

Abstract
Objective
Mutations in leucine-rich repeat kinase 2 (LRRK2) pose a significant genetic risk in familial and sporadic Parkinson's disease (PD). R1441 mutation (R1441G/C) in its GTPase domain is found in familial PD. How LRRK2 interacts with synaptic proteins, and its role in dopamine (DA) homeostasis and synaptic vesicle recycling remain unclear.
Methods
To explore the pathogenic effects of LRRK2R1441G mutation on nigrostriatal synaptic nerve terminals and locomotor activity, we generated C57BL/6N mice with homozygous LRRK2R1441G knockin (KI) mutation, and examined for early changes in nigrostriatal region, striatal synaptosomal [3H]-DA uptake and locomotor activity after reserpine-induced DA depletion.
Results
Under normal conditions, mutant mice showed no differences, (1) in amount and morphology of nigrostriatal DA neurons and neurites, (2) tyrosine hydroxylase (TH), DA uptake transporter (DAT), vesicular monoamine transporter-2 (VMAT2) expression in striatum, (3) COX IV, LC3B, Beclin-1 expression in midbrain, (4) LRRK2 expression in total cell lysate from whole brain, (5) α-synuclein, ubiquitin, and tau protein immunostaining in midbrain, (6) locomotor activity, compared to wild-type controls. However, after a single intraperitoneal reserpine dose, striatal synaptosomes from young 3-month-old mutant mice demonstrated significantly lower DA uptake with impaired locomotor activity and significantly slower recovery from the effects of reserpine.
Interpretation
Although no abnormal phenotype was observed in mutant LRRK2R1441G mice, the KI mutation increases vulnerability to reserpine-induced striatal DA depletion and perturbed DA homeostasis resulting in presynaptic dysfunction and locomotor deficits with impaired recovery from reserpine. This subtle nigrostriatal synaptic vulnerability may reflect one of the earliest pathogenic processes in LRRK2-associated PD.
Introduction
Parkinson's disease (PD) is the second most common neurodegenerative disease, characterized clinically by bradykinesia, rigidity, resting tremor, and pathologically by an irreversible loss of dopaminergic (DA) neurons in substantia nigra pars compacta (SNpc). The underlying mechanism of DA neuronal loss remains unclear but there is evidence to indicate that striatal presynaptic dysfunction associated with defective DA uptake and turnover is observed in patients at the early stages of PD.1–3 DA reuptake and sequestration into synaptic vesicles at striatal nerve terminals facilitate tonically active DA release.4 Perturbation of DA recycling impairing intracellular DA content and synaptic plasticity has been previously observed in experimental mouse models carrying specific gene mutation associated with familial PD.5–7
Most cases of PD occur sporadically and are idiopathic, but causal or predisposing genes have been identified in familial cases.8 Leucine-rich repeat kinase 2 (LRRK2) mutations pose the commonest genetic risk in both familial and sporadic PD.9,10 Three variants of single amino acid substitution at R1441 residue (R1441G/C/H) of LRRK2 in the highly conserved GTPase domain have been described.11 Of these, R1441G substitution is one of the commonest mutations in LRRK2-associated PD,12,13 and has been found in over 13% of Spanish cases from the Basque region.14
LRRK2 is involved in diverse neuronal functions15,16 and is highly expressed in striatum enriched with DA nerve terminals where its expression level is even higher than that in SNpc which contains their DA cell bodies.17 LRRK2 interacts with various synaptic proteins.18,19 Its mutations cause abnormal synaptic dopamine neurotransmission in both LRRK2 knockin and transgenic animal models.20–22 Increased DA turnover was observed in human asymptomatic LRRK2 mutation carriers,23 indicating that perturbation of DA homeostasis may be one of the early events associated with pathogenic LRRK2 mutation. However, how LRRK2R1441G mutation affects synaptic DA homeostasis remains unclear. We hypothesize that LRRK2 plays a regulatory role in presynaptic terminals by modulating DA recycling processes, and such synaptic dysfunction associated with R1441G mutation contributes to the degeneration of nigrostriatal dopaminergic neurons and motor deficits in PD.
Here, we generated homozygous LRRK2R1441G knockin (KI/KI) mice to examine for potential pathophysiological changes in the dopaminergic nigrostriatal pathway. We also explored the susceptibility of our young LRRK2 mutant mice to striatal DA depletion and impaired DA recycling after a single dose of intraperitoneal reserpine, a reversible inhibitor of the vesicular monoamine transporter-2 (VMAT2).
Materials and Methods
Generation of LRRK2R1441G knockin mouse
A targeting cassette spanning exons 28–32 was constructed based on a BAC clone of wild-type mouse Lrrk2 gene (bMQ406i17). A point mutation (c. 4321C>G) was created by mutagenesis, and a neomycin resistance cassette (Neo) flanked by two FRT sites was inserted for positive selection. The targeting cassette was electroporated into mouse embryonic stem (ES) cells (129/S6), and the colonies with the correct homologous recombination were selected by neomycin and verified by Southern blot. The targeting cassette introduced two NcoI restriction sites. Thus, NcoI digestion resulted in fragments with different lengths for wild-type and targeted alleles. These were detected by two external probes (Fig.1A). Positive ES cell colonies with one copy of correctly targeted allele were injected into blastocysts (C57BL/6N) to obtain chimeric mice. Neo was subsequently removed by breeding with Flipper mice. The resultant heterozygous knockin (KI/WT) mice were maintained under the C57BL/6N mouse background by backcrossed to C57BL/6N mice for seven generations and intercrossed to generate homozygous knockin (KI/KI) mice and wild-type littermate controls (WT/WT). The mice were maintained as KI/KI and WT/WT. The primers are listed in Table1. Only male mice were used in subsequent experiments.
