

Abstract
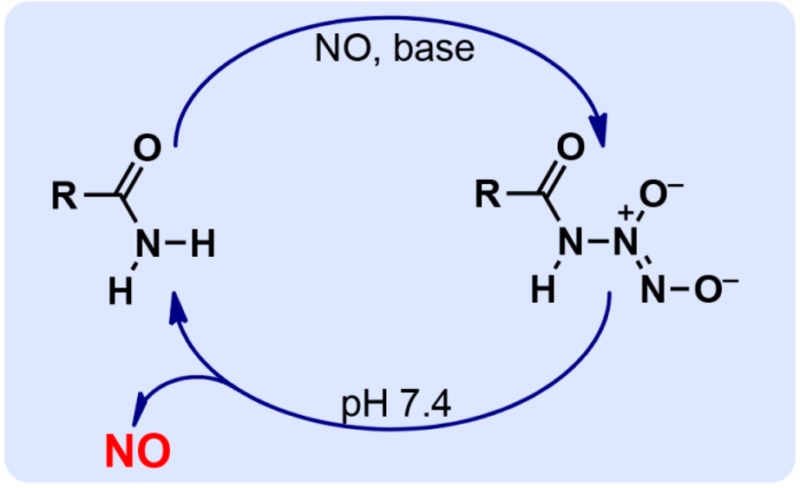
We report the apparently unprecedented direct reaction of nitric oxide (NO) with amides to generate ions of structure R(C=O)NH–N(O)=NO–, with examples including R = Me (1a) or 3-pyridyl (1b). The sodium salts of both released NO in pH 7.4 buffer, with 37 °C half-lives of 1–3 min. As NO-releasing drug candidates, diazeniumdiolated amides would have the advantage of generating only 1 equiv of base on hydrolyzing exhaustively to NO, in contrast to their amine counterparts, which generate 2 equiv of base.
Many secondary and primary amines react with nitric oxide (NO) to generate products of structure R1R2N–N(O)N=O–, known as diazeniumdiolate ions. Extensive structure/reactivity studies involving a wide variety of amines were carried out by the Drago research group a half a century ago,1,2 showing the remarkable generality of this amine-derivatizing reaction. Interestingly, they did not describe any attempts to react the corresponding amides with NO, nor have we seen any such reports in the intervening literature. We have long taken this absence of relevant prior literature as a tacit suggestion that attempts to diazeniumdiolate an amide by directly reacting it with NO would probably be fruitless, until now continuing our focus on the amine/NO adducts that have proven so useful as biomedical research tools and potential clinical entities.3−9
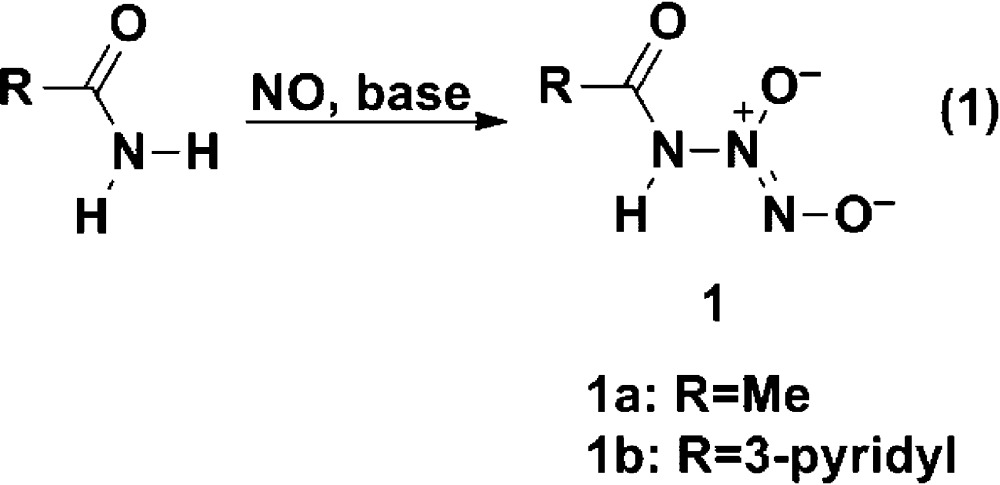
Here we show that this skepticism was unfounded by reporting on the direct reaction of NO with amides under basic conditions to produce novel ionic diazeniumdiolates, an apparently unprecedented transformation (see eq 1).
 |
1 |
Treatment of methanolic acetamide with 3 atm of NO in the presence of sodium methoxide led to deposition of a white solid with an ultraviolet absorbance maximum at 252 nm, consistent with the presence of a diazeniumdiolate group10 [i.e., a group of structure X–N(O)=NO–] and suggesting that the reaction had led to diazeniumdiolate ion 1a, as in eq 1.
