

Abstract
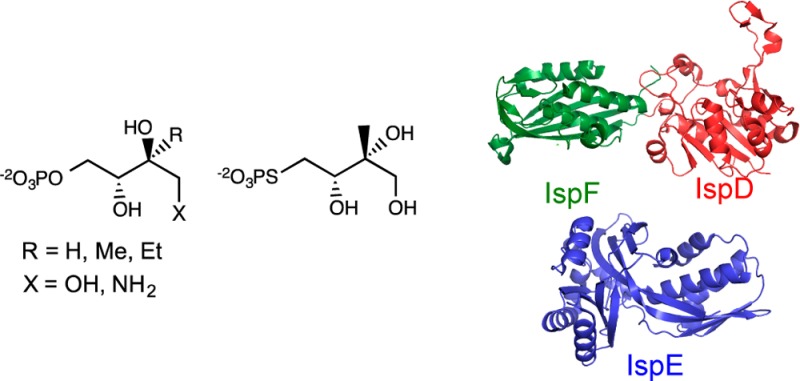
The methylerythritol phosphate biosynthetic pathway, found in most Bacteria, some parasitic protists, and plant chloroplasts, converts d-glyceraldehyde phosphate and pyruvate to isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP), where it intersects with the mevalonate pathway found in some Bacteria, Archaea, and Eukarya, including the cytosol of plants. d-3-Methylerythritol-4-phosphate (MEP), the first pathway-specific intermediate in the pathway, is converted to IPP and DMAPP by the consecutive action of the IspD-H proteins. We synthesized five d-MEP analogues—d-erythritol-4-phosphate (EP), d-3-methylthrietol-4-phosphate (MTP), d-3-ethylerythritol-4-phosphate (EEP), d-1-amino-3-methylerythritol-4-phosphate (NMEP), and d-3-methylerythritol-4-thiolophosphate (MESP)—and studied their ability to function as alternative substrates for the reactions catalyzed by the IspDF fusion and IspE proteins from Agrobacterium tumefaciens, which covert MEP to the corresponding eight-membered cyclic diphosphate. All of the analogues, except MTP, and their products were substrates for the three consecutive enzymes.
Introduction
The isoprenoid biosynthetic pathway produces over 60 000 small-molecule metabolites that perform numerous essential functions in all forms of life.1 A few examples include electron transfer (ubiquinones), cellular membranes (sterols and hopanes), hormones (sterols, sesquiterpenes, diterpenes), pheromones (monoterpenes), photosynthesis (carotenoids and chlorophylls), and signal transduction (isoprenylated proteins). Isoprenoid molecules are synthesized from (R)-mevalonate (MVA)2 or d-methylerythritol phosphate (MEP)3 by two fundamentally different pathways. The MVA pathway provides isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP) for isoprenoid biosynthesis in most Eukarya, including all mammals and the cytosol and mitochondria in plants, Archaea, and a few Bacteria. IPP and DMAPP are synthesized from MEP in most Bacteria and Apicomplexa, a group of parasitic protists.
MEP is synthesized in two steps from pyruvate and d-glyceraldehyde phosphate via d-deoxyxylulose phosphate catalyzed by deoxyxylulose phosphate synthase (DXS) and deoxyxylulose keto-reductase (DXR or IspC), respectively.3MEP is converted to cyclic methylerythritol diphosphate (cMEDP) in three steps catalyzed by diphosphocytidylmethylerythritol (CDP-ME) synthase (IspD), diphosphocytidylmethylerythrityl phosphate (CDP-MEP) synthase (IspE), and cyclomethylerythrityl diphosphate (cMEDP) synthase (IspF). Subsequently, the cyclic diphosphate is opened reductively by hydroxydimethylallyl diphosphate (HDMAPP) synthase (IspG), and the hydroxyl group is removed by a second reduction by IspH to give a mixture of IPP and DMAPP.3 The steps between MEP and IPP/DMAPP and the genes encoding the biosynthetic enzymes are shown in Scheme 1.
Scheme 1. Biosynthesis of IPP and DMAPP from MEP.

The ispD, ispE, and ispF genes encode monofunctional enzymes in most MEP-dependent organisms. However, there are several reports of fused ispD and ispF that encode bifunctional IspDF proteins in bacteria, including several genera of α-proteobacteria belonging to the order Rhizobiales.4 The IspDF protein catalyzes the first and last steps in the conversion of MEP to cMEDP, and the missing ispE activity can be provided in vivo with an enzyme from the same or different organisms. Time course studies with IspDF and IspE from Agrobacterium tumefacians indicate that the enzymes in combination efficiently convert MEP to cMEDP without any evidence for substrate channeling between the individual active sites in the protein complex.5
Since the MEP pathway is orthogonal to the MVA pathway, it is an attractive target for development of small-molecule inhibitors as antibacterial, antiparasitic, and herbicidal agents.6 Most of the reports of inhibitors and alternate substrates are for the DXS,7 DXR,8 IspD,9,10 IspG,11 and IspH9 proteins found in the early and later parts of the pathway. In a recent report, high throughput screening identified a triazolopyrimidine inhibitor of IspD, and subsequent synthetic work provided structurally related submicromolar inhibitors with herbicidal activity.12
Studies with DXR13 indicate that the enzyme accepts a number of analogues as alternate substrates and synthesizes products that are potentially inhibitors of downstream enzymes. We now report the synthesis of five analogues of MEP and showed that four of these molecules and their products are alternate substrates for the three consecutive reactions catalyzed by Agrobacterium tumefaciens IspDF and IspE.
Results and Discussion
Synthesis of MEP Analogues
Five MEP analogues, d-erythritol phosphate (EP), d-methylthreitol phosphate (MTP), d-ethylerythritol phosphate (EEP), d-aminodeoxymethylerythritol phosphate (NMEP), and d-methylerythritol thiophosphate (MESP), were synthesized for this study (Figure 1). EP and EEP were synthesized from 1,3-O-benzylidene-d-erythrulose and 4-(t-butyldimethylsilyl) ether (1) by the dioxanone approach outlined in Scheme 2.14 The five-step route to MTP was described previously.14
Figure 1.

MEP analogues.
Scheme 2. Synthesis of EP and EEP.

The route to EP from dioxanone 1 is shown in Scheme 2. Reduction of 1 with NaBH4 in methanol, followed by desilylation with tetrabutylammonium fluoride, gave diol 3 with NMR spectral data in accord with the literature.15 Phosphorylation of the primary hydroxyl group with dibenzyl chlorophosphate in pyridine and hydrogenolysis of the benzyl and benzylidene protecting groups gave EP, whose NMR parameters matched the reported values.10
EEP was prepared by a similar set of reactions. Treatment of ketone 1 with ethylmagnesium bromide proceeded with highly stereoselective axial-face addition to the carbonyl group. Previously, an “axial”/“equatorial” stereoselectivity of 20:1 was seen for the related addition of methylmagnesium bromide.14 Desilylation with Bu4NF gave diol 6. The diol was lithiated with n-butyllithium and phosphorylated regioselectively at the primary hydroxyl group with freshly prepared dibenzyl chlorophosphate. Catalytic hydrogenolysis of the dibenzyl phosphate and benzylidene ring gave EEP.
NMEP was synthesized from (Z)-4-(tert-butyldimethylsilanyloxy)-3-methyl-but-2-en-1-ol (8)16 (Scheme 3). Our original strategy involved phosphorylation of 8 using the benzyl phosphite-iodine procedure,17 removal of the TBDMS group, and introduction of the amino group by a Mitsunobu displacement18 with phthalimide, followed by hydrolysis of the imide. However, our attempts to hydrolyze the phthalimide group prior to a Sharpless dihydroxylation19 failed, presumably because of the reactivity of the phosphate triester. Alcohol 8 was then protected as a THP ether; the TBDMS protecting group was removed; and nitrogen was introduced at C1 by a Mitsunobu displacement.17 The phthalimide moiety was removed with hydrazine, and the resulting amine was protected as a Boc amide. The THP blocking group was removed, and alcohol 14 was phosphorylated using benzyl phosphite-iodine.17 A Sharpless dihydroxylation19 gave 16 as a 12:1 mixture of 2R,3S and 2S,3R enantiomers, as judged by chiral HPLC. NMEP was obtained after removing the benzyl and Boc protecting groups.
Scheme 3. Synthesis of NMEP.