Figure 1.
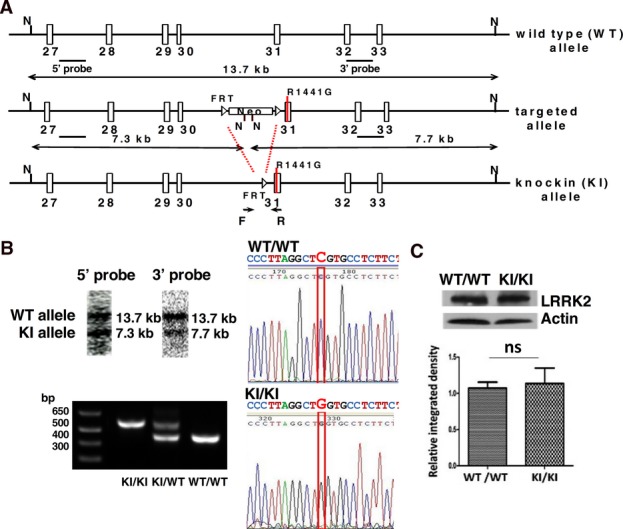
Generation of LRRK2R1441G knockin mouse. (A) Gene targeting and confirmation strategy. N = NcoI. Neo = neomycin resistance cassette. (B) Verification of correct gene targeting (Southern blot), LRRK2 point mutation sequencing (c. 4321 C>G), and genotyping. (C) LRRK2 protein expression was not altered in KI/KI mouse (9 weeks, whole brain lysate; N = 4). Data are presented as mean + SEM. ns: not significant by unpaired Student's t-test.
Table 1.
Primer sequences for embryonic stem (ES) cell colony screening and mouse genotyping.
| Objective | Primer sequences |
|---|---|
| 5′ probe synthesis | F: 5′-GCTGGAGGAGTCGAGAGTTC-3′ |
| R: 5′-GCATGGTGTGCTATAATCC-3′ | |
| 3′ probe synthesis | F: 5′-CGCCGTTCACTTCCTAAATG-3′ |
| R: 5′-GCATATAGATAAGAACCGGTAC-3′ | |
| Routine genotyping | F: 5′-ACGCTGCTGTGTCACACGGTT-3′ |
| R: 5′-TCCGAAGCTTTGCCAGCGCAT-3′ | |
| Sequencing | 5′-ACGCTGCTGTGTCACACGGTT-3′ |
Mouse husbandry and genotyping
All mice were maintained in the Laboratory Animal Unit, University of Hong Kong with accreditation through the Association for Assessment and Accreditation of Laboratory Animal care international (AAALAC). For genotyping, DNA was extracted from ear clips by incubation in 100 μL PBND buffer (50 mmol/L KCl; 10 mmol/L Tris-HCl, pH 8.0; 2.5 mmol/L MgCl2; 0.1 mg/mL Gelatin; 0.45% (v/v) Nonidet P40; 0.45% (v/v) Tween20) with 1 mg/mL proteinase K at 55°C overnight. After boiling for 5 min, the supernatant was used for genotyping by PCR (Fig.1A and Table1).
Locomotor activity test
Locomotor activity of mice was monitored using open-field tests.24 Briefly, the mice were allowed to acclimatize in the behavioral testing room 3 days before the tests. The locomotor recording was started immediately after the mice were placed in the center of the plastic arena (26 × 26 × 40 cm) of the test chamber adapted under dim light. The mice were allowed to move freely and tracked for 60 min (EthoVision3.0, Noldus Information Technology, Wageningen, the Netherlands). The tests were applied similarly to all the parallel groups. Movement duration and distance moved of each mouse was recorded 2 days prior to and 1 day after reserpine (Sigma-Aldrich, St. Louis, MO) [1 mg/kg, intraperitoneal (i.p.)] treatment. Recovery from reserpine treatment was monitored for up to 14 days after injection.
Histology
For morphological examination of nigrostriatal neural network, mice were perfused with cold PBS followed by 4% PFA under anesthesia. Whole brain was removed and postfixed in 4% PFA at 4°C overnight. After dehydration by exposure to serial dilutions of ethanol, the brain was cleared by xylene before paraffin infiltration. Coronal sections (10 μm thick) were cut through the midbrain (including substantia nigra) or striatum and horizontal sections (10 μm thick) through the cerebellum. For immunohistochemistry, endogenous peroxidase activity was blocked by 3% H2O2. After blocking with TBS containing 5% normal serum and 5% BSA for 1 h at room temperature, sections were incubated with primary antibody at 4°C overnight and then a secondary antibody conjugated to HRP. The resultant color development of DAB was monitored under the microscope.25 Stained slides were dehydrated, cleared by xylene, and mounted with Histomount (Invitrogen, Carlsbad, CA). The primary antibodies were: anti-DAT (1:50, Santa-Cruz Biotechnology #sc-32258), anti-TH (1:500, Millipore #AB318), anti-α-synuclein (1:200, Cell Signaling #4179), anti-tau46 (1:400, Cell Signaling #4019), anti-ubiquitin (1:400, Cell Signaling #3933). Dopaminergic neuronal cell bodies from five consecutive sections stained with TH at the comparable SNpc region from three individual mice in each group were counted under blinded conditions by two different researchers independently. Striatal density was assessed using ImageJ software26,27 (http://rsbweb.nih.gov/ij/plugins/track/track.html) to measure TH staining density at the comparable striatal region from four consecutive sections taken from three individual mice in each group.