The product proved relatively stable in basic solutions but decomposed on lowering the pH to 7.4 with a half-life at 37 °C of 1.2 min. Abundant NO was seen as a gaseous product of this reaction. In addition, significant yields of nitrite ion were observed. The results for the nitrogenous products are summarized in Table 1. The organic products were analyzed by NMR of the spent reaction mixture. Acetamide was detected in 78% yield, and the sodium acetate yield was close to 22%.
Table 1. UV Spectral Data Plus Rates and Yields of Nitrogenous Products on Hydrolyzing Ions 1 at pH 7.4 and 37 °C: Comparison with IPA/NO.
| compound | R | NOa | N2Oa | nitritea | t1/2 (min) | λmax (nm) 0.1 M NaOH | ε (mM–1 cm–1) 0.1 M NaOH |
|---|---|---|---|---|---|---|---|
| 1a | Me | 88% (±1%) | <0.01 | 9% (±0.3%) | 1.2 | 252 | 9.7 |
| 1b | 3-pyridyl | 52% (±1%) | 7% (±0.7%) | 10% (±1%) | 2.8 | 250 | 11.8 |
| IPA/NO | 30% | 70% | 3% | 2.3b | 252b | 8.7b |
Yield as a percent of theoretical plus or minus the standard deviation of three independent measurements.
From Maragos et al.12
Similar results were seen when the experiment was repeated using nicotinamide instead of acetamide. Again, the product was stable in base but on neutralization smoothly lost its UV peak at 250 nm, suggestive of the diazeniumdiolate structure 1b with a half-life of 2.8 min at pH 7.4 and 37 °C. The relative distribution of nitrogenous products was similar to that seen in the case of acetamide in that they are primarily NO and nitrite, though in this case, some N2O was seen (Table 1). Nicotinamide was recovered in 55% yield, similarly to that of the NO (52%) and nicotinic acid (45% of theory).
The two ionic amide diazeniumdiolates can be compared to the primary amine analogue IPA/NO (Scheme 1), where the acyl groups of ions 1 are replaced with an alkyl residue. Although the three compounds have similar stability profiles, they differ greatly in the relative yields of their nitrogenous products (Table 1). As presented above, both 1a and 1b primarily generate NO and nitrite with very little N2O production. In contrast, IPA/NO decomposes via two pathways running in parallel; the minor pathway regenerates starting materials from its synthesis (isopropylamine + 2 equiv of NO), while the major pathway produces a mole each of HNO and isopropanediazoate ion.11
Scheme 1. Structural Comparison of Ionic Diazeniumdiolated Amides 1a and 1b to the Primary Amine Diazeniumdiolate IPA/NO.

The ionic primary amine diazeniumdiolate IPA/NO can be dialkylated in two successive steps, first at the terminal O and then at the amine N.13 Ion 1a can also be dialkylated, but it differs from IPA/NO in that the intermediate monoalkyl species could not be isolated. An additional difference between 1a and IPA/NO is that the former could theoretically be alkylated at the amide oxygen, a functionality absent in IPA/NO (Scheme 2). No evidence for alkylation at that position was seen, however, as X-ray crystallography confirmed the O,N-dialkylated structure 2 in Scheme 2.
Scheme 2. Benzylation of Ion 1a Produced the O,N-Dialkylated Derivative 2 (O,O′-Dialkylated Structure Was Not Observed).

