

Abstract
Objective
Congenital cytomegalovirus (CMV) infection is the leading viral cause of neurodevelopmental disorders in humans, with the most severe and permanent sequelae being those affecting the cerebrum. As the fetal immune reactions to congenital CMV infection in the brain and their effects on cerebral development remain elusive, our aim was to investigate primitive innate immunity to CMV infection and its effects on cerebral corticogenesis in a mouse model for congenital CMV infection using a precise intraplacental inoculation method.
Methods
At 13.5 embryonic days (E13.5), pregnant C57BL/6 mice were intraplacentally infected with murine CMV (MCMV). Placentas and fetal organs were collected at 1, 3, and 5 days postinfection and analyzed.
Results
MCMV antigens were found frequently in perivascular macrophages, and subsequently in neural stem/progenitor cells (NSPCs). With increased expression of inducible nitric oxide synthase and proinflammatory cytokines, activated macrophages infiltrated into the infectious foci. In addition to the infected area, the numbers of both meningeal macrophages and parenchymal microglia increased even in the uninfected areas of MCMV-infected brain due to recruitment of their precursors from other sites. A bromodeoxyuridine (BrdU) incorporation experiment demonstrated that MCMV infection globally disrupted the self-renewal of NSPCs. Furthermore, BrdU-labeled neurons, particularly Brn2+ neurons of upper layers II/III in the cortical plate, decreased in number significantly in the MCMV-infected E18.5 cerebrum.
Interpretation
Brain macrophages are crucial for innate immunity during MCMV infection in the fetal brain, while their aberrant recruitment and activation may adversely impact on the stemness of NSPCs, resulting in neurodevelopmental disorders.
Introduction
Intrauterine infection with cytomegalovirus (CMV) is the leading viral cause of developmental disorders of the central nervous system (CNS) in humans,1,2 and more frequently causes disabilities than do other well-known childhood disorders, such as Down syndrome.3 Although human CMV (HCMV) can infect multiple organs, the most severe and permanent sequelae after congenital infection are those affecting the cerebrum, such as microcephaly, epilepsy, and mental retardation.4–7 These sequelae are found more frequently in infants infected during the first trimester, a critical period for cerebral corticogenesis,8 than in those infected during the later trimesters.9 In cerebral corticogenesis, neural stem/progenitor cells (NSPCs) of the ventricular and subventricular zone (VZ and SVZ) generate the cerebral cortex through their “stemness”; that is, through their proliferation, migration, and differentiation.10 Therefore, congenital HCMV infection can be a teratogenic factor for cortical malformations that result in the abovementioned sequelae.
In terms of the direct teratogenic action of CMV, previous in vitro studies have clearly demonstrated that murine and human NSPCs infected with murine CMV (MCMV) and HCMV, respectively, lose their stemness.11–13 Although growing evidence indicates that inflammatory reactions triggered by infectious agents have serious impacts on immature developing brains and are possibly related to brain malformations and neurodevelopmental disorders,14–17 most of the evidence is based on studies using bacterial and viral mimics, such as lipopolysaccharide and polyinocinic-polycytidylic acid.14,17 In other words, fetal immune reactions to neuroinvasion with a live virus and the influence of the immune reactions on cerebral corticogenesis have not yet been well investigated.18
Although studies on human subjects have obvious limitations, and while CMV has strict species specificity, in vivo model systems using mouse embryos and neonates infected with MCMV have nevertheless shed light on the neuropathogenesis of congenital CMV infection.19,20 Previously, we developed a mouse model of intraplacental MCMV infection to simulate transplacental HCMV infection and to investigate CMV neuropathogenesis.21 In this model, neonatal mice infected with MCMV in utero exhibited growth retardation and microcephaly in a fashion similar to that observed in association with congenital CMV infection in humans. However, there have been no precise pathological or immunological analyses of the effects of congenital CMV infection on fetal brain development.
In this study, by using a fine glass micropipette, we improved the accuracy of the intraplacental inoculation with MCMV and made the inoculation process less invasive. The model allowed us to show the transmission of MCMV from placenta to fetal brain as well as the fetal innate immune responses, including macrophage/microglial reactions, and their impact on cerebral corticogenesis.
Materials and Methods
Ethical statement
This study was approved by the Animal Care and Use Committee of Hamamatsu University School of Medicine. All animal experiments were performed in accordance with the “Guidelines for Animal Experiments of Hamamatsu University School of Medicine.” All surgery was performed under anesthesia, and all efforts were made to minimize suffering.