Western blot
Striatum and midbrain were dissected and homogenized in cold lysis buffer containing: 50 mmol/L Tris-Cl, pH 7.4, 150 mmol/L NaCl, 1% sodium deoxycholate, 1 mmol/L EDTA, 1% Triton X-100, 1 mmol/L phenylmethylsulfonyl fluoride, supplemented with protease inhibitor cocktail. Lysates were clarified by centrifugation at 4°C for 15 min at 13,500 g. Protein concentration was determined by the Bradford assay. Equal amounts of protein were boiled for 10 min at 95°C in sample buffer containing: 62.5 mmol/L Tris, pH 6.8, 100 mmol/L DTT, 2% SDS, 10% glycerol, and 0.002% bromophenol blue. Samples were electrophoresed in 12% SDS-polyacrylamide gels and transferred onto nitrocellulose membranes. Resulting blot were blocked with 5% nonfat skimmed milk in TBS and probed with antibodies against LC3B (1:1000, Cell Signaling, Danvers, MA; #3868, 14/16 kD), Beclin-1 (1:1000, Cell Signaling #3495, 60 kD), COX IV (1:2000, Abcam, Cambridge, MA; #ab16056, 17 kD) glycosylated DAT (1:500, Santa-Cruz Biotechnology, Santa Cruz Biotechnology, CA; #sc-32258, 80 kD), nonglycosylated DAT (1:1000, Santa-Cruz Biotechnology #sc-10042, 50 kD), TH (1:2000, Millipore, Bedford, MA; #MAB318, 60 kD), α-synuclein (1:1000, Cell Signaling #417, 18 kD), Tau46 (1:1000, Cell Signaling #4019, 50–80 kD), Ubiquitin (1:1000, Cell Signaling #3933), Neuronal Class III β-Tubulin (Tuj1) (1:2000, Covance, Princeton, NJ; #MRB-435P, 50 kD), Actin (1:500, Santa-Cruz Biotechnology #sc-1615, 43 kD), VMAT2 (1:500, Santa-Cruz Biotechnology #sc-7721R, 63 kD), Synaptophysin (1:2000, Cell Signaling #D35E4, 38 kD). For chemiluminescence detection, blots were incubated with HRP-conjugated secondary antibodies followed by ECL-plus substrate detection. Immunoblots were quantified by computerized scanning densitometry.
Synaptosomal [3H]-dopamine uptake assay
Synaptosomal [3H]-DA uptake assay was performed as previously described with slight modifications.28–30 Briefly, 5 μg synaptosomes were prewarmed and incubated in 200 μL of Krebs–Ringer buffer (120 mmol/L NaCl; 4.8 mmol/L KCl; 1.3 mmol/L CaCl2; 1.2 mmol/L MgSO4; 1.2 mmol/L KH2PO4; 25 mmol/L NaHCO3; 6 mmol/L glucose; pH 7.6) containing 100 nmol/L [3H]-DA for 5 min at 37°C. Nonspecific uptake was determined by a parallel negative control in the presence of 10 μM nomifensine (DAT inhibitor). To test the effect of reserpine, synaptosomes and reserpine (vehicle control, 50 nmol/L, 500 nmol/L) were prewarmed for 2.5 min at 37°C. The reaction was stopped by adding 200 μL cold Krebs–Ringer buffer and mixture was passed through a UniFilter®-96 GF/C filter (PerkinElmer, Norwalk, CT; #6005174) by unifilter-96 harvester (Packard), followed by three washes of cold Krebs–Ringer buffer. The filter was dried at 55°C overnight, and [3H] radioactivity (count per minute, cpm) was measured using the TopCount®NXTTM counter (Packard). Each treatment group was measured at least in duplicate for each experiment.
Statistics
Statistical analyses were carried out using the Prism (GraphPad Software Inc., San Diego, CA). Data are expressed as mean ± standard error mean (SEM). The number of animals and independent experiments are indicated in the figure legends. Student's t-test was used to compare levels of protein expression among groups. Two-way ANOVA with post hoc Bonferroni multiple comparison tests were used to compare [3H]-DA uptake and locomotor activity in the open-field tests. Differences between groups were considered significant at P < 0.05.
Results
Generation of the LRRK2R1441G knockin (KI) mouse
We generated homozygous LRRK2R1441G KI mice carrying a point mutation in exon 31 (c.4321C>G) that causes R1441G substitution in LRRK2 protein. The Southern blot result showed the 13.7 kb band representing the WT allele detected by 5′ and 3′ probes, and the 7.3 kb band by 5′ probe and 7.7 kb band by 3′ probe representing the KI allele (Fig.1A). The presence of mutant murine genomic DNA was verified by Southern blot, genotyping, and sequencing (Fig.1B). The mutant LRRK2 expression is controlled by its endogenous promoter (Fig.1A). Expression of mutant LRRK2 protein in KI/KI mouse brain was similar to wild-type protein in WT/WT control mouse brain (Fig.1C).
Striatum and SNpc were morphologically normal in aged mutant mice
There was no apparent phenotypic difference in KI/KI mice as compared with WT/WT. They were fertile and had normal body weight, brain size, and locomotor activity (Figs4 and S2). Histological examination revealed similar levels of TH and DAT in SNpc and striatum at 3 months and 18–22 months (Fig.2A and B; Fig. S3). There were no significant differences in number of TH+ DA neurons in SNpc and TH staining intensity in striatum (KI/KI vs. WT/WT mice at 18–22 months – Fig.2C). The two independent methods – counting total TH+ cells in SNpc and striatal neurite density gave consistent results, showing no significant difference in the neuronal numbers and neurite density in KI/KI and WT/WT mice, consistent with our Western blot results on TH protein expression. Similarly, glycosylated and nonglycosylated DAT had comparable expression levels in KI/KI and WT/WT in both striatum and midbrain at age 18 months (Fig.2D).
Figure 4.
