

Abstract

In efforts directed toward the synthesis of seco-prezizaane sesquiterpenoids, a stereoselective annulation reaction has been developed between 4-hydroxy-1,6-enynes and TMS-alkynes that delivers cross-conjugated triene-containing hydroindanes. Contrary to previous reports, enyne substrates bearing two propargylic ethers enable the presumed organometallic intermediate to be trapped by double elimination. The tendency of products from this annulation to undergo Diels–Alder-based dimerization was harnessed to accomplish a two-step complexity-generating sequence en route to densely functionalized carbo- and heteorocyclic systems.
Evergreen shrubs/trees of the Illicium genus have been a rich source of structurally unique sesquiterpenoids.1 In particular, a variety of seco-prezizaane-type natural products have been isolated from this source (Figure 1), some of which exhibit potent neurotoxic activities while others act as neurotrophic agents. While subclassification of the group has been adopted based on their varying topographical features (anisatin-, pseudoanisatin-, majucin-, and minwanensin-type), they all share a highly oxygenated and compact carbocyclic skeleton. Perhaps due to the combination of reported biological activities and the apparent molecular complexity of these agents, many have reported studies aimed at accomplishing laboratory syntheses of members of the class as well as synthetic analogs.2 Recognizing that relatively minor structural changes in the known seco-prezizaane sesquiterpenes results in substantial changes in biological activity and that small molecule neurotrophic agents could be valuable in the pursuit of novel therapeutics, we initiated studies aimed at defining an efficient enantioselective entry to a fused carbocycle motif that may serve as an entry point to a range of seco-prezizaane natural and unnatural products. These efforts have resulted in the discovery of a convergent coupling process for the synthesis of highly unsaturated hydroindanes from the union of simple TMS-alkynes and 4-hydroxy-1,6-enynes. These products have been observed to undergo highly stereoselective Diels–Alder cycloaddition to deliver densely functionalized complex carbocyclic and heterocyclic frameworks.
Figure 1.

A representative sample of natural products from Illicium, some of which have been described as effective neurotrophic agents.
Our efforts began with the goal of accessing hydroindane motifs harboring cross-conjugated trienes (2) as potential precursors to the seco-prezizaanes (1, Figure 2A). In efforts to design a coupling reaction suitable to accomplish this goal, we built on our earlier success in developing hydroxyl-directed metallacycle-mediated annulation chemistry3 and imagined that the hydroindane 2 might be accessible from a new metallacycle-mediated cascade reaction between a simple TMS-alkyne (3) and a suitably functionalized polyunsaturated alkoxide such as 4 or 5. While the proposed use of a cumulene-containing enyne 4 in a metallacycle-mediated annulation process was thought to be interesting, our previous experience with allenic alkoxides in related coupling chemistry4 led us to the expectation that annulation chemistry with 4 would be particularly challenging owing to the multiple competing reaction pathways for carbometalation accessible to such substrates. Reaction with the 4-hydroxy-1,6-enyne 5, however, was reasoned to be an attractive alternative en route to 2, where carbocycle annulation could be accompanied by a sequence that concludes by two elimination reactions that result in loss of the propargylic phenyl and methyl ethers. Such a sequence was envisioned to occur as depicted in Figure 2B, where union of enyne 5 with the metallacyclopropene 6 (derived from a TMS-alkyne) would deliver the metallacyclopentadiene 7 with high levels of regioselectivity (with respect to each internal alkyne). This organometallic intermediate has the potential to participate in an elimination reaction of the allylic methyl ether to generate a cumulene 8 (an unproductive pathway)5 or engage the distal alkene of the metallacyclic intermediate in an intramolecular [4 + 2] cycloaddition.3 If cyclization were favored, a densely functionalized bridged bicyclic metallacyclopentene intermediate 9 would result, an intermediate that has the potential to undergo elimination of the phenyl ether to deliver a complex allylic organometallic species (10) that can participate in an additional elimination reaction to generate the desired cross-conjugated triene-containing carbocycle 2. In such a manner, coupling of enyne 5 with a TMS-alkyne 6 would deliver the targeted hydroindane skeleton 2 possessing the desired C3–C4 alkene as part of a cross-conjugated triene motif.
