:1125–1136. doi: 10.1161/CIRCRESAHA.112.273144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blouin A, Cormier Y. Endotracheal intubation in guinea pigs by direct laryngoscopy. Lab Anim Sci. 1987;37:244–245. [PubMed] [Google Scholar]
- 25.Nambiar MP, Gordon RK, Moran TS, et al. A simple method for accurate endotracheal placement of an intubation tube in Guinea pigs to assess lung injury following chemical exposure. Toxicol Mech Methods. 2007;17:385–392. doi: 10.1080/15376510601094131. [DOI] [PubMed] [Google Scholar]