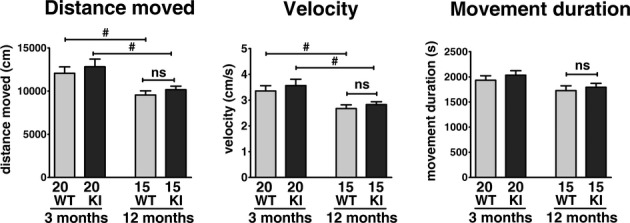
LRRK2R1441G knockin mice have similar locomotor activities as WT/WT in open-field test. Locomotor activity of KI/KI mice was not significantly different from WT/WT at 3 and 12 months of age, although an age-dependent decrease was observed. #P < 0.05; two-way ANOVA with post hoc Bonferroni multiple comparison tests. (3 months: N = 20; 12 months: N = 15; mean + SEM).
Figure 2.
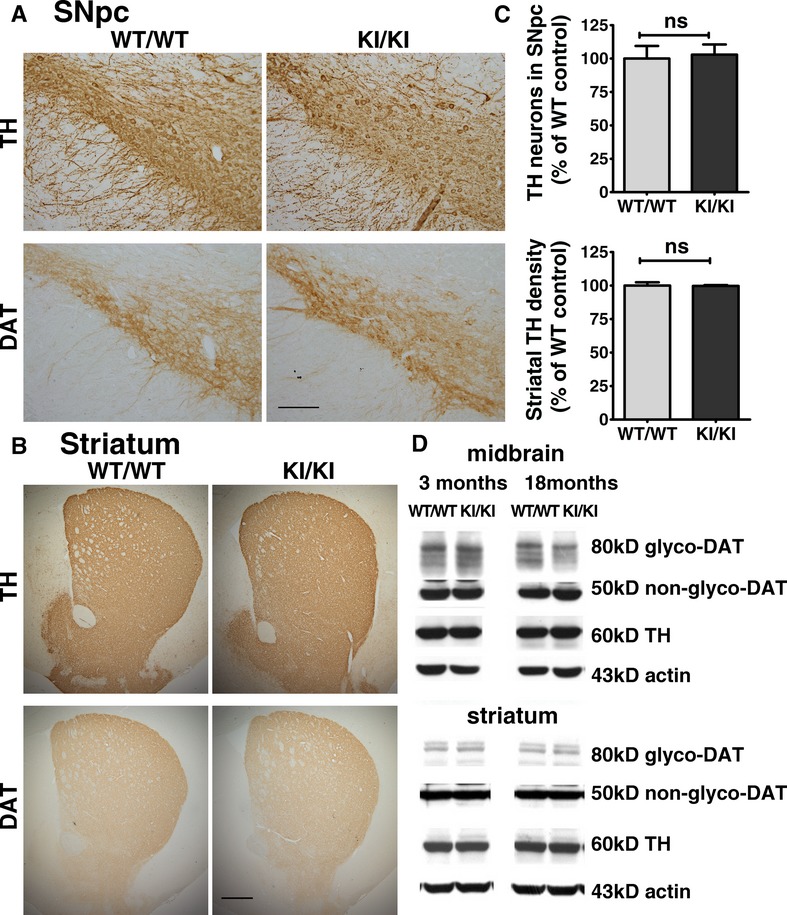
Aged LRRK2R1441G knockin mice have similar amounts of DA neurons and neurites compared to wild-type mice. (A, B) No apparent difference in the expression pattern of TH and DAT in SNpc and striatum between WT/WT and KI/KI mice at 18–22 months (N = 3). Scale = 150 μm in (A) and 200 μm in (B). (C) Statistical analysis of TH+ DA neurons numbers and neurite intensity of stained SNpc and striatum revealed no significant difference in combination of TH expression and cell count (unpaired Student's t-test, N = 3). (D) Western blot showed similar expression levels of TH and DAT (glycosylated and nonglycosylated) in striatum and midbrain in WT/WT and KI/KI mice at 3 and 18 months of age (N = 3).
No apparent difference in degree of protein aggregation in aged mutant mice midbrain compared with wild-type controls
Histological studies revealed similar ubiquitin and tau expression in midbrain, and α-synuclein in midbrain and cerebellum at 18–22 months (Fig.3A). These three proteins had comparable expression levels in KI/KI and WT/WT in midbrain at age 18 months (Western blot; Fig.3B) or at 22 months (immunohistochemistry; Fig.3A). There were no differences in expression levels of COX IV (mitochondrial marker), LC3B and Beclin-1 (autophagy markers) between KI/KI and WT/WT young mice (Fig. S4) and aging mice (Fig.3B).
Figure 3.