Virus, cells, and plaque assay
The wild-type Smith strain of MCMV, MCMV-Smith, which had been passaged in mouse embryonic fibroblasts, was provided by Dr. Y Minamishima (Miyazaki, Japan). Construction of MCMV-EGFP, MCMV expressing enhanced green fluorescent protein (EGFP) under the control of the MCMV e1 promoter, was described previously.22 MCMV-EGFP and MCMV-Smith showed similar viral growth, latency, and pathogenesis.23 Mouse embryonic fibroblasts were prepared from 12.5-day-old embryos (E12.5) of BALB/c mice (SLC Japan, Hamamatsu, Japan), and were grown in Dulbecco's modified Eagle's essential medium (DMEM) containing penicillin (100 U/mL), streptomycin (50 μg/mL), and 10% fetal bovine serum. Viral titers were determined by a plaque assay using mouse embryonic fibroblasts monolayers as described previously.24 Ultraviolet light (UV)-inactivated MCMV that neither replicates nor produces any detectable level of the IE1 protein was used as a mock control.
Intraplacental MCMV infection
Inbred specific pathogen-free pregnant C57BL/6 female mice were purchased from SLC Japan. All mice were housed under specific pathogen-free conditions. Pregnant mice were randomly divided into two groups: the MCMV- and mock-infected groups. MCMV inoculation to the placenta was performed via a fine glass micropipette with a diameter of 40 μm (Fig. 1A), which was made from a borosilicate glass capillary (No. 30-0019, 1.0 mm O.D. × 0.58 mm I.D., Harvard Apparatus, Holliston, MA) using a micropipette puller (Model P-97; Sutter Instrument Company, Novato, CA), as described for transient delivery of serotonin into the placenta.25 In the MCMV-infected group, 1 × 105 plaque-forming unit (PFU) of MCMV-Smith or MCMV-EGFP in 1 μL of DMEM was injected through the uterine wall into the labyrinth region in the placenta. In the mock-infected group, UV-inactivated MCMV virus in 1 μL of DMEM was injected into the placenta in the same way. All surgical procedures were performed under anesthesia induced by an intraperitoneal injection of 0.2–0.3 mL of 6.5 mg/mL sodium pentobarbital. Fetuses were surgically collected at the indicated number of days postinfection (dpi), and the number of live fetuses and dead fetuses were counted, followed by the weighing of the fetuses, placentas, and brains. The placenta, brain, heart, and liver were stored at −80°C and used for plaque assays, real-time polymerase chain reaction (PCR), and real-time reverse transcriptase PCR (RT-PCR). For immunohistochemical analysis and in situ hybridization, the brains were fixed by perfusion with 4% paraformaldehyde (PFA), removed, immersed in 20% sucrose, and quickly frozen in liquid nitrogen. Coronal sections were cut with a cryostat at 12 μm, air dried, and then stored at −80°C. Some of the infected brains were frozen in liquid nitrogen without fixation with PFA, serially cut, then fixed in isopropyl alcohol, and stored at −80°C. For the labeling of proliferating cells, a number of pregnant mice were intraperitoneally injected with bromodeoxyuridine (BrdU, 50 mg/kg; Sigma-Aldrich, St. Louis, MO).
Figure 1.

Intraplacental MCMV infection and development of MCMV-infected fetuses. (A) Inoculation procedure during surgery. The arrowhead and arrow indicate the glass micropipette for injection and the labyrinth region in the placenta, respectively. (B) Survival rates of fetuses infected with MCMV at E13.5 and infection rates of their brains. (C) Fluorescence stereomicroscopic views of MCMV-EGFP infectious foci at 3 dpi in a fetus, its brain, and placenta. A magnified view of an infectious focus in the fetus is also shown in the inner panel. The fluorescence images of brain and placenta were superimposed on their bright field images. The arrowhead in the image of the placenta indicates the injection site. Fetal body, scale bar = 2 mm. Brain and placenta, scale bar = 1 mm. (D) Weights of the fetal body, brain, and placenta at E18.5 (5 dpi). Mean ± standard error of mean (SEM) are shown. *P < 0.05 compared with mock-infected mice. MCMV, murine cytomegalovirus; EGFP, enhanced green fluorescent protein.