Salt 1b could be dialkylated with methyl iodide and benzyl bromide (Scheme 3). To our surprise, the dialkylation products isolated after normal aqueous workup, which we expected to have structure 3, contained no halide ion, as evidenced by mass spectrometry and absence of a precipitate on addition of the silver(I) ion to their aqueous solutions. Molecular weight measurements of the dibenzylation products suggested that the N–H proton had been lost during workup, forming zwitterions 4b and 4b′, as shown in Scheme 3, and this was confirmed by X-ray crystallography of the dibenzyl derivative.
Scheme 3. Diazeniumdiolation of Nicotinamide, Followed by Dialkylation To Form Zwitterions 4.

We were surprised to find no prior literature documenting a successful reaction of NO directly with an amide. The only product of such a hypothetical reaction that we could locate had the constitution of N-diazeniumdiolated acetanilide ion, but it was reportedly generated in a radical-mediated side reaction in the decomposition of N-nitrosoacetanilide.14 (Several N-diazeniumdiolated carbamates have also been prepared by routes that bypassed the ionic structure 1 by first O-alkylating the terminal oxygen of IPA/NO to make a neutral species and then acylating the NH group.)15,16
Mechanistically, by analogy with the pathways established for the primary amine diazeniumdolates, we assume that the decomposition of ions 1 consists of two general mechanisms, one being a major, or in the case of 1a exclusive, pathway leading to regeneration of the two NO molecules, with the starting amide being the organic product. A concurrent pathway, minor for 1b but not seen for 1a, begins with cleavage of HNO (followed by its dimerization to N2O) and production of an acyldiazoate ion (which hydrolyzes to the carboxylic acid/carboxylate with loss of N2), as shown in Scheme 4.
Scheme 4. Proposed Mechanisms of Dissociation of Diazeniumdiolated Amide Ions 1 at Physiological pH (See Salmon et al.11 for a Full Mechanistic Description of IPA/NO Hydrolysis, Including pH Effects).

From the observed stoichiometry, 1a appears to dissociate exclusively by the NO release pathway shown at the top of Scheme 4, as no N2O was detected. The NO and acetamide yields were roughly similar, as anticipated (88 and 78%, respectively). The 22% acetate yield was not expected according to this mechanism but could have arisen by nitrosative deamination of the remaining acetamide or by simple hydrolysis. In contrast, dissociation of 1b did produce some N2O (7%), indicating a role for the lower pathway of Scheme 4; however, the upper route producing NO and nicotinamide remained the dominant path, the products being recovered in yields of 52 and 55%, respectively.
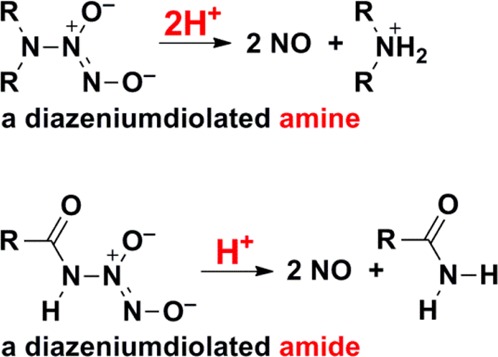
An advantage of diazeniumdiolated amides relative to the widely used secondary amine diazeniumdiolates as NO donors is that the former’s exhaustive decomposition in neutral buffer consumes only 1 equiv of acid while the amine derivatives consume 2 equiv (Scheme 5). Thus, the decomposition of diazeniumdiolated amides produces a nonbasic amide, while exhaustive hydrolysis of secondary amine diazeniumdiolates produces a still basic amine that consumes an additional proton. This property would be especially beneficial in a biomedical setting, where careful pH control is essential.
Scheme 5. Hydrolysis of Diazeniumdiolated Amines To Produce NO Consumes Two Protons, While That of the Corresponding Amide Derivatives Consumes Only One.
The results suggest that a variety of useful NO-releasing hybrid drugs might be forthcoming from a systematic study of reactions of different bioactive amides with NO as described above.
Experimental Section
Caution: IPA/NO and certain other diazeniumdiolates have shown a tendency to dissociate rapidly and unexpectedly and hence should be handled with due care.