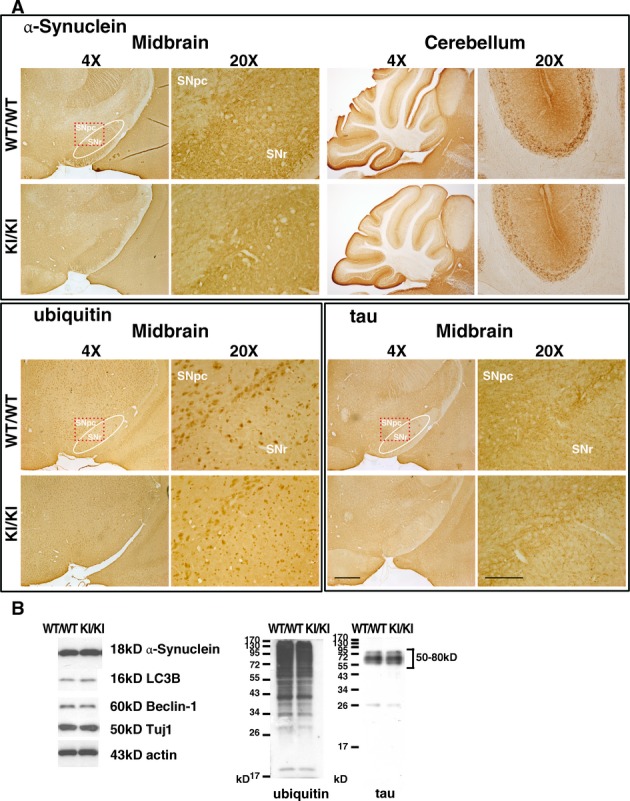
Aged LRRK2R1441G knockin mice have similar α-synuclein, ubiquitin, and tau expression patterns compared to those of WT/WT mice. (A) No apparent difference in the expression patterns of α-synuclein in midbrain and cerebellum, and the similar expression patterns of ubiquitin and tau in midbrain between WT/WT and KI/KI mice at 18–22 months (N = 3). SNpc substantia nigra pars compacta; SNr substantia nigra pars reticulata. Scale = 500 μm in 4× magnification and 150 μm in 20× magnification. (B) Western blot showed similar expression level of α-synuclein, ubiquitin, tau and autophagy makers LC3B and Beclin-1 in midbrain of WT/WT and KI/KI mice at 18 months of age (N = 3).
Mutant mice were more susceptible to reserpine-induced locomotor deficits, and recovered slower
LRRK2 KI/KI mice demonstrated similar locomotor activity as WT/WT at age of 3 and 12 months. Older mice had less distance moved and slower velocity in both WT/WT and KI/KI groups (P < 0.05; Fig.4), but such decrease did not involve a significant interacting effect between genotype (i.e. R1441G) and aging.
Reserpine (1 mg/kg, i.p.) significantly decreased locomotor activity in both 3-month-old WT/WT and KI/KI mice (distance moved; velocity; movement duration: all P < 0.05; N = 8) at 2 days posttreatment, (Fig.5). However, compared to WT/WT mice, mutant mice exhibited a significantly greater decrease in these locomotor parameters after reserpine injection (WT/WT vs. KI/KI; all P < 0.05; Fig.5). Both WT/WT and KI/KI started to recover from their locomotor deficits after day 2 post reserpine. For WT/WT mice, the locomotor parameters recovered to pretreatment levels by day 14 post reserpine. However, KI/KI mice had consistently lower locomotor activity at all three time points (i.e., day 2, 7, and 14 post-reserpine), as compared with WT/WT controls (all P < 0.05; Fig.5).
Figure 5.
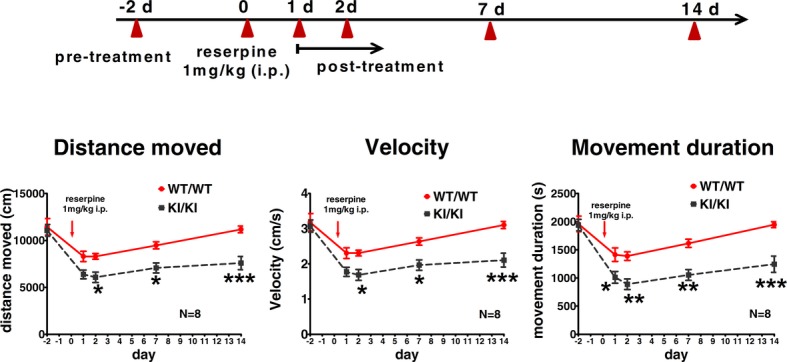
LRRK2R1441G knockin mice are more susceptible to reserpine-induced locomotor deficits. KI/KI mice injected with reserpine (1 mg/kg) demonstrated more severe impairment in locomotor activity compared to that of WT/WT at 3 months of age. Both mutant and control mice recovered after treatment, and KI/KI mice recovered much more slowly than WT/WT mice. *P < 0.05; **P < 0.01; ***P < 0.001; two-way ANOVA with post hoc Bonferroni multiple comparison tests; N = 8; mean ± SEM.
Young mutant mice exhibited more severe reduction in striatal synaptosomal DA uptake than wild-type controls after reserpine treatment
Total [3H]-DA uptake was performed in striatal synaptosomal isolates extracted from both 3 and 18 months old mice. Both WT/WT and KI/KI synaptosomes had similar levels of DA uptake at their corresponding ages (Fig.6A). Similar to the locomotor activity, there was no significant interacting effect between genotype (i.e. R1441G) and aging on [3H]-DA uptake between 3 and 18 month-old mice (Fig.6A).
Figure 6.
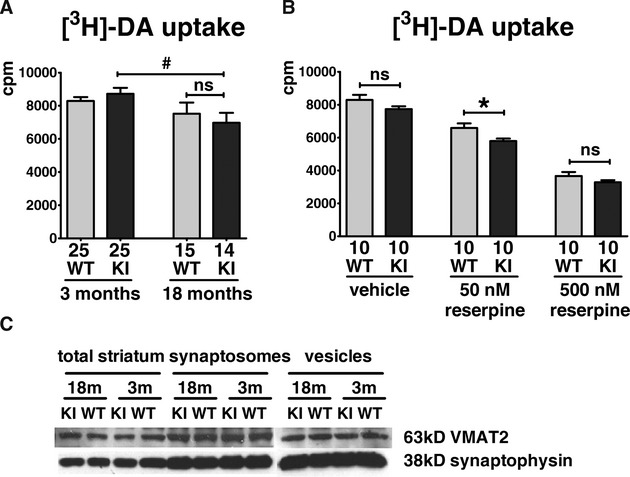
Effects of aging and reserpine on striatal synaptosomal [3H]-DA uptake. (A) There was no significant difference in [3H]-DA uptake between WT/WT and KI/KI mice at 3 or 18 months old, although an age-dependent decrease was observed in both genotypes (#P < 0.05; two-way ANOVA with post hoc Bonferroni multiple comparison tests; N = 25 for each 3 months old group; N ≥ 14 for each 18 months old group; mean + SEM). (B) [3H]-DA uptake in KI/KI mice had greater decrease with reserpine (50 nmol/L) than WT/WT mice at 3 months of age. *P < 0.05; two-way ANOVA with post hoc Bonferroni multiple comparison tests; N = 10; mean + SEM. Numbers below each bar represent number of independent measurements. (C) Expression levels of VMAT2 in total cell lysate, synaptosomes, and synaptic vesicles from striatum were similar in WT/WT and KI/KI mice at 3 and 18 months of age (N = 3).