The synthesis of MESP is outlined in Scheme 4. Ester 17(16) was asymmetrically dihydroxylated as described for 15.19 Diol 18 was first protected as an orthoester. Although the reaction proceeded in high yield as judged from an NMR spectrum of the crude product, the orthoester was unstable on a silica column and was isolated in 51% yield. Reduction of the methyl ester with LiBH4 proceeded in excellent yield (NMR), but again the resulting alcohol was unstable on silica and was obtained in 61% yield after purification. To improve yields, we omitted purification steps for reactions 2 and 3, and pure tosylated orthoester (steps 2–4) was obtained in an overall 89% yield. The orthoester protecting group was removed in two steps by a mild treatment with aq. HCl to give a formate ester, followed by hydrolysis with ammonia in CH3OH to give diol tosylate 22. Treatment of 22 with tBuOK and removal of the silyl blocking group gave epoxide 24, which was opened with inorganic thiophosphate to give MESP. An NMR analysis of the C1 Mosher’s ester of diol 24 indicated a 17:1 ratio of 2S,3R and 2R,3S enantiomers.20
Scheme 4. Synthesis of MESP.

Evaluation of EP, MTP, and EEP as Alternate Substrates for IspDF and IspE
EP, MTP, and EEP, along with MEP as a control, were incubated with different combinations of [α-32P]CTP, [γ-32P]ATP, IspDF, and IspE. The labeling patterns expected from these experiments are shown in Scheme 5. The reaction mixtures were analyzed by TLC,4,5 and the results are shown in Figure 2.
Scheme 5. Predicted Pattern for Incorporation of 32P into the Products from Incubations with IspDF and IspE.
Figure 2.

TLC plate showing the different products following 1 h incubations with MEP, EP, MTP, or EEP (500 μM) with IspDF (4.8 μM) and [α-32P]CTP (150 μM, 40 μCi/μmol) or IspDF, IspE (6.2 μM), [γ-32P]ATP (150 μM, 320 μCi/μmol), and [α-32P]CTP (150 μM, 40 μCi/μmol). The reactions were quenched with methanol. Lane 1: MEP, CTP (control); lane 2: MEP, ATP (control); lane 3: MEP, IspDF, CTP; lane 4: MEP, IspDF, IspE, ATP, CTP; lane 5: EP, IspDF, CTP; lane 6: EP, IspDF, IspE, ATP, CTP; lane 7: MTP, IspDF, CTP; lane 8: MTP, IspDF, IspE, ATP, CTP; lane 9: EEP, IspDF, CTP; lane 10: EEP, IspDF, IspE, ATP, CTP.
Incubation of IspDF with MEP and [α-32P]CTP gave CDP-ME as the only radioactive product, as expected for catalysis by the IspD active site in the IspDF fusion protein4 (see lane 3). A similar incubation with IspDF and IspE with MEP, [α-32P]CTP, and [γ-32P]ATP gave two 32P-labeled products, cMEDP (radiolabel from [γ-32P]ATP) and CMP (radiolabel from [α-32P]CTP) (see lane 4).4 A spot with a smaller Rf in the region expected for CDP-MEP was not seen.
Related incubations with EP indicated that IspDF gave CDP-E (lane 5) and IspDF/IspE gave cEDP and CMP (lane 6), although the reactions appeared to be slower and a spot was seen for CDP-E. The TLC profiles for EEP suggest that CDP-EE and CMP have similar Rf values. Incubation of IspDF with EEP and [α-32P]CTP (lane 9) gives a single spot as expected for formation of labeled CDP-EE. While a similar incubation with IspDF/IspE with EEP, [α-32P]CTP, and [γ-32P]ATP gives two spots, as expected, however the spot assigned to cEEDP has the same Rf value as CMP (lane 10). No spots were seen with expected Rf values for CDP-EP or CDP-EEP.
The formation of CDP-EE was established by a series of incubations with EEP and different combinations of labeled and unlabeled ATP and CTP (Figure 3). To provide a basis for comparison, we incubated MEP with IspDF, IspE, [α-32P]CTP, and [γ-32P]ATP, which resulted in well-resolved spots for CMP and cMEDP (lane 7), while incubation with IspDF and [α-32P]CTP gave CDP-ME with an Rf distinctive from those of CMP and cMEDP (lane 8). Similar experiments with EEP, visualized in lanes 3 and 1, respectively, show similar patterns, except that the putative spot for CDP-EE has the same Rf value as CMP. Overlapping Rf’s for CDP-EE and CMP were established by the experiments visualized in lanes 2, 4, and 5. In lane 2, EEP was incubated with IspDF, [α-32P]CTP, and [γ-32P]ATP to give [32P]CDP-EE, with the same Rf for [32P]CMP from incubation of EEP with IspDF, IspE, [α-32P]CTP, and ATP seen in lane 4. The spot for [32P]CMP is absent when EEP is incubated with IspDF, IspE, unlabeled CTP, and [γ-32P]ATP (lane 5), establishing that CMP and CDP-EE comigrate. Thus, our TLC studies indicate that the alternate substrates EP and EEP behave similar to MEP(5) for the three consecutive reactions catalyzed by IspDF and IspE. No clear evidence was seen for the mandatory CDP-EP and CDP-EEP, although spots indicative of the intermediates were seen at the leading edge for the spot for ATP in time course experiments (also see Figures S1 and S2, Supporting Information). Difficulty in detecting CDP-EP and CDP-EEP is not surprising. CDP-MEP migrates at a poorly defined spot at the leading edge of that for ATP and only constitutes a maximum of ∼10% of the total radioactivity during time course measurements.
Figure 3.

TLC plate showing the different products following 1 h incubations of different combinations of MEP and EEP (500 μM), IspDF (4.8 μM), IspE (6.2 μM), unlabeled ATP, [γ-32P]ATP (150 μM, 320 μCi/μmol), unlabeled CTP, and [α-32P]CTP (150 μM, 40 μCi/μmol). The reactions were quenched with methanol. Lane 1: EEP, [α-32P]CTP, IspDF; lane 2: EEP, [α-32P]CTP, [γ-32P]ATP, IspDF; lane 3: EEP, [α-32P]CTP, [γ-32P]ATP, IspDF, IspE; lane 4: EEP, [α-32P]CTP, ATP, IspDF, IspE; lane 5: EEP, CTP, [γ-32P]ATP, IspDF, IspE; lane 6: EEP, [γ-32P]ATP, IspDF; lane 7: MEP, [α-32P]CTP, [γ-32P]ATP, IspDF, IspE; lane 8: MEP, [α-32P]CTP, IspDF; lane 9: EEP, [γ-32P]ATP; lane 10: EEP, [α-32P]CTP.
Negative-ion LC–MS analyses of incubation mixtures similar to those described in Figures 2 and 3 were performed using single ion monitoring, as previously described for MEP.4,5 They support the assignments for the products from incubations with EP or EEP. The reaction mixture from incubation of EP with CTP and IspDF gave a peak at m/z 506 and expected for CDP-E (Figure 4A). A similar analysis of the incubation of EP with CTP, ATP, IspDF, and IspE gave peaks m/z 263 for cEDP (Figure 4B) and m/z 322 for CMP (data not shown). Related incubations with EEP gave peaks at m/z 534 for CDP-EE (Figure 4C), m/z 291 for cEEDP (part D), and m/z 322 for CMP (data not shown).
Figure 4.

LC–MS chromatograms of products detected by single-ion monitoring of products from the following incubations: (A) EP, CTP, IspDF; (B) EP, CTP, ATP, IspDF, IspE; (C) same as A with EEP as a substrate; (D) same as B with EEP as a substrate.
In contrast to EP and EEP, MTP was not a substrate for IspD (Figure 2). In addition, MTP at concentrations up to 5 mM did not inhibit turnover of MEP. Thus, it appears that the threitol diastereomer is not recognized by the active site of IspD.
Evaluation of NMEP and MESP as Alternate Substrates for IspDF and IspE
MEP and NMEP were incubated with IspDF, IspE, [α-32P]CTP, and [γ-32P]ATP under conditions similar to those shown in Figures 2 and 3. TLC analysis of the products from MEP(5) showed the expected pattern of spots (Figure S3, Supporting Information, lanes 1 and 2). TLC analysis of a similar experiment with NMEP showed the formation of new radioactive materials with smaller Rf’s than seen for the corresponding MEP derivatives that were not resolved into individual peaks (Figure S3, Supporting Information, lane 4). Related experiments with MEP where IspDF was replaced with H281S, a mutant which does not have IspF activity, gave spots for CDP-ME and CDP-MEP but not cMEDP, as expected (Figure S3, Supporting Information, lane 3). A related incubation of NMEP with [H281S]IspDF and IspE (Figure S3, Supporting Information, lane 6) and with [H281S]IspDF (Figure S3, Supporting Information, lane 5) again gave unresolved spots at lower Rf’s. The product mixtures were then analyzed by negative ion LC–MS. Selective ion monitoring at m/z 519 (CDP-NME), m/z 599 (CDP-NMEDP), and m/z 276 (cNMEDP) gave major peaks at 9.02 min (CDP-NME), 37.06 min (CDP-NMEDP), and 9.37 min (cNMEDP) (Figures S4 and S5, Supporting Information).