Figure 2.
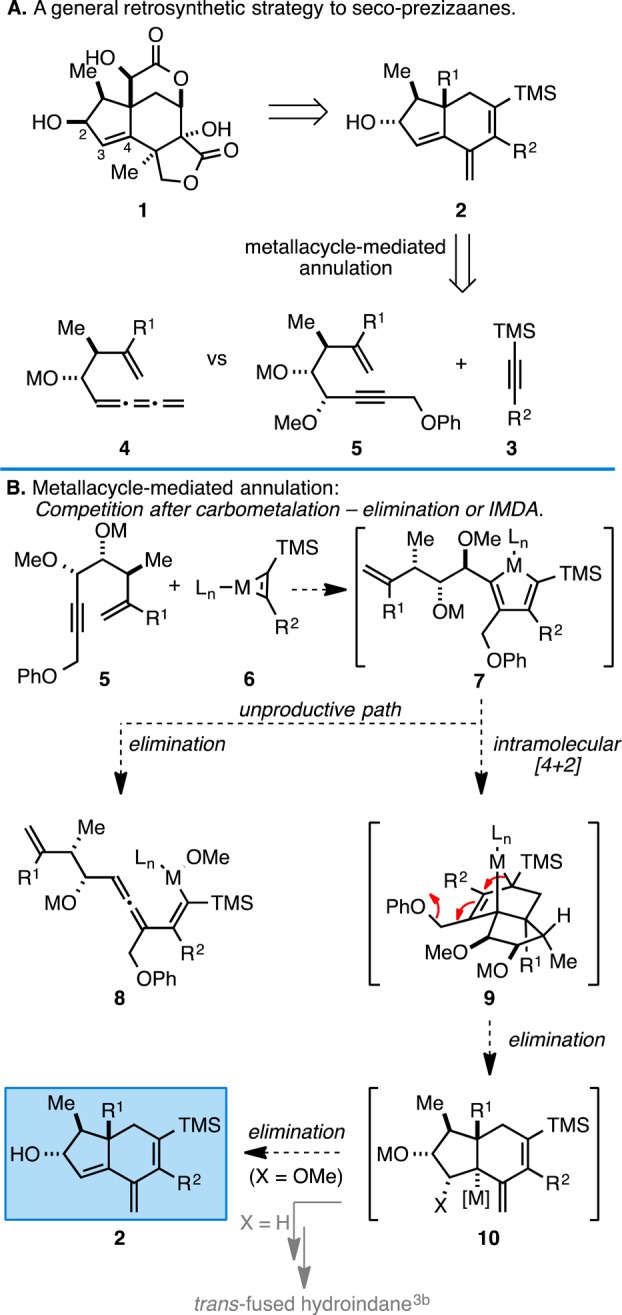
Retrosynthetic approach to the secoprezizaane core and competing reaction paths in a complex metallacycle-mediated annulation cascade.
Our initial experiments aimed at exploring this hypothesis began by preparing a model enyne substrate. As illustrated in Figure 3A, a sequence of Sharpless asymmetric epoxidation (for kinetic resolution of the enyne),6 methylation, and regioselective nucleopheolic addition7 was successful for generating 4-hydroxy-1,6-enyne substrates 15 and 19. With these substrates in hand, we began to explore the proposed metallacycle-mediated coupling/cycloaddition/double elimination cascade. As illustrated in Figure 3B, and to our great delight, Ti(Oi-Pr)4/2BuLi-mediated8 cross-coupling of enyne 15 with TMS-propyne (20) led to the formation of hydroindane 21 (Figure 3B). This observation is consistent with the conclusion that intramolecular [4 + 2] of the metallacyclopentadiene intermediate (7 → 9; Figure 2B) is faster than direct elimination of the allylic methyl ether (7 → 8). While we were able to acquire spectroscopic evidence for the formation of 21, attempted characterization was complicated due to the presence of a “decomposition” pathway that proceeded through dimerization via highly regio- and stereoselective intermolecular [4 + 2] cycloaddition (→22). Recognizing that the cross-conjugated triene system embedded in the hydroindane structure was particularly reactive in cycloaddition chemistry, we subjected the crude product from the Ti-mediated annulation to N-phenylmaleimide (0 °C, PhMe), conditions that led to the formation of tetracycle 23 in 49% yield (over two steps). Consistent with earlier results that demonstrated high regio- and stereoselectivity associated with hydroxyl-directed Ti-mediated annulation reactions for hydroindane synthesis,3 and the expected high endo selectivity of the intermolecular [4 + 2], no isomeric products were observed from this tandem sequence.