Following reserpine treatment, [3H]-DA uptake into striatal synaptosomes (50 nmol/L reserpine) was significantly lower (-12%) in isolates from KI/KI mice compared with WT/WT even at age 3 months (WT/WT: 6583 ± 283 cpm; KI/KI: 5786 ± 157 cpm; N = 10; P < 0.05; Fig.6B). A dose-dependent decrease in DA uptake was observed with reserpine treatment, but the difference in DA uptake between KI/KI and WT/WT became insignificant at the excessive dose of 500 nmol/L (Fig.6B). Using two-way ANOVA analysis, the mutant genotype (P < 0.01) and reserpine (P < 0.01) were independently associated with reduction in DA uptake.
Striatal DAT and VMAT2 protein expression, which may affect total synaptosomal DA uptake, were similar between KI/KI and WT/WT mice (Figs2D, 6C).
Discussion
We describe here a mutant LRRK2R1441G KI/KI mouse expressing physiological levels of the variant protein. Unlike mice overexpressing mutant LRRK2 which showed obvious parkinsonian phenotype,31,32 LRRK2R1441G KI/KI mutation had no apparent abnormal phenotype, or abnormal parameters in dopaminergic function and protein aggregation even in aged mice (Figs3, 4, 6A). The number and morphology of DA neurons were similar between KI/KI and WT/WT in aged mice up to 22 months (Fig.2). This is in contrast to the LRRK2R1441G transgenic mouse which showed severe loss of DA dendrites and morphological abnormalities of DA neurons in SNpc at 9–10 month of age, age-dependent motor deficits, and decreased spontaneous DA release.32 Nevertheless, the R1441G mutant protein in our mutant mice is expressed at physiological level comparable to the WT protein, and we demonstrated that these mutant KI/KI mice were more susceptible to synaptic DA depletion and motor deficits associated with impaired DA uptake induced by reserpine. We believe that the KI/KI mice more closely mimic the disease condition in humans.
Reserpine produces behavioral deficits resembling PD motor symptoms33 through its catecholamine-depleting effects on nerve terminals.34 It inhibits vesicular monoamine transferase-2 (VMAT2) and blocks sequestration of cytosolic DA into synaptic vesicles. With reserpine (50 nmol/L), [3H]-DA uptake was significantly reduced in young KI/KI striatal synaptosomes (Fig.6B). This was not due to expression level of DAT or VMAT2 as their protein expression was not altered in striatum (Fig.2D, 6C). The higher dose of reserpine (500 nmol/L) caused an even greater reduction in synaptosomal DA uptake. However, the difference in [3H]-DA uptake between KI/KI and WT/WT mice was not statistically significant (Fig.6B) because the toxic effects of high dose reserpine probably overwhelmed the effects of the LRRK2 mutation on DA uptake. Extracellular [3H]-DA uptake through DAT into synaptosomes is suppressed by abnormally high cytosolic DA concentration induced by reserpine.35 High concentrations of cytosolic DA can reduce DAT function in vitro.36 The significantly decreased [3H]-DA uptake in reserpine-treated KI/KI striatal synaptosomes (Fig.6B) indicates that LRRK2R1441G mutation potentiates DA depletion in striatum by suppressing uptake of extracellular DA in mutant presynaptic terminals. The significantly reduced locomotor activity in young KI/KI mice after reserpine (Fig.5) indicates that these mutant mice are more sensitive to perturbed DA homeostasis by reserpine than WT/WT. Furthermore, these young mutant mice recovered significantly slower in terms of their locomotor activity suggesting that they are more vulnerable to further stress on their DA uptake system, compared to WT/WT mice which recovered back to baseline levels within 2 weeks. The functional deficits in locomotor activity and synaptosomal DA uptake in these young mutant mice which recovered slower after a single stressor event on DA uptake may have important pathogenic implications in terms of adverse cumulative effects over their lifespan.
Despite the apparent lack of abnormal phenotype in our mutant mice, our results show that a functional susceptibility can still exist. This is illustrated in asymptomatic human LRRK2 mutation carriers who have abnormal putaminal DA turnover shown using positron emission tomography neuroimaging techniques.23 Similarly, DA neurons and locomotor activity appeared normal in LRRK2R1441C KI mice, but they had impaired DA neurotransmission.20 The higher susceptibility to reserpine in our KI/KI mice indicates very early striatal synaptic dysfunction before any structural abnormalities could possibly be observed. Our results implicate the notion that the pathogenic process in PD starts from dysfunction of striatal nerve terminals proceeding in a retrograde dying-back process37–39 rather than a compensatory mechanism to counteract early dopaminergic neuronal loss,40 because there was no significant striatal neuronal loss observed even in our aged mutant mice. How presynaptic defects and abnormal DA homeostasis lead to neuronal cell death associated with LRRK2 mutations require further investigations.