Similar experiments were performed for MESP. In this case, TLC analysis of the products from an incubation with IspDF, IspE, [α-32P]CTP, and [γ-32P]ATP gave spots for cMESDP and CMP (Figure 5, lane 1). The Rf of CDP-MES was established by incubating MESP with [32P]CTP, [32P]ATP, and [H281S]IspDF, which only catalyzes the conversion of MESP to CDP-MES (lane 2), while those for cMESDP and CMP were established by comparing an incubation with [α-32P]CMP and [γ-32P]ATP (lane 1) with an incubation with [α-32P]CMP and unlabeled ATP (lane 3). LC–MS analyses gave negative ion electrospray spectra for CDP-MES (m/z at 536), CDP-MESP (m/z at 616), and cMESDP (m/z 293).
Figure 5.

TLC plate showing the different products formed following a 1 h incubation of MESP (500 μM), IspDF (4.8 μM), or [H281S]IspDF, IspE (6.2 μM), unlabeled ATP or [γ-32P]ATP (150 μM, 320 μCi/μmol), and [α-32P]CTP (150 μM, 40 μCi/μmol). The reactions were quenched with methanol. Lane 1: MESP, [32P]CTP, [32P]ATP, IspDF, IspE; lane 2: MESP, [32P]CTP, [32P]ATP, [H281S]IspDF; lane 3: MESP, [32P]CTP, ATP, IspDF, IspE.
Conclusions
Five MEP analogues were synthesized in this study. Four of these and their products were substrates for the three consecutive reactions catalyzed by IspDF and IspE. EP, EEP, NMEP, and MESP have topologies that are modestly different from that of MEP. In EP and EEP, a hydrogen atom and an ethyl group, respectively, replace the methyl group at C3 in MEP, and both analogues are converted to the corresponding cyclic diphosphates by ispDF and ispE. From a topological perspective, cEDP and its subsequent metabolites are likely alternate substrates for IspG and IspH. However, the resulting nor-analogues of IPP and DMAPP would be unreactive competitive inhibitors of IPP isomerase and farnesyl diphosphate (FPP) synthase, the next two enzymes in the isoprenoid pathway.21,22 In contrast, EEP would give ethyl analogues of IPP and DMAPP, which in turn are substrates for IPP isomerase and farnesyl diphosphate (FPP) synthase.22 Related reactions are used to construct the carbon skeletons of insect juvenile hormones.23MTP, the threo diastereomer of MEP, is not a substrate for ispD and does not inhibit the enzyme. Conversion of cNMEDP to the amino analogue of HDMAPP by IspG would produce a potent nanomolar inhibitor of IspH.24cMESDP contains a highly conservative replacement of oxygen by sulfur that should not substantially impede its conversion to the thiolo analogue of DMAPP, at which point it becomes a low micromolar inhibitor of FPP synthase.25 Thus, the ability of IspDF and IspE to process analogues of MEP presents an opportunity for in vivo synthesis of inhibitors of downstream enzymes and new metabolites.
Experimental Section
Mass analyzers used for HRMS were TOF (pure samples) or Quad/TOF (HPLC/MS).
Synthesis of Alternate Substrates
1,3-Benzylidene-d-erythritol, 4-(t-Butyldimethylsilyl) Ether (2)
A solution of dioxanone 1 (2.17 g, 6.73 mmol)14 in MeOH (22 mL) was stirred and cooled at 0 °C as NaBH4 (305 mg, 8.08 mmol) was added portionwise over 10 min. The cooling bath was removed, and the reaction mixture was allowed to stir 1 h at rt. The reaction was neutralized by the addition of satd. NH4Cl (10 mL) and H2O (10 mL). The aqueous reaction mixture was extracted with CH2Cl2 (2 × 100 mL). The organic extracts were combined, dried (MgSO4), and evaporated to give 2.03 g of crude oil that was an 8:1 mixture (1H NMR) of monosilyl ether 2 and starting material. Column purification (EtOAc:hexane 21:79) gave two fractions. After solvent was removed, the first contained 0.58 g of colorless oil that was a mixture of alcohol 2 (∼40%) and dioxanone 1 (∼40%), based on 1H NMR analysis, and the second contained 1.18 g of 2 (60%, Corr. for Rec. SM) as a colorless oil: TLC Rf = 0.68 (50:50, EtOAc:hexane); [α]D = 32.5 (c = 1.0 in MeOH); 1H NMR (500 MHz, C6D6) δ −0.01 (s, 6H), 0.89 (s, 9H), 2.45 (d, 1H, J = 2.8 Hz, OH, Exch. D2O), 3.46 (t, 1H, J = 10.5 Hz), 3.53 (ddd, 1H, J = 9.0, 6.2, 5.1 Hz), 3.77–3.82 (m, 2H), 3.89 (dd, 1H, J = 10.5, 4.8 Hz), 4.22 (dd, 1H, J = 10.7, 5.4 Hz), 5.31 (s, 1H), 7.10 (t, 1H, J = 7.4 Hz), 7.18 (t, 2H, J = 7.6 Hz), 7.61 (d, 2H, J = 7.5 Hz); 13C NMR (126 MHz, C6D6) δ −5.5, 18.3, 25.9, 65.3, 65.5, 71.0, 80.6, 101.2, 126.7, 128.9, 138.7; IR 3456 (OH), 2930, 2857, 1463, 1254, 1089, 837. No physical data were reported for this known compound.26 However, the physical data were similar to those reported by Fukumoto27 for the enantiomer (1H NMR, CDCl3).
1,3-Benzylidene-d-erythritol (3)
A solution of silyl ether 2 (0.97 g, 2.99 mmol) in THF (5 mL) was stirred and cooled at 0 °C as 1.0 M Bu4NF (3.3 mL, 3.29 mmol) was added dropwise over 2 min. The cooling bath was removed, and the reaction mixture was allowed to stir at room temperature for 15 min. The reaction solution was diluted with H2O (5 mL) and Et2O (70 mL). Following extraction of the aqueous layer with Et2O (70 mL), the ethereal layers were combined, dried (MgSO4), and evaporated to give 1.67 g of an oil. Column purification (EtOAc:hexane 70:30) gave 0.50 g (79%) of a white solid: TLC Rf = 0.47 (EtOAc); 1H NMR (500 MHz, C5H5N) δ 3.95 (t, 1H, J = 10.4 Hz), 4.15 (ddd, 1H, J = 9.4, 5.4, 1.7 Hz), 4.33–4.39 (m, 2H), 4.49 (d, 1H, J = 11.8 Hz), 4.59 (dd, 1H, J = 10.7, 5.4 Hz), 5.82 (s, 1H), 6.69 (br s, 1H, OH, Exch. D2O), 7.22 (d, 1H, J = 5.8 Hz), 7.36 (m, 2H), 7.74 (m, 2H); 13C NMR (126 MHz, C5H5N) δ 61.9, 62.3, 72.3, 84.9, 101.5, 127.2, 128.4, 129.0, 139.5. The NMR data agree with those reported by Pinto in the literature (1H NMR CD3OD).15
1,3-O-Benzylidene-d-erythritol 4-Dibenzylphosphate (4)
The phosphorylation was carried out as described by MacDonald et al.28 A solution of dibenzyl chlorophosphate was prepared by stirring a mixture of N-chlorosuccinimide (76 mg, 0.57 mmol) and dibenzyl phosphite (150 mg, 0.57 mmol) in benzene (2 mL) at room temperature for 1 h. The precipitate was separated by centrifugation and the supernatant added to a solution of 1,3-O-benzylidene-d-erythritol 3 (100 mg, 0.48 mmol) in dry pyridine (4 mL) at 0 °C. The resultant solution was stirred for 24 h at 0 °C and 24 h at room temperature. Another portion of freshly prepared dibenzyl chlorophosphate (0.57 mmol) in benzene (1 mL) was added, and stirring was continued for 48 h. The reaction was quenched with ice (∼1 g), and volatiles were removed under reduced pressure. The residue was diluted with water (30 mL) and extracted with DCM (3 × 30 mL). The combined organic layers were dried over Na2SO4, and the solvent was evaporated. Purification of the residue by chromatography (silica gel benzene/ethyl acetate 1:1) yielded 26 mg of a 1:1 mixture of 4 (18 mg, ca. 8%) and starting material (8 mg) and 27.4 mg (12%) of 4 as a colorless oil: TLC Rf 0.24 (hexane/ethyl acetate 4:6), 0.26 (benzene/ethyl acetate 1:1); 1H NMR (500 MHz, C6D6) δ 3.6 (t, 2H, J = 10.5 Hz), 4.01 (br, 1H), 4.30 (t, 1H, J = 10.5 Hz), 4.35 (dd, 1H, J = 5.4, 10.5 Hz), 4.47 (m, 1H), 4.74 (br s, 1H), 4.81–4.93 (m, 4H), 5.31 (s, 1H), 6.99–7.18 (m, 13H), 7.59 (m, 2H); 13C NMR (126 MHz, C6D6) δ 29.2, 61.4, 67.1, 69.8, 71.3, 81.6, 101.4, 126.8, 136.1, 138.6; 31P NMR (202 MHz, C6D6) δ 1.35.