Figure 3.
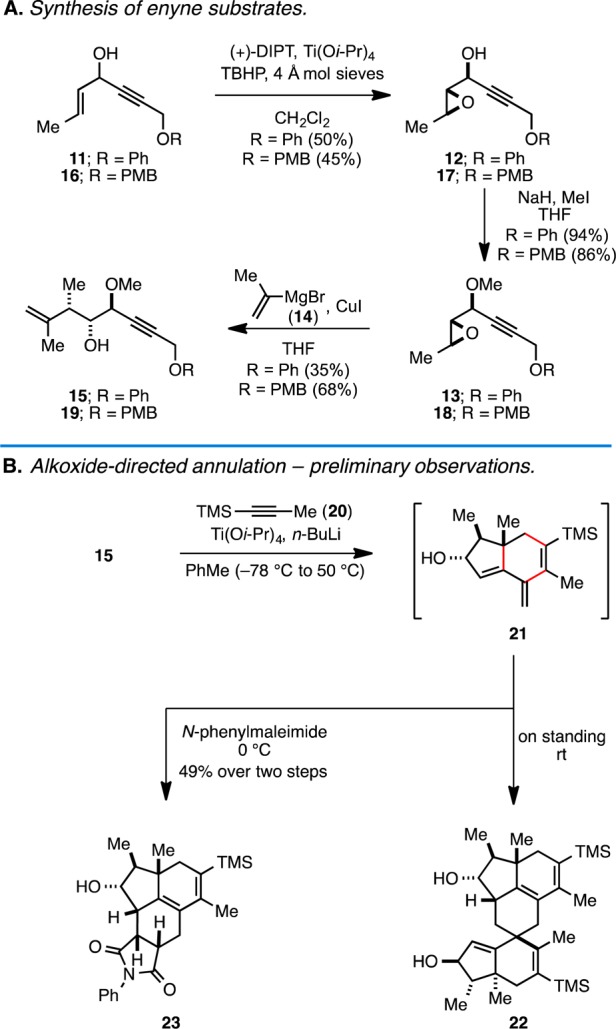
Synthesis of enyne substrates and Ti-mediated annulation/[4 + 2].
With this foundation of data to support the feasibility of our proposed alkoxide-directed carbometalation/intramolecular [4 + 2]/double elimination, and the observation that the cross-conjugated triene-containing hydroindane products possess substantial reactivity as dienes for intermolecular Diels–Alder reactions,9 we explored the generality of this two-step process for the asymmetric construction of densely functionalized carbo- and heterocyclic systems. First, as illustrated in eq 1 of Figure 4, we questioned whether the distal Ph-ether of the enyne 15 was required for the process. Here, the related structure bearing a distal PMB ether was effectively coupled to TMS-propyne 20 and N-phenylmaleimide 24, delivering tetracycle 23 in 45% yield (over two steps). In further exploration of structural variation in the enyne and TMS-alkyne, eqs 2–4 show that the two-step three-component coupling process is effective for enynes bearing allylic phenyl substituents (25 and 31) and TMS-alkynes bearing aliphatic (20, 26) and aromatic (29) substituents. In each case densely functionalized heterocycles 28, 30, and 32 were observed as the only isomeric products in 40–45% yield.
Figure 4.

Examples of intermolecular metallacycle-mediated annulation followed by intermolecular [4 + 2]. (a) Reaction conditions: alkyne, Ti(Oi-Pr)4, n-BuLi, PhMe (−78 to 50 °C), cool to −78 °C, and add Li-alkoxide of enyne (−78 to 50 °C), aqueous workup; then dienophile, PhMe (see Supporting Information for details).