Acknowledgments
This study was supported by the Henry G. Leong Professorship in Neurology (SLH), the Donation Fund for Neurology Research (SLH), Health and Medical Research Fund (SLH), and the Research Grants Council (ZJZ; RGC CRF/HKU3/07C). We gratefully acknowledge Prof. Sookja Chung and Dr. Patrick KK Yeung, Department of Anatomy, University of Hong Kong, for the use of the open-field locomotor assessment system.
Conflict of Interest
None declared.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1. LRRK2 protein structure and PD related R1441 mutations.
Figure S2. Gross morphology and body weight curve.
Figure S3 Young LRRK2R1441G mutant mice have similar amount of DA neuron and neurites compared to wild type.
Figure S4. Expression level of related proteins.
References
- Mo SJ, Linder J, Forsgren L, et al. Pre- and postsynaptic dopamine SPECT in the early phase of idiopathic parkinsonism: a population-based study. Eur J Nucl Med Mol Imaging. 2010;37:2154–2164. doi: 10.1007/s00259-010-1520-3. [DOI] [PubMed] [Google Scholar]
- Sossi V, de la Fuente-Fernandez R, Schulzer M, et al. Dopamine transporter relation to dopamine turnover in Parkinson's disease: a positron emission tomography study. Ann Neurol. 2007;62:468–474. doi: 10.1002/ana.21204. [DOI] [PubMed] [Google Scholar]
- Sossi V, de la Fuente-Fernandez R, Holden TE, et al. Changes of dopamine turnover in the progression of Parkinson's disease as measured by positron emission tomography: their relation to disease-compensatory mechanisms. J Cereb Blood Flow Metab. 2004;24:869–876. doi: 10.1097/01.WCB.0000126563.85360.75. [DOI] [PubMed] [Google Scholar]
- Bisaglia M, Greggio E, Beltramini M, et al. Dysfunction of dopamine homeostasis: clues in the hunt for novel Parkinson's disease therapies. FASEB J. 2013;27:2101–2110. doi: 10.1096/fj.12-226852. [DOI] [PubMed] [Google Scholar]
- Goldberg MS, Pisani A, Haburcak M, et al. Nigrostriatal dopaminergic deficits and hypokinesia caused by inactivation of the familial Parkinsonism-linked gene DJ-1. Neuron. 2005;45:489–496. doi: 10.1016/j.neuron.2005.01.041. [DOI] [PubMed] [Google Scholar]
- Kitada T, Pisani A, Karouani M, et al. Impaired dopamine release and synaptic plasticity in the striatum of parkin−/− mice. J Neurochem. 2009;110:613–621. doi: 10.1111/j.1471-4159.2009.06152.x. [DOI] [PubMed] [Google Scholar]
- Cheng F, Vivacqua G, Yu S. The role of alpha-synuclein in neurotransmission and synaptic plasticity. J Chem Neuroanat. 2011;42:242–248. doi: 10.1016/j.jchemneu.2010.12.001. [DOI] [PubMed] [Google Scholar]
- Trinh J, Farrer M. Advances in the genetics of Parkinson disease. Nat Rev Neurol. 2013;9:445–454. doi: 10.1038/nrneurol.2013.132. [DOI] [PubMed] [Google Scholar]
- Lesage S, Brice A. Parkinson's disease: from monogenic forms to genetic susceptibility factors. Hum Mol Genet. 2009;18:R48–R59. doi: 10.1093/hmg/ddp012. [DOI] [PubMed] [Google Scholar]
- Di Fonzo A, Rohe CF, Ferreira J, et al. A frequent LRRK2 gene mutation associated with autosomal dominant Parkinson's disease. Lancet. 2005;365:412–415. doi: 10.1016/S0140-6736(05)17829-5. [DOI] [PubMed] [Google Scholar]
- Lee BD, Dawson VL, Dawson TM. Leucine-rich repeat kinase 2 (LRRK2) as a potential therapeutic target in Parkinson's disease. Trends Pharmacol Sci. 2012;33:365–373. doi: 10.1016/j.tips.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marti-Masso JF, Ruiz-Martinez J, Bolano MJ, et al. Neuropathology of Parkinson's disease with the R1441G mutation in LRRK2. Mov Disord. 2009;24:1998–2001. doi: 10.1002/mds.22677. [DOI] [PubMed] [Google Scholar]
- Yue Z, Lachenmayer ML. Genetic LRRK2 models of Parkinson's disease: dissecting the pathogenic pathway and exploring clinical applications. Mov Disord. 2011;26:1386–1397. doi: 10.1002/mds.23737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorostidi A, Ruiz-Martinez J, Lopez de Munain A, et al. LRRK2 G2019S and R1441G mutations associated with Parkinson's disease are common in the Basque Country, but relative prevalence is determined by ethnicity. Neurogenetics. 2009;10:157–159. doi: 10.1007/s10048-008-0162-0. [DOI] [PubMed] [Google Scholar]
- Berwick DC, Harvey K. LRRK2 signaling pathways: the key to unlocking neurodegeneration? Trends Cell Biol. 2011;21:257–265. doi: 10.1016/j.tcb.2011.01.001. [DOI] [PubMed] [Google Scholar]
- Mata IF, Wedemeyer WJ, Farrer MJ, et al. LRRK2 in Parkinson's disease: protein domains and functional insights. Trends Neurosci. 2006;29:286–293. doi: 10.1016/j.tins.2006.03.006. [DOI] [PubMed] [Google Scholar]
- Mandemakers W, Snellinx A, O'Neill MJ, et al. LRRK2 expression is enriched in the striosomal compartment of mouse striatum. Neurobiol Dis. 2012;48:582–593. doi: 10.1016/j.nbd.2012.07.017. [DOI] [PubMed] [Google Scholar]
- Hehnly H, Stamnes M. Regulating cytoskeleton-based vesicle motility. FEBS Lett. 2007;581:2112–2118. doi: 10.1016/j.febslet.2007.01.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabo SL, McAllister AK. Mobility and cycling of synaptic protein-containing vesicles in axonal growth cone filopodia. Nat Neurosci. 2003;6:1264–1269. doi: 10.1038/nn1149. [DOI] [PubMed] [Google Scholar]