General
Ultraviolet (UV) spectra were recorded on a diode array spectrophotometer. Nuclear magnetic resonance (NMR) spectra were collected with a 400 MHz spectrometer using appropriate deuterated solvents with chemical shifts reported in parts per million downfield from tetramethylsilane or 2,2,3,3-tetradeuterotrimethylsilylpropionic acid. Ions for high-resolution mass spectrometry were generated with electrospray ionization and detected with a quadrupole time-of-flight mass analyzer.
Purity Measurements for Salts 1a and 1b
Ionic diazeniumdiolates are notoriously difficult to purify, so it is correspondingly problematic to provide quantitative data on the properties of the above-identified products. Nevertheless, rough purity estimates could be derived from NMR experiments using internal standards in alkaline solutions. The result for 1a using potassium phthalimide as internal standard was a purity level of 50%, with the remainder presumably consisting of NaOH and other UV- and NMR-silent constituents. The purity of the 1b sample was similarly determined to be 80% using tert-butyl alcohol as internal standard. These values were used as normalization factors to establish the extinction coefficients as well as theoretical yields of the various products summarized in Table 1.
Sodium 1-(N-Acetylamino)diazen-1-ium-1,2-diolate (1a)
A solution of 5.9 g (0.1 mol) of acetamide in 21.7 mL (0.1 mol) of 4.6 M methanolic sodium methoxide, 10 mL of methanol, and 50 mL of diethyl ether was charged with 40 psi of nitric oxide and stirred under pressure at room temperature. A precipitate began to be visible within 2 h; stirring was continued overnight, giving a thick white precipitate. The precipitate was collected by filtration, washed with acetonitrile, and dried under vacuum to give 3.07 g of a white powder that was 50% pure according to the NMR/internal standard method described above (10% yield). As a further purification step, 2 g of crude 1a (50% pure, 7.1 mmol) was dissolved in 100 mL of methanol and stirred at room temperature. The methanolic solution was treated with 100 mL of ether, giving a white solid that was collected by filtration and triturated with acetonitrile, then dried in vacuo to give 1.7 g of product. This process removed most of the acetamide contaminant: UV (in 0.01 M NaOH) λmax 252 nm (9.7 mM–1 cm–1); 1H NMR (D2O/NaOD) δ 1.68 (s); 13C NMR (D2O/NaOD) δ 20.8, 177.1.
Sodium 1-(N-Nicotinoylamino)diazen-1-ium-1,2-diolate (1b)
A solution of 9.1 g (0.075 mol) of nicotinamide in 30 mL of methanol was treated with 15.2 mL (0.07 mol) of 4.6 M methanolic sodium methoxide. To the solution was added 50 mL of ether, after which the vessel was degassed and charged with 50 psi of nitric oxide. The solution was stirred at room temperature; after 72 h, the pressure was released and the white precipitate was collected by filtration. The solid was washed with acetonitrile and dried under vacuum to give 3.91 g of 1b (80% pure by the internal standard NMR method described above, 20% yield) as a powder: UV (0.01 M NaOH) λmax 250 nm (11.8 mM–1 cm–1); 1H NMR (D2O/NaOD) δ 7.50–7.56 (m, 1H), 8.23–8.28 (m, 1H), 8.59–8.63 (m, 1H), 8.93–8.96 (m, 1H); 13C NMR (D2O/NaOD) δ 123.8, 132.6, 136.4, 147.7, 150.0, 171.4.
Reaction of 1a with Benzyl Bromide To Form 2
To a slurry of 252 mg of 1a (0.89 mmol) and 250 mg of anhydrous sodium carbonate in 10 mL of DMF at 0 °C was added 438 μL (3.6 mmol) of benzyl bromide; the resulting slurry was stirred at 0 °C with gradual warming to room temperature. After being stirred overnight, the reaction mixture was cooled to −80 °C and placed in a lyophilizer; on evaporation, the solid residue was extracted with dichloromethane. The organic layer was dried over sodium sulfate, filtered through a layer of magnesium sulfate, and evaporated to give 229 mg of an oil. The crude product was chromatographed on a 25 g silica gel column and eluted with a gradient from 100% methylene chloride to 70:30 methylene chloride/ethyl acetate to give 102 mg (38%) of pure 2: mp 57–58 °C; UV (ethanol) λmax 244 nm (11.1 mM–1 cm–1); 1H NMR (CDCl3) δ 7.35 (s, 3H), 4.92 (s, 2H), 5.18 (s, 2H), 7.23–7.34 (m, 10H) ; 13C NMR (CDCl3) δ 19.8, 49. 8, 76.7, 128.3, 128.6, 128.6, 128.7, 128.9, 129.0, 133.8, 134.9, 169.1; HRMS (ESI) m/z calculated for C16H18N3O3+m/z (M+ + H)+ 300.13427, measured 300.1350. Anal. Calcd for C16H17N3O3: C, 64.20; H, 5.72; N, 14.04. Found: C, 64.18; H, 5.81; N, 14.29.