d-Erythritol 4-Phosphate, Ammonium Salt (EP)
Deprotection of the dibenzyl phosphate (4, 24 mg, 0.05 mmol) was accomplished by hydrogenation using 20% Pd(OH)2/C (12 mg) in MeOH (3 mL) with magnetic stirring at 1 atm of H2 for 24 h. The catalyst was filtered off, and the solvent was evaporated. The residue was dissolved in water (3 mL) followed by dropwise addition of 10% NH4OH (1 mL). Lyophilization gave 12 mg (100%) of a flocculent amorphous solid: TLC Rf 0.2 (acetonitrile/isopropyl alcohol/10% aq. NH4OH 1:1:1); 1H NMR (500 MHz, D2O) δ 3.62 (dd, 1H, J = 6.9, 11.4 Hz), 3.67 (br, 1H), 3.74 (br t, 1H, J = 6.9 Hz), 3.81 (d, 1H, J = 6.2), 3.90 (br, 2H); 13C NMR (126 MHz, D2O) δ 32.3, 65.5, 67.5, 74.1; 31P NMR (202 MHz, D2O) δ 5.36. The data agree with those reported by Lillo et al.10
1,3-O-Benzylidene-2-C-ethyl-d-erythritol 4-(t-butyldimethylsilyl) Ether (5)
A solution of 1,3-O-benzylidene-d-erythrulose and 4-(t-butyldimethylsilyl) ether (1, 900 mg, 2.79 mmol)14 in dry ether (20 mL) was stirred and cooled at −78 °C as EtMgBr (1.4 mL, 4.2 mmol, 3 M in Et2O) was added dropwise. After 45 min, the reaction was quenched with MeOH (5 mL) and allowed to warm to room temperature. Satd. aq. NH4Cl (15 mL) and water (15 mL) were added, and the product was extracted with ether (3 × 30 mL). The combined organic layers were washed with satd. NaHCO3, dried (MgSO4), and evaporated to give 1.1 g of crude material. Purification by flash chromatography (silica gel, hexane/ethyl acetate 95:5) afforded 810 mg (82%) of a viscous colorless oil: TLC Rf 0.42 (hexane/ethyl acetate 8:2); 1H NMR (500 MHz, C6D6) δ −0.044 (s, 3H), −0.036 (s, 3H), 0.86 (s, 9H), 1.10 (t, 3H, J = 7.5 Hz), 1.60 (app sextet, 1H, J = 7.5 Hz), 2.24 (app sextet, 1H, J = 7.5 Hz), 2.86 (s, 1H), 3.46 (d, 1H, J = 11 Hz), 3.78 (dd, 1H, J = 7.8, 4.0 Hz), 3.87 (m, 2H), 4.26 (d, 1H, J = 11 Hz), 5.40 (s, 1H), 7.11–7.20 (m, 3H), 7.60 (m, 2H); 13C NMR (126 MHz, C6D6) δ −5.8, −5.7, 18.1, 24.0, 25.8, 63.0, 68.1, 72.2, 82.2, 102.0, 126.7, 128.5, 128.9, 138.8.
1,3-O-Benzylidene-2-C-ethyl-d-erythritol (6)
A solution of silyl ether 5 (780 mg, 2.21 mmol) in anhydrous THF (8 mL) was stirred at 0 °C as Bu4NF (2.7 mL, 1 M in THF) was added. After 15 min at room temperature, H2O (5 mL) was added; the product was extracted with ether (80 mL); and the ethereal extracts was dried (MgSO4) and evaporated. Purification by flash chromatography (silica gel, hexane/ethyl acetate 4:6) afforded 486 mg (92%) of a viscous oil: TLC Rf 0.24 (hexane/ethyl acetate 4:6); 1H NMR (500 MHz, C6D6) δ 0.92 (t, 3H, J = 7.5 Hz), 1.35 (app sextet, 1H, J = 7.5 Hz), 2.09 (app sextet, 1H, J = 7.5 Hz), 3.25 (d, 1H, J = 11 Hz), 3.58 (dd, 1 H, J = 6.5, 10.4 Hz), 3.67 (t, 1H, J = 6.5 Hz), 3.73 (dd, 1H, J = 5.9, 10.4 Hz), 4.08 (d, 1H, J = 11 Hz), 5.34 (s, 1H), 7.10–7.19 (m, 13H), 7.54 (m, 2H); 13C NMR (126 MHz, C6D6) δ 23.8, 61.4, 67.7, 72.3, 83.9, 102.1, 126.8, 138.7.
1,3-O-Benzylidene-2-C-ethyl-d-erythritol 4-Dibenzylphosphate (7)
A solution of diol 6 (245 mg, 1.03 mmol) in THF (7 mL) was stirred and cooled at −78 °C as an aliquot of 1.6 M BuLi in hexane (0.96 mL, 1.54 mmol) was added dropwise. After 5 min, a solution of dibenzyl chlorophosphate (1.54 mmol; freshly prepared from dibenzyl phosphite) in benzene (5 mL) prepared as described for 4 was added. Stirring was continued for 10 min at −78 °C and 15 min at room temp. Ether (60 mL) and water (2 mL) were added, and the ethereal layer was dried (MgSO4) and evaporated to give 520 mg of crude product. Purification by flash chromatography (silica gel, hexane/ethyl acetate 4:6) afforded 450 mg (88%) of an oil (14): TLC Rf 0.3 (hexane/ethyl acetate 4:6); 1H NMR (500 MHz, C6D6) δ 0.99 (t, 3H, J = 7.5 Hz), 1.37 (app sextet, 1H, J = 7.5 Hz), 2.14 (app sextet, 1H, J = 7.5 Hz), 3.55 (d, 1H, J = 11 Hz), 4.16 (s, 1 H), 4.26 (d, 1H, J = 11 Hz), 4.25–4.33 (m, 1H), 4.89 (d, 1H, J = 8.2 Hz), 4.86–4.90 (m, 4H), 5.48 (s, 1H), 6.99–7.18 (m, 13H), 7.57 (m, 2H); 13C NMR (126 MHz, C6D6) δ 23.6, 67.0, 69.6, 69.6, 72.5, 84.9, 102.0, 126.9, 128.5, 128.7, 128.9, 136.20, 138.7; 31P NMR (202 MHz, C6D6) δ 0.21.
2-C-Ethyl-d-erythritol 4-Phosphate, Ammonium Salt (EEP)
Compound 7 (410 mg, 0.82 mmol) was hydrogenated (1 atm, −10 °C) using 10% Pd/C (82 mg) in MeOH (10 mL) for 30 min. The mixture was filtered and solvent evaporated to give 270 mg of an oil. The oil was treated with 20% Pd(OH)2/C (130 mg) in MeOH (7 mL) at 1 atm of H2 for 24 h. The catalyst was removed by filtration; solvent was evaporated; and the residue was purified by chromatography (silica gel, acetonitrile/isopropyl alcohol/10% aq. NH4OH 1:1:1). Finally, lyophilization of the eluate afforded 205 mg (94%) of an amorphous solid: TLC Rf 0.24 (acetonitrile/isopropyl alcohol/10% aq. NH4OH 1:1:1); 1H NMR (500 MHz, D2O) δ 0.88 (t, 3H, J = 7.6 Hz), 1.61 (AB part of ABX, 2H), 3.59 and 3.60 (ABq, 2H, J = 11.9 Hz), 3.81–3.85 (m, 2H), 3.93–3.98 (m, 1H); 13C NMR (126 MHz, D2O) δ 27.8, 65.9, 67.4, 76.1, 78.8; 31P NMR (202 MHz, D2O) δ 4.94.