Real-time PCR for quantification of the MCMV copy number
DNA was extracted from tissue homogenates with Trizol (Invitrogen, Carlsbad, CA) according to the manufacturer's protocol. All real-time PCRs were performed using TaqMan Gene Expression Master Mix in an ABI7500 Fast System using the standard curve (absolute quantitation) assay (Applied Biosystems, Foster City, CA). One hundred ng of DNA was added as template DNA to a reaction mixture that contained 2× TaqMan Reaction Mixture, 900 nmol/L primers, and 250 nmol/L TaqMan MGB probe. The MCMV genome was quantified by amplification of the major immediate-early promoter (MIEP) gene using the primers 5′-GGTGGTCAGACCGAAGACT and 5′-GCTGAGCTGCGTTCTACGT, and the probe 5′-FAM-CTGGTCGCGCCTCTTA.26 As a cellular gene, the β-actin gene was quantified using primers 5′-CGTTCCGAAAGTTGCCTTTTA, 5-′GCCGCCGGGTTTTATAGG, and the probe 5′-FAM-CTCGAGTGGCCGCTG. The thermal cycling conditions were one cycle at 50°C for 2 min and at 95°C for 10 min followed by 50 cycles of 95°C for 15 sec and 60°C for 1 min. Serial dilutions of pSM3fr bacterial artificial chromosome (BAC) containing the MCMV whole genome, which was kindly provided by Dr. UH Koszinowski (Munich, Germany),27 were used as the quantification standard for MCMV.
Immunohistochemistry
Mouse monoclonal antibodies (mAbs) specific for the MCMV-IE1 nuclear antigen (Ag) (1:2000, kindly provided by Dr. S Jonjic, Rijeka, Croatia)28 and the MCMV-E1 nuclear Ag (1:5)29 were used for detection of MCMV-infected cells.
For immunoperoxidase staining, after microwave treatment, fetal brain sections were reacted with anti-IE1 mAb and then with horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG polyclonal antibody (pAb) (N-Histofine simple stain mouse MAX PO; Nichirei Bioscience, Tokyo, Japan), and colored with 3,3-diaminobenzidine tetrahydrochloride (DAB).
For immunofluorescence staining, in addition to mAb for MCMV-Ag, the following primary antibodies were used: inducible nitric oxide synthase (iNOS, rabbit pAb, 1:100; Abcam, Cambridge, UK), BrdU (rat mAb, 1:200; Abcam), Brn2 (goat pAb, 1:100; Santa Cruz, Dallas, TX), Tbr1 (rabbit pAb, 1:100; Chemicon, Billerica, MA), Iba-1 (rabbit pAb, 1:500, Wako, Osaka, Japan), SOX2 (rabbit pAb, 1:200; MBL, Nagoya, Japan), F4/80 (rat mAb, 1:20; AbD Serotec, Oxford, UK), CD45 (rabbit pAb, 1:100; Abcam), major histocopatibility complex (MHC) class II (rat mAb, 1:100; BioLegend, San Diego, CA), CD31 (rabbit pAb, 1:50; Abcam), and NG2 (rabbit pAb, 1:200; Millipore, Billerica, MA). The following secondary antibodies were also used: Alexa Fluor 488 anti-mouse IgG1 or IgG2a, Alexa Fluor 546 anti-rabbit, -rat, or -goat IgG, or Alexa Fluor 647 anti-rabbit IgG (Invitrogen). Nuclei were stained with 4′,6-diamidino-2-phenylindole, dihydrochloride (DAPI) (Sigma-Aldrich). For BrdU staining, sections were incubated in 2 N HCl followed by 0.1 mol/L sodium borate after microwave treatment.
Confocal images were captured using an Olympus FV1000 microscope with the latest Fluoview software Ver. 3.1 (Olympus, Tokyo, Japan). A stack of five images from 0.5-μm step slices was combined for the analysis. At least three different animals in each group were used for the analysis, and sections from three fields in each brain were evaluated.
In situ hybridization for MCMV DNA detection
Using the PFA-fixed sections adjacent to the sections used for immunostaining, chromogenic in situ hybridization was performed for the detection of MCMV DNA as described previously.30 The DNA probe was made from pSM3fr BAC labeled with digoxigenin (DIG)-11-dUTP using a nick translation kit (Roche, Mannheim, Germany). After hybridization, the sections were subsequently incubated with HRP-conjugated anti-DIG Fab fragments (1:100; Roche) and colored with DAB, followed by hematoxylin counterstaining.
Flow cytometry
At 3 and 5 dpi, mock- and MCMV-EGFP-infected fetuses were perfused through the left cardiac ventricle with 5 mL of ice-cold phosphate-buffered saline (PBS), containing 2 U/mL heparin. Each cerebrum with meninges and the choroid plexus was minced into small fragments and then incubated in the presence of 25 U/mL DNase I and 0.01% collagenase II at 37°C for 30 min. The cells were filtered through a nylon screen (70 μm; BD Bioscience, San Diego, CA) to remove cell aggregates, centrifuged, and then twice washed with RPMI 1640 medium. The cells were incubated with antibodies against CD45-APC (1:100), CD11b-PE/Cy7 (1:100), NK1.1-APC/Cy7 (1:20), Ly-6G/Ly-6C (Gr1)-PacificBlue (1:50), Cd45R/B220-PerCP/Cy5.5 (1:100), and CD3-Alexa Flour 700 (1:100) (BioLegend) for 30 min. After collection, rinsing with RPMI 1640 and filtration through a nylon screen (40 μm), the cells were sorted and analyzed on a FACSAria system (BD Bioscience). Isotype antibodies were used as controls.