Reaction of 1b with Benzyl Bromide To Form 4b
A slurry of 1.54 g of 80% pure 1b (6.1 mmol) in 20 mL of N,N-dimethylformamide was reacted with benzyl bromide (1.19 mL, 0.01 mol) under conditions similar to those used to prepare 2 (previous paragraph) to give 420 mg (19%) of pale brown solid. The product was recrystallized from hot water to give white X-ray quality crystals of 4b: mp 236–8 °C; UV (in 1:1 H2O/CH3CN) λmax 239 nm (11.9 mM–1 cm–1); 1H NMR (DMSO-d6) δ 5.07 (s, 2H), 5.90 (s, 2H), 7.33–7.52 (m, 10H), 8.10 (t, 1H) J = 7.4 Hz, 8.88 (d, 1H) J = 7.8 Hz; 13C NMR (DMSO-d6) δ 63.1, 73.0, 127.7, 127.9, 128.1, 128.3, 128.8, 129.3, 129.3, 134.4, 137.1, 139.0, 143.7, 143.8, 144.7, 162.6; HRMS (ESI) m/z calculated for C20H19N4O3+, 363.14517, measured m/z 363.14567. Anal. Calcd for C20H18N4O3·0.25H2O: C, 65.47; H, 5.08; N, 15.27. Found: C, 65.62; H, 4.84; N, 15.54.
Reaction of 1b with Dimethyl Sulfate To Form 4b′
Compound 1b (1.34 g, 80% pure, 5.3 mmol) was dissolved in 50 mL of cold methanol (0 °C) and reacted with dimethyl sulfate (1.52 mL, 16 mmol) as above to give 1.8 g of an orange oil. The resulting crude product was chromatographed on silica gel using a gradient from 100% dichloromethane to 50:50 dichloromethane/methanol as the eluent. A fraction containing 296 mg of product was concentrated, whereupon the residual oil crystallized on standing. Recrystallization from methanol gave 259 mg (23%) of analytically pure 4b′: mp 202–205 °C; UV (H2O) λmax 239 nm (4.9 mM–1 cm–1) and λmax 268 nm (3.3 mM–1 cm–1); 1H NMR (D2O) δ 4.11 (s, 3H), 4.62 (s, 3H), 8.11 (br s, 1H), 8.89 (br s, 2H), 9.21 (br s, 1H); 13C NMR (D2O) δ 51.2, 64.0, 130.6, 138.9, 146.5, 147.6, 149.2, 170.0; HRMS (ESI) m/z calculated for C8H11N4O3+m/z 211.08257, measured 211.08338. Anal. Calcd for C8H10N4O3·0.25H2O: C, 44.75; H, 4.93; N, 26.10. Found: C, 44.62; H, 4.85; N, 25.75.
Kinetic Studies
Kinetic experiments were performed at 37 °C using a standard UV–visible spectrophotometer. Reactions were initiated by addition of substrate to a preheated cuvette containing the buffer. Analyte concentration was measured in 100 μM 0.1 M phosphate buffer, pH 7.4, containing 50 μM diethylenetriamine pentaacetic acid (DTPA). In each experiment, the data were analyzed at the λmax and the rate was derived by fitting the data to an exponential curve typical for first-order processes.
Analysis for NO
Chemiluminescence detection and quantification of NO evolving from the reactions were conducted using a commercial nitric oxide analyzer. A pH 7.4 solution of 0.1 M phosphate buffer with 50 μM DTPA was sparged with inert gas until a steady detector response was established. Compound 1a, 1b, or IPA/NO was added to a final concentration of 100 μM, and the NO release profile was followed over time after injection. The resulting curve was integrated to quantify the amount of NO released/mol of compound.