- Tong Y, Pisani A, Martella G, et al. R1441C mutation in LRRK2 impairs dopaminergic neurotransmission in mice. Proc Natl Acad Sci USA. 2009;106:14622–14627. doi: 10.1073/pnas.0906334106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melrose HL, Dachsel JC, Behrouz B, et al. Impaired dopaminergic neurotransmission and microtubule-associated protein tau alterations in human LRRK2 transgenic mice. Neurobiol Dis. 2010;40:503–517. doi: 10.1016/j.nbd.2010.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Huang C, Tong J, et al. Temporal expression of mutant LRRK2 in adult rats impairs dopamine reuptake. Int J Biol Sci. 2011;7:753–761. doi: 10.7150/ijbs.7.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sossi V, de la Fuente-Fernandez R, Nandhagopal R, et al. Dopamine turnover increases in asymptomatic LRRK2 mutations carriers. Mov Disord. 2010;25:2717–2723. doi: 10.1002/mds.23356. [DOI] [PubMed] [Google Scholar]
- Zhang X, Yeung PK, McAlonan GM, et al. Transgenic mice over-expressing endothelial endothelin-1 show cognitive deficit with blood-brain barrier breakdown after transient ischemia with long-term reperfusion. Neurobiol Learn Mem. 2013;101:46–54. doi: 10.1016/j.nlm.2013.01.002. [DOI] [PubMed] [Google Scholar]
- Tieu K, Perier C, Caspersen C, et al. D-beta-hydroxybutyrate rescues mitochondrial respiration and mitigates features of Parkinson disease. J Clin Investig. 2003;112:892–901. doi: 10.1172/JCI18797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozerdem U, Wojcik EM, Barkan GA, et al. A practical application of quantitative vascular image analysis in breast pathology. Pathol Res Pract. 2013;209:455–458. doi: 10.1016/j.prp.2013.03.004. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caudle WM, Richardson JR, Delea KC, et al. Polychlorinated biphenyl-induced reduction of dopamine transporter expression as a precursor to Parkinson's disease-associated dopamine toxicity. Toxicol Sci. 2006;92:490–499. doi: 10.1093/toxsci/kfl018. [DOI] [PubMed] [Google Scholar]
- Marazziti D, Mandillo S, Di Pietro C, et al. GPR37 associates with the dopamine transporter to modulate dopamine uptake and behavioral responses to dopaminergic drugs. Proc Natl Acad Sci USA. 2007;104:9846–9851. doi: 10.1073/pnas.0703368104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drenan RM, Grady SR, Steele AD, et al. Cholinergic modulation of locomotion and striatal dopamine release is mediated by alpha6alpha4* nicotinic acetylcholine receptors. J Neurosci. 2010;30:9877–9889. doi: 10.1523/JNEUROSCI.2056-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramonet D, Daher JP, Lin BM, et al. Dopaminergic neuronal loss, reduced neurite complexity and autophagic abnormalities in transgenic mice expressing G2019S mutant LRRK2. PLoS ONE. 2011;6:e18568. doi: 10.1371/journal.pone.0018568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Liu W, Oo TF, et al. Mutant LRRK2(R1441G) BAC transgenic mice recapitulate cardinal features of Parkinson's disease. Nat Neurosci. 2009;12:826–828. doi: 10.1038/nn.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betarbet R, Sherer TB, Greenamyre JT. Animal models of Parkinson's disease. BioEssays. 2002;24:308–318. doi: 10.1002/bies.10067. [DOI] [PubMed] [Google Scholar]
- Bezard E, Przedborski S. A tale on animal models of Parkinson's disease. Mov Disord. 2011;26:993–1002. doi: 10.1002/mds.23696. [DOI] [PubMed] [Google Scholar]
- Metzger RR, Brown JM, Sandoval V, et al. Inhibitory effect of reserpine on dopamine transporter function. Eur J Pharmacol. 2002;456:39–43. doi: 10.1016/s0014-2999(02)02647-x. [DOI] [PubMed] [Google Scholar]
- Berman SB, Zigmond MJ, Hastings TG. Modification of dopamine transporter function: effect of reactive oxygen species and dopamine. J Neurochem. 1996;67:593–600. doi: 10.1046/j.1471-4159.1996.67020593.x. [DOI] [PubMed] [Google Scholar]
- Cheng HC, Ulane CM, Burke RE. Clinical progression in Parkinson disease and the neurobiology of axons. Ann Neurol. 2010;67:715–725. doi: 10.1002/ana.21995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolam JP, Pissadaki EK. Living on the edge with too many mouths to feed: why dopamine neurons die. Mov Disord. 2012;27:1478–1483. doi: 10.1002/mds.25135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz-Schaeffer WJ. Neurodegeneration in Parkinson disease: moving Lewy bodies out of focus. Neurology. 2012;79:2298–2299. doi: 10.1212/WNL.0b013e318278b6a7. [DOI] [PubMed] [Google Scholar]
- Adams JR, van Netten H, Schulzer M, et al. PET in LRRK2 mutations: comparison to sporadic Parkinson's disease and evidence for presymptomatic compensation. Brain. 2005;128(Pt 12):2777–2785. doi: 10.1093/brain/awh607. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. LRRK2 protein structure and PD related R1441 mutations.
Figure S2. Gross morphology and body weight curve.
Figure S3 Young LRRK2R1441G mutant mice have similar amount of DA neuron and neurites compared to wild type.
Figure S4. Expression level of related proteins.