(Z)-tert-Butyl-dimethyl-[2-methyl-4-(tetrahydro-pyran-2-yloxy)-but-2-enyloxy]silane (9)
3,4-Dihydro-2H-pyran (126 mg, 1.5 mmol) and PPTS (25 mg, 0.1 mmol) were added to a solution of alcohol 8(16) (217 mg, 1 mmol) in CH2Cl2 at rt. After stirring overnight, the solution was washed with saturated NaHCO3 (10 mL) and extracted with CH2Cl2 (3 × 10 mL). The combined organic extracts were dried over Na2SO4. The solvent was removed at reduced pressure, and the residue was chromatographed on silica gel with gradient elution by hexanes/ethyl acetate (0% to 10% ethyl acetate) to give 295 mg (98%) of a colorless oil: Rf 0.87 (hexanes:ethyl acetate 1:1); 1H NMR (CDCl3, 300 MHz) 0.07 (6H, s), 0.90 (9H, s), 1.49–1.85 (9H, m), 3.47–3.54 (1H, m), 3.83–3.91 (1H, m), 3.99–4.06 (1H, s), 4.18 (2H, s), 4.21–4.28 (1H, m), 4.61 (1H, t, J = 2.9 Hz), 5.42 (1H, m); 13C NMR (CDCl3, 75 MHz) −5.3, 18.3, 19.5, 21.0, 25.5, 25.9, 30.6, 61.8, 62.2, 62.9, 97.9, 122.4, 139.7; HRMS (MALDI) calcd for C16H32O3SiNa [M + Na+] 323.2013, found 323.2001.
(Z)-2-Methyl-4-(tetrahydro-pyran-2-yloxy)-but-2-en-1-ol (10)
A solution of 1 M TBAF in THF (4.36 mL, 4.36 mmol) was added dropwise to a solution of 9 (437 mg, 1.45 mmol) in THF (15 mL) at 0 °C. The temperature was allowed to rise to rt. After stirring for 3 h, the reaction was quenched with brine (15 mL). The layers were separated, and the aqueous layer was extracted with ether (3 × 10 mL). The combined organic extracts were dried over Na2SO4. The solvent was removed at reduced pressure, and the residue was chromatographed on silica gel with gradient elution by hexanes/ethyl acetate (0% to 30% ethyl acetate) to give 315 mg (94%) of a colorless oil: Rf 0.25 (hexanes:ethyl acetate 1:1); 1H NMR (CDCl3, 300 MHz) 1.52–1.86 (9H, m), 2.59 (1H, broad s), 3.50–3.57 (1H, m), 3.82–3.90 (1H, m), 4.00–4.27 (4H, s), 4.70 (1H, t, J = 2.9 Hz), 5.51 (1H, t, J = 7.2 Hz); 13C NMR (CDCl3, 75 MHz) 19.1, 21.9, 25.3, 30.4, 61.4, 62.0, 62.4, 97.0, 123.3, 141.0; HRMS (MALDI) calcd for C10H18O3Na [M + Na+] 209.1148, found 209.1158.
(Z)-2-[2-Methyl-4-(tetrahydropyran-2-yloxy)but-2-enyl]isoindole-1,3-dione (11)
A solution of DIAD (223 mg, 1.1 mmol) in THF (1 mL) was added dropwise to a solution of an alcohol 10 (187 mg, 1 mmol), phthalimide (162 mg, 1.1 mmol), and PPh3 (289 mg, 1.1 mmol), in THF (4 mL) at rt. After stirring overnight, the solvent was removed at reduced pressure. The residue was chromatographed on silica with gradient elution by hexanes/ethyl acetate (0% to 25% ethyl acetate) to give 241 mg (76%) of a white solid: Rf 0.63 (hexanes:ethyl acetate 1:1); mp = 77–78 °C; 1H NMR (CD2Cl2, 300 MHz) 1.48–1.83 (9H, m), 3.47–3.54 (1H, m), 3.83–3.91 (1H, m), 4.21–4.28 (1H, m), 4.32 (2H, s), 4.38–4.45 (1H, m), 4.65–4.67 (1H, m), 5.53–5.59 (1H, m), 7.70–7.76 (2H, m), 7.80–7.84 (2H, m); 13C NMR (CD2Cl2, 75 MHz) 20.1, 21.9, 26.1, 31.3, 38.8, 62.7, 63.5, 98.6, 123.6, 126.7, 132.6, 134.3, 134.5, 168.7; HRMS (MALDI) calcd. for C18H21NO4Na [M + Na+] 338.1363, found 338.1357.
(Z)-2-Methyl-4-(tetrahydropyran-2-yloxy) But-2-enylamine (12)
To a solution of phthalimide 11 (318 mg, 1 mmol) in EtOH (9 mL) was added hydrazine hydrate (150 mg, 3 mmol) in EtOH (1 mL). The solution was warmed to 50 °C, stirred for 30 min, and heated at reflux for 2 h. The solution was cooled and filtered. The filter cake was washed with EtOH (3 × 10 mL), and solvent was removed at reduced pressure. The residue was dissolved in ether (20 mL), and 1 M NaOH (5 mL) was added to adjust the pH to 12. The aqueous layer was saturated with NaCl and extracted with ether. The combined organic extracts were dried over Na2SO4. The solvent was removed at reduced pressure to give 144 mg (77%) of a yellow liquid, which was used in the next step without further purification: 1H NMR (CD2Cl2, 300 MHz) 1.1.40–1.81 (11H, m), 3.23 (2H, s), 3.44–3.50 (1H, m), 3.78–3.86 (1H, m), 3.95–4.02 (1H, m), 4.15–4.21 (1H, m), 4.57–4.60 (1H, m), 5.34–5.41 (1H, m); 13C NMR (CD2Cl2, 75 MHz) 20.1, 22.1, 26.1, 31.2, 42.9, 62.6, 63.1, 98.2, 122.6, 142.4.
(Z)-[2-Methyl-4-(tetrahydropyran-2-yloxy) but-2-enyl] Carbamic Acid tert-Butyl Ester (13)
Amine 12 (600 mg, 3.24 mmol) was dissolved in iPrOH/H2O (3:1, v/v, 140 mL), and solid Na2CO3 (3.24 g) was added. The solution was cooled to 0 °C. A solution of di-tert-butyl dicarbonate (1.46 g, 6.48 mmol) in iPrOH/H2O (3:1, v/v, 20 mL) was added. The mixture was stirred overnight at rt. iPrOH was removed at reduced pressure, and the aqueous layer was extracted with ether. The combined organic extracts were dried over Na2SO4. The solvent was removed at reduced pressure, and the residue was chromatographed on silica with gradient elution by hexanes/ethyl acetate (0% to 15% ethyl acetate) to give 640 mg (69%) of a colorless oil: Rf 0.56 (hexanes:ethyl acetate 1:1); 1H NMR (CDCl3, 300 MHz) 1.26–1.89 (18H, m2), 3.49–3.56 (1H, m), 3.78 (2H, d, J = 5.9 Hz), 3.83–3.90 (1H, m), 4.00–4.07 (1H, m), 4.21–4.27 (1H, m), 4.63–4.65 (1H, m), 4.75 (1H, broad s), 5.51 (1H, t, J = 7 Hz); 13C NMR (CDCl3, 75 MHz) 19.4, 22.0, 25.34, 28.3, 30.5, 40.8, 62.2, 62.7, 79.1, 97.7, 124.1, 138.0, 156.0; HRMS (MALDI) calcd for C15H27NO4Na [M + Na+] 308.1828, found 308.1828.
(Z)-(4-Hydroxy-2-methylbut-2-enyl)carbamic Acid tert-Butyl Ester (14)
PPTS (25 mg, 0.1 mmol) was added to the solution of 13 (286 mg, 1 mmol) in EtOH (10 mL). The solution was stirred at 55 °C for 1 h and allowed to cool to rt. The solvent was removed at reduced pressure, and the residue was chromatographed on silica with gradient elution by hexanes/ethyl acetate (0% to 40% ethyl acetate) to give 198 mg (98%) of a colorless oil: Rf 0.56 (hexanes:ethyl acetate 1:1); 1H NMR (CDCl3, 300 MHz) 1.43 (9H, s), 1.76 (3H, t, J = 0.7 Hz), 3.19 (1H, broad s), 3.74 (2H, dd, J1 = 5.6 Hz, J2 = 0.7 Hz), 4.14 (2H, t, J = 6.5 Hz), 4.91 (1H, broad s), 5.67 (1H, t, J = 7.3 Hz); 13C NMR (CDCl3, 75 MHz) 21.6, 28.3, 40.7, 57.5, 79.9, 127.2, 136.2, 156.2; HRMS (MALDI) calcd for C10H19NO3Na [M + Na+] 224.1263, found 224.1268.