To sort the mononuclear cells from each cerebrum with meninges, cells were stained with a combination of CD45-APC and CD11b-PE/Cy7, and then separated into CD45high/CD11b+ (isolated cell numbers: 10,835 ± 5593 cells in the mock-infected brain at 5 dpi, n = 6; 15,000 ± 0 cells in the MCMV-infected brain at 5 dpi, n = 4), CD45low/CD11b+ (14,314 ± 6408 cells in the mock-infected brain at 5 dpi, n = 6; 15,520 ± 1804 cells in the MCMV-infected brain at 5 dpi, n = 4), CD45high/CD11b+ (1235 ± 286.0 cells in the mock-infected brain at 5 dpi, n = 6; 1541 ± 234 cells in the MCMV-infected brain at 5 dpi, n = 4), and CD45low/CD11b+ populations (cell sorting was stopped at 100,000 cells in both the mock- and MCMV-infected brains). The sorted cellular populations were stored at −80°C in Trizol and used later for RNA isolation.
Real-time RT-PCR for quantification of mRNA expression of inflammatory mediators
Total RNA was isolated from the frozen samples, purified with an RNeasy Mini Kit (Qiagen, Hilden, Germany) and treated with RNase-free DNase I (Qiagen), according to the manufacturer's instructions. One hundred nanograms of cDNA synthesized using a SuperScript III First Strand Synthesis kit with random primers (Invitrogen) was used for each PCR reaction. TaqMan assay mixtures for IL-1β (Mm00434228), INF-α4 (Mm00833969), INF-β1 (Mm00439552), IL-10 (Mm00439614), and monocyte chemotactic protein-1 (MCP-1)/chemokine (C-C motif) ligand 2 (CCL2) (Mm00441242) were obtained from Applied Biosystems. The following primer pairs and probes were synthesized commercially (Sigma-Aldrich): iNOS, 5′-CAGCTGGGCTGTACAAACCTT, 5′-CATTGGAAGTGAAGCGTTTCG, and 5′-FAM-CGGGCAGCCTGTGAGACCTTTGA; IL-1β, 5′-CAACCAACAAGTGATATTCTCCATG, 5′-GATCCACACTCTCCAGCTGCA, and 5′-FAM-CTGTGTAATGAAAGACGGCACACCCACC; TNF-α, 5′-CATCTTCTCAAAATTCGAGTGCAA, 5′-TGGGAGTAGACAAGGTACAACCC, 5′-FAM-CACGTCGTAGCAAACCACCAAGTGGA; IL-6, 5′-GAGGATACCACTCCCAACAGACC, 5′-AAGTGCATCATCGTTGTTCATACA, and 5′-FAM-CAGAATTGCCATTGCACAACTCTTTTCTCA; INF-γ, 5′-TCAAGTGGCATAGATGTGGAAGAA, 5′-TGGCTCTGCAGGATTTTCATG, and 5′-FAM-TCACCATCCTTTTGCCAGTTCCTCCAG; glyceraldehyde-3-phosphate dehydrogenase (GAPDH), 5′-TCACCACCATGGAGAAGGC, 5′-GCTAAGCAGTTGGTGGTGCA, and 5′-FAM-ATGCCCCCATGTTTGTGATGGGTGT.
Real-time PCR was performed using the StepOne Plus System (Applied Biosystems). The GAPDH housekeeping gene was used as a control for all experiments. The GAPDH expression level from the mock-infected animals was taken as 1 and the gene expression levels of the target genes were expressed as relative values.
Statistical analysis
Numerical results were expressed as the mean ± standard error of mean (SEM) obtained from at least three independent experiments. Statistical analysis was performed using an unpaired two-tailed Student's t-test. Probability values (P) <0.05 were considered to be significant.