Griess Assay Test for Nitrite Detection
Each reaction was allowed to proceed to completion, according to the protocol above, in the absence of purging with inert gas. We then added 100 μL of Griess reagent, 300 μL of sample, and 2.6 mL of deionized water together in a spectrophotometer cuvette. We incubated the mixture for 30 min at room temperature and prepared a reference sample by mixing 100 μL of Griess reagent and 2.9 mL of deionized water. The absorbance of the nitrite-containing sample at 548 nm relative to the reference sample was converted to nitrite concentrations using a calibration curve.
N2O Measurements by Gas Chromatography
Reactions were run according to the conditions stated above. The gas chromatography was performed using an electron capture detector, equipped with a 63Ni 370 MBq source. A packed column (2 m × 2.0 mm i.d.) was used with helium as the carrier gas. The GC operation conditions were as follows: injector and detector temperatures were at 250 °C; oven temperature was programmed from 90 to 200 °C at 20 °C/min and held at 200 °C for 1.1 min; helium flow was 30 mL/min; and nitrogen was used as makeup gas at 2 mL/min.
Organic Product Analysis
The organic products of the aqueous decomposition of 1a and 1b were analyzed by NMR upon exhaustion of the nitrogenous products in a deuterated phosphate buffer under the conditions listed above.
X-ray Crystal Data on Compounds 2 and 4b
Single-crystal X-ray diffraction data on compounds 2 and 4b were collected using Mo Kα radiation and a Bruker APEX-2 CCD area detector. Crystals were prepared for data collection by coating with high viscosity microscope oil. The oil-coated crystal was mounted on a micromesh mount and transferred to the diffractometer. The structures were solved by direct methods and refined by full-matrix least-squares on F2 values using the programs found in the SHELXTL suite (Bruker, SHELXTL v6.10, 2000, Bruker AXS Inc., Madison, WI). Corrections were applied for Lorentz, polarization, and absorption effects. Parameters refined included atomic coordinates and anisotropic thermal parameters for all non-hydrogen atoms. Hydrogen atoms on carbons were included using a riding model (coordinate shifts of C applied to H atoms) with the C–H distance set at 0.96 Å. Complete information on data collection and refinement is available in the Supporting Information.
For compound 2, a 0.256 × 0.047 × 0.011 mm3 crystal was prepared for data collection and a data set collected at 150 K. The crystal was triclinic in space group P1̅, with unit cell dimensions of a = 5.429(4) Å, b = 8.743(5) Å, c = 16.342(11) Å, α = 96.30(3)°, β = 96.60(3)°, and γ = 92.33(3)°. Data were 92.5% complete to 23.30° θ (∼0.90 Å, the limit of diffraction for this crystal) with an average redundancy of 1.35. The final anisotropic full-matrix least-squares refinement on F2 with 201 variables converged at R1 = 7.72% for the observed data and wR2 = 24.35% for all data.
For compound 4b, a 0.258 × 0.184 × 0.058 mm3 crystal was prepared for data collection and a data set collected at 150 K. The crystal was monoclinic in space group C2/c, with unit cell dimensions of a = 18.3109(19) Å, b = 10.8087(9) Å, c = 20.0132(16) Å, and β = 109.785(4)°. Data were 99.6% complete to 28.33° θ (∼0.73 Å) with an average redundancy of 4.22. The final anisotropic full-matrix least-squares refinement on F2 with 244 variables converged at R1 = 4.54% for the observed data and wR2 = 14.23% for all data.
Atomic coordinates for 2 and 4b have been deposited with the Cambridge Crystallographic Data Centre (deposition numbers 1015513 and 1015514, respectively). Copies of the data can be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge, CB2 1EZ, UK [fax: +44(0)-1223-336033 or e-mail: deposit@ccdc.cam.ac.uk.
Acknowledgments
We thank Dr. Sergey Tarasov and Ms. Marzena A. Dyba (Biophysics Resource in the Structural Biophysics Laboratory, Frederick National Laboratory for Cancer Research) for assistance with HRMS. This project was funded by the National Cancer Institute (NCI), National Institutes of Health (NIH), under Contract HHSN261200800001E, and by the Intramural Research Program of the NIH, NCI, Center for Cancer Research. The X-ray crystallographic work was supported by NIDA through Interagency Agreement #Y1-DA1101 with the Naval Research Laboratory (NRL).