(Z)-[4-(Bisbenzyloxyphosphoryloxy)-2-methylbut-2-enyl]carbamic Acid tert-Butyl Ester (15)
A solution of I2 (609 mg, 2.4 mmol) in CH2Cl2 (30 mL) was added to a solution of P(OBn)3 in CH2Cl2 (20 mL) at −40 °C. The mixture was stirred for 15 min, and the cooling bath was removed. After 30 min the solution became colorless and was cannulated to a mixture of alcohol 14 (200 mg, 1.2 mmol) and pyridine (570 mg, 7.2 mmol) in CH2Cl2 (30 mL) at −40 °C over 30 min. After the addition was complete, the mixture was stirred for 1 h at rt, and the solvents were removed at reduced pressure. The residue was dissolved in ether (50 mL) and washed with 0.3 M KHSO4 (30 mL), satd. NaHCO3 (30 mL), and brine (30 mL). The combined organic extracts were dried over Na2SO4, and solvent was removed at reduced pressure. The residue was chromatographed on silica gel with gradient elution by hexanes/ethyl acetate (0% to 30% ethyl acetate) to give 320 mg (70%) of a colorless oil: Rf 0.48 (hexanes:ethyl acetate 1:1); 1H NMR (CDCl3, 300 MHz) 1.44 (9H, s), 1.74 (3H, t, J = 0.6 Hz), 3.71 (2H, d, J = 6.2 Hz), 4.54 (2H, dd, J1 = 7.7 Hz, J2 = 2.3 Hz), 4.94 (1H, broad s), 5.02 (2H, dd, J1 = 6.5 Hz, J2 = 1.7 Hz), 5.43 (1H, t, J = 7 Hz), 7.35 (10H, s); 13C NMR (CDCl3, 75 MHz) 21.8, 28.4, 40.4, 63.4, 63.4, 69.1, 69.2, 121.8, 121.9, 127.9, 128.0, 128.5, 128.6, 128.7, 135.8, 135.9, 140.5, 156.1; 31P NMR (CDCl3, 125 MHz) 0.68; HRMS (MALDI) calcd for C24H32NO6PNa [M + Na+] 484.1865, found 484.1872.
1-Deoxy-1-boc-amino-4-(dibenzyloxyphosphoryloxy)-3-methyl-d-erythritol (16)
A solution of AD mix β (3.14 g), NaHCO3 (565 mg, 6.73 mmol), and CH3SO2NH2 (160 mg, 1.68 mmol) in tBuOH:H2O (1:1 v/v, 12 mL) was cooled to 0 °C, and alkene 15 (250 mg, 0.56 mmol) was added. The mixture was stirred overnight at 0 °C before Na2SO3 (3.36 g) was added. The temperature was allowed to rise to rt as the solution was stirred for 1 h. The layers were separated, and the aqueous layer was extracted with CH2Cl2 (3 × 20 mL). The combined organic extracts were dried over Na2SO4, and solvent was removed at reduced pressure. The residue was chromatographed on silica with gradient elution by hexanes/ethyl acetate (0% to 30% ethyl acetate) to give 172 mg (64%) of a colorless oil: Rf 0.25 (hexanes:ethyl acetate 1:1); [α]D20 + 11.7 (c 0.75, CHCl3); 1H NMR (CDCl3, 300 MHz) 0.04 (3H, s), 1.43 (9H, s), 2.96–3.05 (2H, m), 3.33–3.40 (1H, m), 3.68–3.72 (1H, m), 3.98–4.07 (1H, m), 4.33 (1H, t, J = 10.5 Hz), 4.69 (2H, s), 4.98–5.09 (4H, m), 5.36 (1H, broad s), 7.34 (10H, s); 13C NMR (CDCl3, 75 MHz) 19.7, 28.3, 48.4, 69.3, 69.4, 69.6, 69.7, 73.3, 73.8, 80.4, 128.1, 128.1, 128.6 135.2, 135.6, 158.3; 31P NMR (CDCl3, 125 MHz) 0.72; HRMS (MALDI) calcd for C24H34NO8PNa [M + Na+] 518.1909, found 518.1909.
1-Amino-3-methyl-d-erythritol-4-phosphate, Ammonium Salt (NMEP)
A suspension of 16 (100 mg, 0.2 mmol) and Pd/C (8 mg) in CH3OH (5 mL) was flushed with H2 and was allowed to stir for 1 h under a balloon of H2. The mixture was concentrated at reduced pressure. An 1H NMR spectrum showed that no benzyl groups remained. The residue was dissolved in CH3OH (4 mL), and 3 M HCl (1 mL) was added. After 30 min, the mixture was concentrated at reduced pressure, and the residue was chromatographed on silica eluted with a iPrOH:H2O:NH4OH (6:0.5:2.5 v/v/v) mixture to give 41 mg (89%) of a white solid: Rf 0.38 (iPrOH:H2O:NH4OH (6:1:3 v/v/v)); [α]D20 + 15.4 (c 0.65, D2O); 1H NMR (D2O, 300 MHz) 1.26 (3H, s), 3.07 (1H, d, J = 13.2 Hz), 3.25 (1H, d, J = 13.2 Hz), 3.75–3.77 (1H, m), 3.81–3.91 (1H, m), 3.97–4.04 (1H, m); 13C NMR (D2O, 75 MHz) 20.4, 46.2, 64.5, 71.4, 75.8 (J = 7 Hz); 31P NMR (D2O, 125 MHz) 4.20; HRMS (MALDI) calcd for C5H13NO6P [M – H+] 214.0486, found 214.0489.
(2S,3S)-4-(tert-Butyldimethylsilanyloxy)-2,3-dihydroxy-3-methylbutyric Acid Methyl Ester (18)
Alkene 17(16) (300 mg, 1.16 mmol) was asymmetrically dihydroxylated following the procedure described for 16. The residue was chromatographed on silica with gradient elution by hexanes/ethyl acetate (0% to 15% ethyl acetate) to give 250 mg (73%) of a colorless oil: Rf 0.14 (hexanes:ethyl acetate 4:1); [α]D20 +26.2 (c 1.8, CHCl3); 1H NMR (CDCl3, 300 MHz) 0.09 (6H, s), 0.91 (9H, s), 1.17 (3H, d, J = 0.6 Hz), 3.10 (1H, s), 3.34–3.37 (1H, m), 3.48 (1H, d, J = 10 Hz), 3.68 (1H, d, J = 10 Hz), 3.81 (3H, s), 4.11 (1H, d, J = 8 Hz); 13C NMR (CDCl3, 75 MHz) −5.6, −5.6, 18.3, 19.64, 25.8, 52.5, 68.0, 73.3, 75.0, 173.7; HRMS (MALDI) calcd for C12H26O5SiNa [M + Na+] 301.1435, found 301.1435.
(4S,5S)-5-(tert-Butyldimethylsilanyloxymethyl)-2-methoxy-1,3-dioxolane-4-carboxylic Acid Methyl Ester (19)
To a solution of trimethyl orthoformate (291 mg, 2.74 mmol) and diol 18 (200 mg, 0.69 mmol) in CH2Cl2 (9 mL) was added CSA (16 mg, 0.07 mmol) in CH2Cl2 (1 mL). After stirring overnight, the solution was washed with saturated NaHCO3; the layers were separated; and the aqueous layer was extracted with CH2Cl2 (3 × 20 mL). The combined organic extracts were dried over Na2SO4. The solvent was removed at reduced pressure to give 230 mg (100%) of a colorless oil. This material partially decomposed when chromatographed on silica and was used in the next reaction without purification. A small sample was isolated by chromatography for characterization: Rf 0.63 (hexanes:ethyl acetate 3:2); dr 87:13 (by 1H NMR analysis of the orthoester); for major diastereomer 1H NMR (CDCl3, 300 MHz) 0.02 (6H, s), 0.86 (9H, s), 1.49 (3H, s), 3.34 (3H, s), 3.56 (2H, dd, J1 = 18 Hz, J2 = 10.6 Hz), 4.40 (1H, s), 5.88 (1H, s); 13C NMR (CDCl3, 75 MHz) −5.7, −5.6, 18.3, 22.4, 25.7, 51.6, 52.1, 65.6, 79.0, 84.7, 116.0, 168.5; HRMS (MALDI) calcd for C14H28O6SiNa [M + Na+] 343.1547, found 343.1558.