Results
Establishment of a congenital infection model using intraplacental MCMV inoculation
As described in the Materials and Methods section, all pregnant C57BL/6 mice were treated surgically and intraplacentally infected with 1 × 105 PFU of MCMV, mock infected, or inoculated with DMEM using a fine glass micropipette (Fig. 1A). In the first set of mice, inoculation with MCMV-Smith was performed at E11.5, E12.5, and E13.5, and the fetuses were collected at 3 dpi. The survival rates of the fetuses in dams infected with MCMV-Smith were 25% (2/8), 35% (13/37), and 67% (10/15) at E11.5, E12.5, and E13.5, respectively. Those in dams infected with UV-inactivated virus (mock infected) were 50% (10/20), 63% (10/16), and 89% (8/9) at E11.5, E12.5, and E13.5, respectively. Similar results were obtained using dams that had undergone a similar surgical procedure but were inoculated with DMEM. Therefore, it is likely that fetuses at E11.5 and E12.5 do not readily develop in utero after surgery, and fetuses at E13.5 were used for the subsequent studies.
Next, pregnant mice were infected intraplacentally at E13.5 as described above, and the fetuses were collected at 1, 3, and 5 dpi. Although the survival rates of the fetuses gradually decreased, the infection rates (the detection limit was 1 viral DNA copy/million cells) in the brains were 30–40% (Fig. 1B). Amplification by PCR did not reveal any MCMV genomic DNA in the placentas or brains of the mock-infected fetuses.
Pregnant mice were also infected intraplacentally with MCMV-EGFP, MCMV expressing EGFP during the viral replicative cycle, as we previously demonstrated that MCMV-EGFP grows in the same way as MCMV-Smith.23 Fluorescence stereomicroscopic views of the placentas and fetuses at 3 dpi demonstrated multiple infectious foci, consisting of infected cells in a “green mirror ball” arrangement, in the fetal body surface, fetal brain, and placenta (Fig. 1C). Although the body weight of the MCMV-EGFP-infected fetuses at 5 dpi was 15% less than that of mock-infected fetuses (Fig. 1D), both mock- and MCMV-infected mice had no macroscopic morphological abnormalities and no significant differences in the weight of the placenta and brain were observed at any time points.
Dynamics of MCMV infection and distribution of MCMV-infected cells in the fetal cerebrum
The viral titers of MCMV-Smith in the placenta peaked at 3 dpi, while the titers in the other organs increased until 5 dpi (Fig. 2A). The viral DNA copy numbers in all organs increased in parallel (Fig. 2B). Viral loads in the placenta, liver, and heart exceeded those in the brain, suggesting that MCMV replication in the placenta and peripheral organs preceded viral spread to the brain. As there were no substantial differences in the viral titer kinetics and DNA copy numbers between the wild-type MCMV-Smith and MCMV-EGFP even in this study (data not shown), all further experiments were performed using MCMV-EGFP.
Figure 2.

Infectious dynamics and intracerebral distribution of MCMV in the fetus. Viral loads in the brain, placenta, heart, and liver determined by infectious viral titers (A) and DNA copy numbers (B). The dotted line in (A) indicates the detection limit. Mean ± SEM of five mice in each group are shown. (C) Distribution of MCMV-IE1 Ag+ cells in the fetal cerebrum at 3 and 5 dpi. The number of MCMV-IE1 Ag+ cells in each anatomical region was counted for 10 coronal sections sequentially cut at intervals of 240 μm. Mean ± SEM in each anatomical region of six MCMV-infected fetal cerebra are shown. (D) A representative coronal section of MCMV-infected cerebrum at 3 dpi (left panel, scale bar = 0.5 mm) and its high-power views (middle panel, scale bar = 50 μm). Arrowheads indicate MCMV-IE1 Ag+ infectious foci. Detection of MCMV DNA in serial sections by in situ hybridization (ISH) is shown in the right panels. MCMV, murine cytomegalovirus.
MCMV-antigen (Ag)+ cells in the serial sections prepared from more than 30 MCMV-infected fetal cerebra at each time point were detected using monoclonal antibodies (mAbs) against the MCMV IE1 nuclear protein, the antigen expressed immediately after viral infection, and against the E1 protein, the antigen expressed only during the viral productive phase. As immunohistological staining with both antibodies yielded the same images of MCMV-infected cells in the fetal cerebra, we describe these as “MCMV-Ag+ cells” hereafter. MCMV-Ag+ cells were found most frequently in the meninges, and then focally in the periventricular area surrounding the lateral ventricles, the third ventricle, and the cerebral aqueduct of the cerebral parenchyma (Fig. 2C and D). There were no significant differences in the numbers or distribution of the MCMV-Ag+ cells in the brains at 3 and at 5 dpi (Fig. 2C). Signals obtained in the in situ hybridization assay for detection of replicated viral DNA corresponded to MCMV-Ag+ cells identified in the adjacent section (Fig. 2D, right panel). Thus, congenital infection in our MCMV model resulted in replicative infection in the fetal cerebrum.