Supporting Information Available
Full 1H and 13C NMR spectra for new compounds and crystallographic data for compounds 2 and 4b. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Drago R. S.; Karstetter B. R. J. Am. Chem. Soc. 1961, 83, 1819–1822. [Google Scholar]
- Drago R. S.Reactions of Nitrogen(II) Oxide. In Free Radicals in Inorganic Chemistry (Advances in Chemistry Series Number 36); American Chemical Society: Washington, DC, 1962; pp 143–149. [Google Scholar]
- Scatena R.; Bottoni P.; Pontoglio A.; Giardina B. Curr. Med. Chem. 2010, 17, 61–73. [DOI] [PubMed] [Google Scholar]
- Keefer L. K. ACS Chem. Biol. 2011, 6, 1147–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basudhar D.; Bharadwaj G.; Cheng R. Y.; Jain S.; Shi S.; Heinecke J. L.; Holland R. J.; Ridnour L. A.; Caceres V. M.; Spadari-Bratfisch R. C.; Paolocci N.; Velazquez-Martinez C. A.; Wink D. A.; Miranda K. M. J. Med. Chem. 2013, 56, 7804–7820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu J.; Liu L.; Huang Z.; Lai Y.; Ji H.; Peng S.; Tian J.; Zhang Y. J. Med. Chem. 2013, 56, 4641–4655. [DOI] [PubMed] [Google Scholar]
- Yepuri N. R.; Barraud N.; Mohammadi N. S.; Kardak B. G.; Kjelleberg S.; Rice S. A.; Kelso M. J. Chem. Commun. 2013, 49, 4791–4793. [DOI] [PubMed] [Google Scholar]
- Worley B. V.; Slomberg D. L.; Schoenfisch M. H. Bioconjugate Chem. 2014, 25, 918–927. [DOI] [PubMed] [Google Scholar]
- Dharmaraja A. T.; Ravikumar G.; Chakrapani H. Org. Lett. 2014, 16, 2610–2613. [DOI] [PubMed] [Google Scholar]
- Keefer L. K.; Flippen-Anderson J. L.; George C.; Shanklin A. P.; Dunams T. M.; Christodoulou D.; Saavedra J. E.; Sagan E. S.; Bohle D. S. Nitric Oxide 2001, 5, 377–394. [DOI] [PubMed] [Google Scholar]
- Salmon D. J.; Torres de Holding C. L.; Thomas L.; Peterson K. V.; Goodman G. P.; Saavedra J. E.; Srinivasan A.; Davies K. M.; Keefer L. K.; Miranda K. M. Inorg. Chem. 2011, 50, 3262–3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maragos C. M.; Morley D.; Wink D. A.; Dunams T. M.; Saavedra J. E.; Hoffman A.; Bove A. A.; Isaac L.; Hrabie J. A.; Keefer L. K. J. Med. Chem. 1991, 34, 3242–3247. [DOI] [PubMed] [Google Scholar]
- Saavedra J. E.; Bohle D. S.; Smith K. N.; George C.; Deschamps J. R.; Parrish D.; Ivanic J.; Wang Y.-N.; Citro M. L.; Keefer L. K. J. Am. Chem. Soc. 2004, 126, 12880–12887. [DOI] [PubMed] [Google Scholar]
- Tan C.-C.; Ruchardt C.; Binsch G.; Hofner D.; Huisgen R.; Nakaten H. Chem. Ber. 1975, 108, 1027–1035. [Google Scholar]
- Nandurdikar R. S.; Maciag A. E.; Cao Z.; Keefer L. K.; Saavedra J. E. Bioorg. Med. Chem. 2012, 20, 2025–2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z.; Kaur J.; Bhardwaj A.; Alsaleh N.; Reisz J. A.; DuMond J. F.; King S. B.; Seubert J. M.; Zhang Y.; Knaus E. E. J. Med. Chem. 2012, 55, 10262–10271. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.