(4S,5S)-5-[tert-Butyldimethylsilanyloxymethyl)-2-methoxy-5-methyl[1,3]dio-xolan-4-yl]methanol (20)
A solution of 2 M LiBH4 in THF (0.6 mL, 1.2 mmol) was added dropwise by a syringe to a solution of 19 (200 mg, 0.6 mmol) in ether (20 mL) at rt. The resulting solution was stirred for 1 h before EtOH (5 mL) and brine (5 mL) were added. The organic layer was separated, and the aqueous layer was extracted with ether (3 × 10 mL). The combined organic extracts were dried over Na2SO4. The solvent was removed at reduced pressure to give 175 mg (quant) of a colorless oil. This material partially decomposed when chromatographed on silica and was used in the next reaction without purification. A small sample was isolated by chromatography for characterization: Rf 0.63 (hexanes:ethyl acetate 3:2); dr 87:13 (by 1H NMR analysis of the orthoester); for major diastereomer 1H NMR (CDCl3, 300 MHz) 0.11 (6H, s), 0.91 (9H, s), 1.45 (3H, s), 3.32 (3H, s), 3.71–4.09 (5H, m), 5.67 (1H, s); 13C NMR (CDCl3, 75 MHz) −5.7, −5.7, 18.1, 22.8, 25.7, 51.7, 60.3, 65.0, 81.3, 82.6, 114.8; HRMS (FTMS) calcd for C12H25O4Si [M – OCH3] 261.1517, found 261.15164.
(4S,5S)-5-[tert-Butyldimethylsilanyloxymethyl)-2-methoxy-5-methyl[1,3]dio-xolan-4-yl]methyl p-Toluenesulfonate (21)
Alcohol 20 (60 mg, 0.21 mmol) and DMAP (75 mg, 0.62 mmol) were dissolved in CH2Cl2 (5 mL), and TsCl (78 mg, 0.41 mmol) in CH2Cl2 (2 mL) was added by syringe. The mixture was allowed to stir overnight. The solvents were removed at reduced pressure, and the residue was chromatographed on silica gel with gradient elution by hexanes/ethyl acetate (0% to 10% ethyl acetate) to give 90 mg (98%) of a colorless oil: Rf 0.59 (hexanes:ethyl acetate 3:2); dr 87/13 (by 1H NMR analysis of the orthoester); [α]D20 +10.2 (c 0.9, CHCl3); for the major diastereomer 1H NMR (CDCl3, 300 MHz) 0.82 (9H, s), 1.34 (3H, s), 2.45 (3H, s), 3.29 (3H, s), 3.32 (1H, d, J = 10.5 Hz), 3.50 (1H, d, J = 10.5 Hz), 4.10–4.18 (2H, m), 4.33–4.41 (1H, m), 5.68 (1H, s), 7.13 (2H, d, J = 8 Hz), 7.80 (2H, d, J = 8 Hz); 13C NMR (CDCl3, 75 MHz) −5.9, −5.7, 17.9, 21.6, 25.6, 51.5, 65.1, 68.4, 79.7, 82.5, 115.4, 128.0, 129.9, 132.7, 144.9; HRMS (FTMS) calcd for C19H31O6SSi [M – OCH3] 415.1605, found 415.16047.
(2R,3S)-4-(tert-Butyldimethylsilanyloxy)-2,3-dihydroxy-3-methylbutyl Tosylate (22)
A solution of 21 (230 mg, 0.54 mmol) in THF (6 mL, 3 M HCl:THF, 1:25) was allowed to stir for 30 min. The mixture was cooled to 0 °C; 6 mL of ammonia in MeOH were added; and the solution was allowed to stir for 15 min at rt. The solvent was removed at reduced pressure, and the residue was chromatographed on silica with gradient elution by hexanes/ethyl acetate (0% to 30% ethyl acetate) to give 160 mg (76%) of a colorless oil; Rf 0.36 (hexanes:ethyl acetate 1:1); [α]D20 +18.9 (c 1.7, CHCl3); 1H NMR (CDCl3, 300 MHz) 0.06 (6H, s), 0.88 (9H, s), 1.07 (3H, s), 2.45 (3H, d, J = 0.3 Hz), 2.71–2.74 (2H, broad), 3.38 (1H, d, J = 10 Hz), 3.67 (1H, d, J = 10 Hz), 3.82 (1H, dd, J1 = 6 Hz, J2 = 2.5 Hz), 4.09 (1H, dd, J1 = 8 Hz, J2 = 2.0 Hz), 4.32 (1H, dd, J1 = 8.0 Hz, J2 = 2.5 Hz), 7.36 (2H, d, J = 8 Hz), 7.81 (2H, d, J = 8 Hz); 13C NMR (CDCl3, 75 MHz) −5.6, 18.1, 19.0, 21.6, 25.8, 67.6, 72.0, 72.7, 72.9, 128.0, 129.9, 132.6, 145.0; HRMS (FTMS) calcd. for C18H33O6SSi [M+H+] 405.17616, found 405.17603.
(2S)-1-(tert-Butyl-dimethylsilanyloxy)-2-[(S)-1-oxiranyl]propan-2-ol (23)
To a solution of tosylate 22 (367 mg, 0.91 mmol) in THF (6 mL) at 0 °C was added tBuOK (112 mg, 1.0 mmol) in THF (4 mL). The solution was allowed to stir for 1 h at 0 °C; the solvent was removed at reduced pressure; and the residue was chromatographed on silica with gradient elution by hexanes/ethyl acetate to give 196 mg (93%) of a colorless oil: Rf 0.36 (hexanes:ethyl acetate 1:1); [α]D20 +15.0 (c 2.6, CHCl3); 1H NMR (CDCl3, 300 MHz) 0.08 (6H, s), 0.91 (9H, s), 1.18 (3H, s), 2.36 (1H, s), 2.74 (1H, dd, J1 = 4.0 Hz, J2 = 1.0 Hz), 2.83 (1H, dd, J1 = 2.8 Hz, J2 = 2.2 Hz), 3.01 (1H, dd, J1 = 2.8 Hz, J2 = 1.0 Hz), 3.51 (1H, d, J = 9.8 Hz), 3.61 (1H, d, J = 9.8 Hz); 13C NMR (CDCl3, 75 MHz) −5.6, 18.2, 20.9, 25.8, 44.0, 55.7, 68.4, 70.2; HRMS (FTMS) calcd for C11H25O3Si [M + H+] 233.15675, found 233.15712.
(2S)-2-[(S)-1-Oxiranyl]propane-1,2-diol (24)
The complex Et3N–3HF (1.1 g, 6.9 mmol) in THF (2 mL) was added dropwise to a solution of silyl ether 23 (160 mg, 0.69 mmol) in THF (8 mL) at rt. After stirring overnight, Et3N (5 mL) was added; solvent was removed at reduced pressure, and the residue was chromatographed on silica with gradient elution by hexanes/ethyl acetate (0% to 30% ethyl acetate) to give 68 mg (83%) of a colorless oil: Rf 0.29 (hexanes:ethyl acetate 1:1); er 17:1 (as determined by 19F NMR analysis of the Mosher’s ester); [α]D20 +17.4 (c 0.65, CHCl3); 1H NMR (CDCl3, 300 MHz) 1.26 (3H, s), 2.36 (1H, broad), 2.50 (1H, s), 2.81–2.88 (2H, m), 3.04 (1H, dd, J1 = 2.9 Hz, J2 = 1.1 Hz), 3.49–3.62 (2H, m); 13C NMR (CDCl3, 75 MHz) 22.3, 44.5, 56.6, 67.2, 70.3; HRMS (FTMS) calcd for C5H9O2 [M – OH] 101.05971, found 101.06000.
3-Methyl-d-erythritol-4-thiolophosphate, Ammonium Salt (MESP)
To epoxide 50 (34 mg, 0.29 mmol) was added trisodium thiophosphate (126 mg, 0.32 mmol) in H2O (0.6 mL) at rt. The solution was allowed to stir overnight and then lyophilized. The residue was chromatographed on cellulose to give 25 mg of a white powder (32%): Rf 0.42 (iPrOH:H2O:NH4OH 6/1/3 v/v/v); [α]D20 +16.3 (c 0.53, D2O); 1H NMR (D2O, 300 MHz) 1.09 (3H, s), 2.67–2.78 (1H, m), 2.96–3.05 (1H, m), 3.51 (2H, dd, J1 = 17.2 Hz, J2 = 11.8 Hz), 3.72 (1H, dd, J1 = 8.4 Hz, J2 = 2.2 Hz); 13C NMR (D2O, 75 MHz) 17.7, 31.5 (J = 3 Hz), 66.7, 75.0, 75.5 (J = 3 Hz); 31P NMR (D2O, 125 MHz) 17.82; HRMS (FTMS) calcd for C5H14O6PS [M + H+] 233.02432, found 233.02482.