MCMV invades the fetal brain via the bloodstream and targets macrophages and NSPCs in the cerebrum
The type of MCMV-infected cells in the fetal cerebrum was determined by immunohistochemical analysis for MCMV-Ag and various cell markers. In the meninges (Fig. 3A–D), MCMV-Ag+ cells were distributed along the CD31+ blood vessels and involved CD31+ endothelial cells in some cases (Fig. 3A, arrow head), while over half of the MCMV-Ag+ cells were F4/80+ macrophages (Fig. 3B, arrow head). F4/80+ macrophages existed in the infectious foci twinning around CD31+ endothelial cells (Fig. 3C). In the brain parenchyma (Fig. 3E–P), MCMV-Ag+ cells were detected along the CD31+ blood vessels penetrating into the brain parenchyma from the lateral ventricle (LV) (Fig. 3E). F4/80+ macrophages infiltrated into the infectious foci (Fig. 3F), and existed alongside the CD31+ blood vessels (Fig. 3G). In addition to F4/80+ macrophages, CD31+ endothelial cells (Fig. 3I, arrow head) and NG2+ perivascular cells (pericytes) (Fig. 3M, arrow head) were occasionally infected with MCMV. Regardless of the number of MCMV-infected cells in the meninges, MCMV-Ag was detected only occasionally in the parenchyma just below the infected meninges. These results indicate that MCMV invasion into the parenchyma occurs via a transvascular process, not as the result of direct invasion from the meninges.
Figure 3.
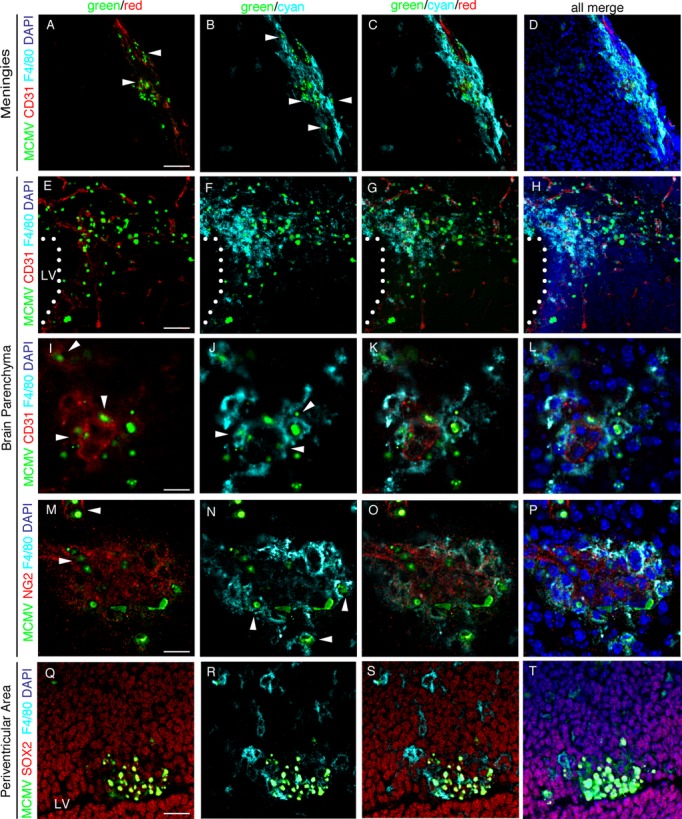
Identification of MCMV-Ag+ cell types in MCMV-infected fetal cerebrum. Detection of MCMV-E1 Ag (green) and various cell markers in the infectious foci in the sections of MCMV-infected fetal cerebrum at 3 dpi by immunofluorescence staining using primary antibodies against macrophage/microglia (F4/80, in cyan), endothelial cells (CD31, in red), perivascular cells (NG2, in red), and NSPCs (SOX2, in red), respectively. Nuclei were stained with DAPI (in blue). Meninges (A–D), parenchyma (E–P), and the VZ/SVZ of the periventricular area (Q–T) stained with the indicated antibodies are shown. Arrowheads in (A) and (I), those in (B), (J), and (N), and those in (M) indicate endothelial cells, macrophages, and perivascular cells, respectively. Dotted lines in (E–H) indicate the interface between the lateral ventricle (LV) and parenchyma. Scale bars = 50 μm (A–D, Q–T), 100 μm (E–H), and 20 μm (I–P). MCMV, murine cytomegalovirus; NSPC, neural stem/progenitor cells; VZ, ventricular zone; SVZ, subventricular zone.