As depicted in eq 5, the terminal intermolecular [4 + 2] cycloaddition can be conducted with an unsymmetrical dienophile. Here, the union of enyne 19 with alkyne 20 and dienophile 33 delivered cycloadduct 34 in 43% yield as a single isomeric product.
The cycloaddition was also effective with other classes of highly activated dienophiles. For example, use of napthoquinone as the dienophile resulted in formation of the complex pentacyclic carbocycle 37 in 54% yield (eq 6). When a triazolene dione (39) was employed as the dienophile, a 2:1 adduct was formed in what appears to be a highly stereoselective fashion, albeit delivering the complex heterocycle 40 in a modest 36% isolated yield.9
Finally, while most attempted coupling reactions did proceed to deliver cross-conjugated triene-containing hydroindanes that were successfully trapped by an intermolecular Diels–Alder reaction, we were unable to achieve this annulation process with enyne 41 (eq 8). Here, attempted coupling resulted only in the formation of the tetraene 43, a compound derived from simple protonation of the metallacyclopentadiene product of alkyne–alkyne coupling (i.e., 7; Figure 2B).10
In conclusion, we have developed a new metallacycle-mediated cascade process that produces hydroindane structures containing an embedded cross-conjugated triene. This reaction was designed based on our previously described Ti-mediated hydroxyl-directed [2 + 2 + 2] annulation that was proposed to proceed by way of regioselective alkyne–alkyne coupling and stereoselective intramolecular [4 + 2] cycloaddition.3 The present reaction process diverges from these recently discovered carbocycle-forming processes by diverting the reactivity of the presumed bridged tricyclic metallacyclopentene intermediate (i.e., 9, Figure 2B) down a pathway for double elimination. While we were successful in realizing the proposed cascade, the cross-conjugated triene-containing carbocyclic products were particularly reactive. If not trapped by intermolecular [4 + 2] cycloaddition with an activated dienophile, these products dimerized on standing. While future studies will aim to divert the reactivity of these products toward a pathway of potential utility for the synthesis of seco-prezizaane sesquiterpenes, we recognize that the highly selective annulation and subsequent intermolecular [4 + 2] cycloaddition represents a powerful asymmetric entry to densely functionalize carbo- and heterocyclic systems.
Acknowledgments
We thank the NIH NIGMS (GM080266) for financial support.
Supporting Information Available
Procedures and spectroscopic data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- For a recent review, see:Liu Y.-N.; Su X.-H.; Huo C.-H.; Zhang X.-P.; Shi Q.-W.; Gu Y.-C. Chem. Biodiversity 2009, 6, 963–989. [DOI] [PubMed] [Google Scholar]
- For a review of laboratory syntheses of natural products from Illicium species, see:; a Urabe D.; Inoue M. Tetrahedron 2009, 65, 6271–6289. [Google Scholar]; For recent studies, see:; b Shi L.; Meyer K.; Greaney M. F. Angew. Chem., Int. Ed. 2010, 122, 9250–9253. [DOI] [PubMed] [Google Scholar]; c Harada K.; Imai A.; Uto K.; Carter R. G.; Kubo M.; Hioki H.; Fukuyama Y. Org. Lett. 2011, 13, 988–991. [DOI] [PubMed] [Google Scholar]; d Trzoss L.; Xu J.; Lacoske M. H.; Mobley W. C.; Theodorakis E. A. Org. Lett. 2011, 13, 4554–4557. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Xu J.; Trzoss L.; Chang W. K.; Theodorakis E. A. Angew. Chem., Int. Ed. 2011, 50, 3672–3676. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Yang Y.; Fu X.; Chen J.; Zhai H. Angew. Chem., Int. Ed. 2012, 51, 9825–9828. [DOI] [PubMed] [Google Scholar]; g Chen J. W.; Gao P.; Yu F. M.; Yang Y.; Zhu S. Z.; Zhai H. B. Angew. Chem., Int. Ed. 2012, 51, 5897–5899. [DOI] [PubMed] [Google Scholar]; h Ogura A.; Yamada K.; Yokoshima S.; Fukuyama T. Org. Lett. 2012, 14, 1632–1635. [DOI] [PubMed] [Google Scholar]; i Trzoss L.; Xu J.; Lacoske M. H.; Mobley W. C.; Theodorakis E. A. Chem.—Eur. J. 2013, 19, 6398–6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Greszler S. N.; Reichard H. A.; Micalizio G. C. J. Am. Chem. Soc. 2012, 134, 2766–2774. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Jeso V.; Aquino C.; Cheng X.; Mizoguchi H.; Nakashige M.; Micalizio G. C. J. Am. Chem. Soc. 2014, 136, 8209–8212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Shimp H. L.; Micalizio G. C. Chem. Commun. 2007, 4531–4533. [DOI] [PMC free article] [PubMed] [Google Scholar]; b McLaughlin M.; Shimp H. L.; Navarro R.; Micalizio G. C. Synlett 2008, 735–738. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Shimp H. L.; Hare A.; McLaughlin M.; Micalizio G. C. Tetrahedron 2008, 64, 6831–6837. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Barlan A. U.; Micalizio G. C. Tetrahedron 2010, 66, 4775–4783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Yamashita K.; Sato F. Tetrahedron Lett. 1996, 37, 7275–7278. [Google Scholar]; b Fukuhara K.; Okamoto S.; Sato F. Org. Lett. 2003, 5, 2145–2148. [DOI] [PubMed] [Google Scholar]; c Tanaka R.; Sasaki M.; Sato F.; Urabe H. Tetrahedron Lett. 2005, 46, 329–332. [Google Scholar]; For examples of elimination reactions of Ti–alkene and Ti–alkyne complexes, see:; d Kasatkin A.; Nakagawa T.; Okamoto S.; Sato F. J. Am. Chem. Soc. 1995, 117, 3881–3882. [Google Scholar]; e Lee J.; Cha J. K. Tetrahedron Lett. 1996, 37, 3663–3666. [Google Scholar]; f Okamoto S.; An D. K.; Sato F. Tetrahedron Lett. 1998, 39, 4551–4554. [Google Scholar]
- Katsuki T.; Martin V. S. Org. React. 1996, 48, 1–299. [Google Scholar]
- For a related example of the regioselective opening of a disubstituted epoxide in the context of the total synthesis of macbecin I, see:Baker R.; Castro J. L. J. Chem. Soc. Chem. Commun. 1989, 378–381. [Google Scholar]
- For an early example of forming Ti–alkyne and Ti–imine complexes from Ti(Oi-Pr)4/n-BuLi, see:; a Obora Y.; Moriya H.; Tokunaga M.; Tsuji Y. Chem. Commun. 2003, 2820. [DOI] [PubMed] [Google Scholar]; b Tarselli M. A.; Micalizio G. C. Org. Lett. 2009, 11, 4596–4599. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Rassadin V. A.; Six Y. Tetrahedron 2014, 70, 787–794. [Google Scholar]
- For an early example of the use of cross-conjugated trienes in diene-transmissive Diels–Alder reactions, see:; a Blomquist A. T.; Verdol J. A. J. Am. Chem. Soc. 1955, 77, 81–83. [Google Scholar]; For more recent examples, see:; b Spino C.; Liu G.; Tu N.; Girard S. J. Org. Chem. 1994, 59, 5596–5608. [Google Scholar]; c Woo S.; Squires N.; Fallis A. G. Org. Lett. 1999, 1, 573–575. [Google Scholar]; d Kwon O.; Park S. B.; Schreiber S. L. J. Am. Chem. Soc. 2002, 124, 13402–13404. [DOI] [PubMed] [Google Scholar]
- Ryan J.; Micalizio G. C. J. Am. Chem. Soc. 2006, 128, 2764–2765. [DOI] [PubMed] [Google Scholar]; While not yet fully understood, it appears that nonbonded steric interactions resulting from the two aryl substituents apparently dissuade intramolecular [4 + 2] cycloaddition. Previous studies3a have shown that conjugated enynes are viable coupling partners in related Ti-mediated annulation chemistry, and the attempted reaction of substrate 41 with TMS-alkyne 26 also led to a triene product without the observation of an intramolecular [4 + 2] cycloaddition.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.