Product Studies
TLC Analysis
Enzymatic reactions were carried out at 37 °C in 0.1 mM Tris·HCl buffer, pH 7.6, containing 5 mM DTT, 10 mM MgCl2, 150 μM CTP, 150 μM ATP, 500 μM MEP (racemic), d-EP or d-MTP or d-EEP, 4.8 μM IspDF, 6.2 μM IspE (where applicable) in a final volume of 50 μL. [32P]NTPs were diluted from 5 mM stock solutions of 40 μCi/μmol [α-32P]CTP and 320 μCi/μmol [γ-32P]ATP. Reactions were initiated by addition of CTP. After 1 h, the reactions were quenched with 60 μL of methanol and were put on ice. TLC analysis (Polygram Sil N-HR; Macherey and Nagel) was performed by spotting 4.5 μL of the reaction mixture and developing the plates with n-propanol/ethyl acetate/H2O (6:1:3, v/v/v). Radioactivity was quantified by phosphorimaging.
LC–MS Analysis
LC–MS analyses for incubations with EP, EEP, NMEP, and MESP similar to those described in the TLC analyses were carried out on a 1 mM scale using unlabeled CTP (0.75 mM), unlabeled ATP (0.75 mM), IspDF (4.8 μM), and IspE (6.2 μM). The reactions were incubated for 1 h and were centrifuged through a membrane (10 kDa cutoff) to remove the enzymes. Products were detected by negative-ion electrospray LC–MS using a Phenomex Prodigy 5 μ ODS(3) 100A (250 × 4.60 mm 5 μM) column eluted with 20 mM N,N′-dimethylhexylamine in 10% methanol, pH 7.0, adjusted with formic acid (Buffer A) and 2 mM N,N′-dimethylhexylamine in 50% methanol, pH 7.0 (Buffer B), as previously described.4
Time Course Studies
A solution of 500 μM EP (or EEP), 4.8 μM IspDF, and 6.2 μM IspE in 0.1 M Tris·HCl buffer, pH 7.6 (37 °C), 10 mM MgCl2, containing 5 mM DTT, in a final volume of 100 μL was preincubated for 10 min at 37 °C. γ-[32P]ATP (150 μM) and α-[32P]CTP (150 μM, 40 μCi/μmol) were added sequentially to initiate the reaction. At various times, 6 μL portions of the mixture were removed and quenched with 6 μL of methanol. After 61 min, an additional 2 μg portions of each enzyme were added to the reaction mixture.
Inhibition Studies with MTP
A solution of 500 μM MEP and 0.36 μM IspDF in 0.1 M Tris·HCl buffer, pH 7.6 (37 °C), containing 5 mM DTT and 10 mM MgCl2 was preincubated for 10 min. α-[32P]CTP (150 μM) was added to initiate the reaction. At various times, 6 μL portions of the mixture were removed and quenched with 6 μL of methanol. The samples were analyzed by TLC.4 The same experiment was performed except MTP was added to 5 mM. No change was seen in the rate of formation of CDP–ME.
Acknowledgments
We are grateful to the National Institute of General Medical Sciences (GM 25521 to CDP and GM 13956 to RMC) for financial support of this research.
Supporting Information Available
Additional figures for time course TLC and LC–MS chromatograms. NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org/
Author Present Address
§ (S.G.K.) Twenty-six Corporate Circle, Albany Molecular Research Inc., Albany, N.Y. 12212.
Author Present Address
⊥ (C.E.D.) Goodwin Procters’s Technology and Life Sciences Group, Exchange Place, 53 State St., Boston, MA 01209.
Author Present Address
# (C.L.) Université de Toulouse, UPS, CNRS, Laboratoire de synthèse et physio-Chimie de Molécules d’Intérêt Biologique, LSPCMIB, 118 route de Narbonne F-31062, Toulouse cedex 9, France.
The authors declare no competing financial interest.
Author Status
∥ (M.U.) Deceased.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Bochar D. A.; Freisen J. A.; Stauffacher C. V.; Rodwell V. W.. Biosynthesis of Mevalonic acid from Acetyl Co-A). In Comprehensive Natural Products Chemistry; Cane D., Ed.; Pergamon Press: Oxford, 1999; Vol. 2, pp 15–44. [Google Scholar]
- Miziorko H. M. Arch. Biochem. Biophys. 2012, 505, 131–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L.; Chang W.-C.; Xiao Y.; Liu H.-W.; Liu P. Annu. Rev. Biochem. 2013, 82, 497–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Testa C. A.; Lherbet C.; Pojer F.; Noel J. P.; Poulter C. D. Biochim. Biophys. Acta 2006, 1764, 85–96. [DOI] [PubMed] [Google Scholar]
- Lherbet C.; Pojer F.; Richard S. B.; Noel J. P.; Poulter C. D. Biochemistry 2006, 45, 3548–3553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter W. N. Curr. Top. Med. Chem. 2011, 11, 2048–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris F.; Vierling R.; Boucher L.; Bosch J.; Freel Meyers C. L. ChemBioChem. 2013, 14, 1309–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponaire S.; Zingle C.; Tritsch D.; Grosdemange-Billiard C.; Rohmer M. Eur. J. Med. Chem. 2012, 51, 277–285. [DOI] [PubMed] [Google Scholar]
- Majumdar A.; Shah M. H.; Bitok J. K.; Hassis-LeBeau M. E.; Meyers C. L. F. Mol. Biosys. 2009, 5, 935–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lillo A. M.; Tetzlaff C. N.; Sangari F. J.; Cane D. E. Bioorg. Med. Chem. Lett. 2003, 13, 737–739. [DOI] [PubMed] [Google Scholar]
- Wang W.; Oldfield E. Angew. Chem., Int. Ed. 2014, 53, 4294–4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witschel M. C.; Hçffken H. W.; Seet M.; Parra L.; Mietzner T.; Thater F.; Niggeweg R.; Rçhl F.; Illarionov B.; Rohdich F.; Kaiser J.; Fischer M.; Bacher A.; Diederich F. Angew. Chem., Int. Ed. 2011, 50, 7931–7935. [DOI] [PubMed] [Google Scholar]
- Fox D. T.; Poulter C. D. J. Org. Chem. 2005, 70, 1978–1985. [DOI] [PubMed] [Google Scholar]
- Lagisetti C.; Urbansky M.; Coates R. M. J. Org. Chem. 2007, 72, 9886–9895. [DOI] [PubMed] [Google Scholar]
- Ghavami A.; Johnston B. D.; Pinto B. M. J. Org. Chem. 2001, 66, 2312–2317. [DOI] [PubMed] [Google Scholar]
- Koppisch A. T.; Blagg B. S. J.; Poulter C. D. Org. Lett. 2000, 2, 215–217. [DOI] [PubMed] [Google Scholar]
- Gefflaut T.; Lemaire M.; Valentin M.-L.; Bolte J. J. Org. Chem. 1997, 62, 5920–5922. [Google Scholar]
- Mitsunobu O. Synthesis 1981, 1, 1–28. [Google Scholar]
- Kolb H. C.; VanNieuwenhze M. S.; Sharpless K. B. Chem. Rev. 1994, 94, 2483–547. [Google Scholar]
- Hoye T. R.; Jeffrey C. D.; Shao F. Nat. Protoc. 2007, 2, 2451–2458. [DOI] [PubMed] [Google Scholar]
- Nishino T.; Ogura K.; Seto S. Biochim. Biophys. Acta 1971, 235, 322–325. [DOI] [PubMed] [Google Scholar]
- Poulter C. D.; Rilling H. C.. Prenyl Transferases and Isomerase. In Biosynthesis of Isoprenoid Compounds; Porter J. W., Ed.; John Wiley & Sons: New York, 1981; Vol. 1, Chapter 4, pp 162–224. [Google Scholar]
- Poulter C. D.; Argyle J. C.; Mash E. A.; Laskovics G. M.; Wiggins P. L.; King C. R.. Inhibitors of Isoprenoid Biosynthesis. In Regulation of Insect Development and Behavior; Chemistry and Biochemistry of Juvenile Hormones; Wroclaw Technical University Press: Wroclaw, Poland, 1981; Chapter 3, pp 149–162. [Google Scholar]
- Janthawornpong K.; Krasutsky S.; Chaignon P.; Rohmer M.; Poulter C. D.; Seemann M. J. Am. Chem. Soc. 2013, 135, 1816–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan R. M.; Poulter C. D. J. Org. Chem. 2001, 66, 6705–6710. [DOI] [PubMed] [Google Scholar]
- Barker S. A.; Foster A. B.; Haines A. H.; Lehmann J.; Webber J. M.; Zweifel G. J. Chem. Soc. 1963, 4161–4167. [Google Scholar]
- Ihara M.; Takino Y.; Tomotake M.; Fukumoto K. J. Chem. Soc., Perkin Trans. 1 1990, 8, 2287–2292. [Google Scholar]
- MacDonald D. L.; Fischer H. O. L.; Ballou C. E. J. Am. Chem. Soc. 1956, 78, 3720–3722. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.