In the infectious foci of the cerebral periventricular area (Fig. 3Q–T) and periaqueductal gray matter, most of MCMV-Ag+ cells were SOX-2+ NSPCs (Fig. 3Q). Uninfected F4/80+ macrophages crowded around the infectious foci of the NSPCs (Fig. 3R), and infiltrated into the foci (Fig. 3S). The infiltration of uninfected F4/80+ macrophages was found at all infectious foci, and they outnumbered the MCMV-infected macrophages.
Thus, in the fetal cerebrum, the major target cells allowing MCMV replication are macrophages and NSPCs.
MCMV infection induces macrophage infiltration and aberrant distribution of microglia in the fetal cerebrum
As we observed infiltration of uninfected macrophages into the infectious foci, cell numbers and distributions of Iba1+ brain macrophages were compared between the fetal cerebra obtained from mock- and MCMV-infected dams. Iba1+ brain macrophages, including microglia, increased in the cerebra at both E16.5 (3 dpi) (Fig. 4A) and at E18.5 (5 dpi) (Fig. 4B) in the infected fetuses. These increases in Iba1+ macrophages were observed predominantly not only around the infectious foci in the meninges and parenchyma (Fig. 4A – arrow head – and C), but also in the uninfected area of the MCMV-infected cerebrum. At 5 dpi (E18.5), the number of Iba1+ cells in the uninfected area were significantly increased in the meninges, cerebral cortex, and hippocampus in MCMV-infected cerebra compared with the mock-infected cerebra (Fig. 4D). Interestingly, in the MCMV-infected cerebra, microglia without viral infection aberrantly appeared in the cortical plate (CP) (Fig. 4B, upper panel), where microglia are usually absent during the fetal period in the mice brain.31
Figure 4.

Increase in the number of brain macrophages in MCMV-infected fetal cerebrum. (A) Representative corresponding coronal sections in mock- (left) and MCMV-infected (right) E16.5 (3 dpi) fetal cerebra. The sections were stained doubly with mAbs against MCMV-E1 Ag (green) and Iba-1 (red). Arrowheads indicate MCMV infectious foci. Scale bar = 0.5 mm. (B) Representative corresponding coronal sections of uninfected cerebral cortex in mock- (left) and MCMV-infected (right) E18.5 (5 dpi) fetal cerebra. Mock- or MCMV-infected pregnant mice were pulsed intraperitoneally with BrdU at E18.5 and the brains of their fetus were removed 3 h later. The sections were stained doubly with mAbs against Iba-1 (red) and BrdU (cyan). Arrowheads indicate Iba-1 and BrdU double-positive cells. Arrows indicate autofluorescence of red blood cells. Scale bar = 50 μm. (C) The numbers of Iba1+ brain macrophages in 0.4 mm2 areas of the cerebrum which contained one infectious focus and those in the corresponding cerebral areas of mock-infected fetuses at E16.5 (3 dpi). Mean ± SEM of three mice under each set of conditions is shown. *P < 0.05 compared with mock-infected mice. (D) The number of Iba1+ meningeal macrophages and microglia cells in uninfected cerebral areas in MCMV-infected E18.5 (5 dpi) fetuses. The numbers of Iba1+ cells were also counted in the corresponding anatomical areas of mock-infected cerebra. Mean ± SEM of three mice are shown. *P < 0.05 compared with mock-infected mice. (E) The numbers of Iba-1 and BrdU double-positive cells in the uninfected areas of MCMV-infected E18.5 (5 dpi) fetal cerebra. The numbers of Iba1 and BrdU double-positive cells were counted in the corresponding anatomical areas in mock-infected cerebra. Mean ± SEM of three mice are shown. *P < 0.05 compared with mock-infected mice. MCMV, murine cytomegalovirus.
Aberrant increase in macrophages/microglia is not due to their in situ proliferation
To determine whether the aberrant increase in uninfected Iba-1+ brain macrophages, including microglia, in the uninfected areas was due to in situ proliferation or recruitment from other sites, a BrdU incorporation assay for proliferating cells was performed at 5 dpi (Fig. 4B). Although MCMV infection induced a significant increase in the number of fetal brain macrophages in the uninfected areas of the brain except for the marginal zone (MZ) and intermediate zone (IZ) (Fig. 4D), the numbers of BrdU-labeled proliferating macrophages in the uninfected areas of the MCMV-infected fetuses were significantly reduced in comparison with those in the mock-infected fetuses (Fig. 4E). Therefore, the aberrant increase in macrophages in the MCMV-infected animals is thought not to be due to cell proliferation, but is likely the result of the recruitment of macrophages from other sites.
MCMV infection increased the number of macrophages and microglia in the fetal cerebrum
To characterize the types of cells showing increased numbers, a flow cytometric analysis based on the expression levels of CD45 and CD11b were performed.32 CD11b+ populations were divided into three fractions, namely, CD45high/CD11b+, CD45int/CD11b+, and CD45low/CD11b+ populations, which correspond to the monocyte/macrophage, intermediate microglia and microglia populations, respectively (Fig. 5A and B). CD11b+ cells in the MCMV-infected cerebra both at 3 dpi and 5 dpi were significantly increased by ∼1.6 times those in the mock-infected cerebra, and accounted for ∼3% of the total number of cerebral cells. The monocyte/macrophage population in the MCMV-infected cerebra was increased by nearly twice of that in the mock-infected ones. The microglia population in the infected cerebra was also increased by ∼1.5 times. In both mock- and MCMV-infected cerebra, the percentages of microglia were larger at 3 dpi, but smaller at 5 dpi, than those of monocyte/macrophages. Intermediate microglia accounted for only ∼6% of the CD11b+ population, and no significant difference was observed between the populations in mock- and MCMV-infected cerebra. The proportions of the other CD45high leukocytes, including natural killer (NK) cells, T cells, B cells, and granulocytes, in the all cerebral cells were very small in comparison with the monocyte/macrophage and microglia populations and again no significant differences were observed in their populations between the mock- and MCMV-infected cerebra at 3 dpi (Fig. 5C). In addition, ∼90% of the monocyte/macrophage population in both mock- and MCMV-infected cerebra clearly expressed both F4/80 and Iba-1 antigens (data not shown). This suggests that the monocyte proportion of the monocyte/macrophage population was very low as they usually express CD11b but not Iba-1.33 Thus, the flow cytometric data are consistent with the histological findings showing the MCMV-induced dramatic and aberrant increases in brain macrophages, including microglia.
Figure 5.
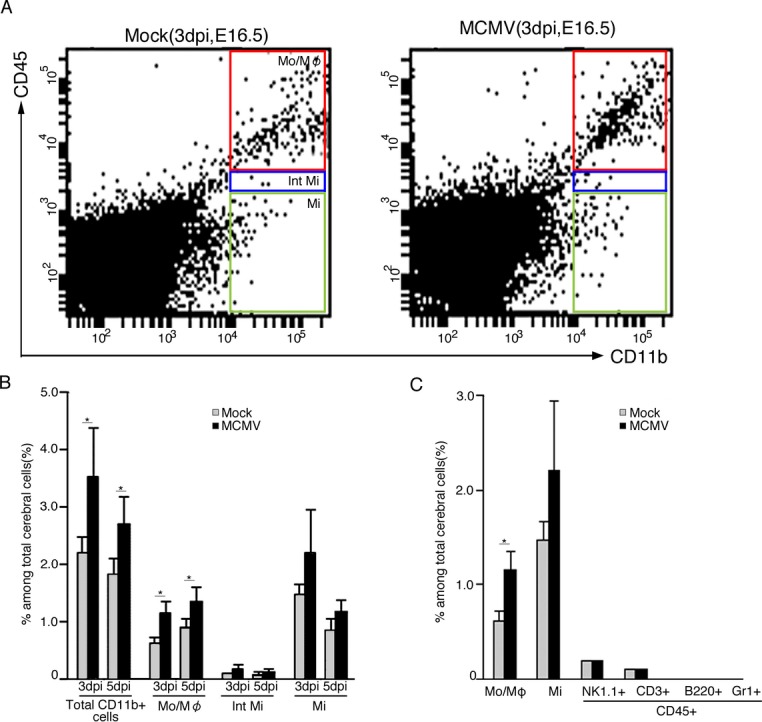
Flow cytometric analysis of cells in the fetal cerebra with meninges of mock- and MCMV-infected fetuses at E16.5 (3 dpi) and E18.5 (5 dpi). Total leukocytes were gated for CD45 expression and further characterized for CD11b, NK1.1 (NK cells), CD3 (T cells), B220 (B cells), and Gr1 (neutrophils). The analysis was performed in triplicate using a total of nine mock- and four MCMV-infected fetuses at each time point. (A) Representative flow cytometric plots separating monocyte/macrophage (CD45high/CD11b+, Mo/Mφ), intermediate microglia (CD45int/CD11b+, Int Mi), and microglia (CD45low/CD11b+, Mi) populations of mock- (left panel) or MCMV-infected (right panel) cerebra at E16.5 (3 dpi). (B) The percentages of total CD11b+ cells, Mo/Mφ, Int Mi, and Mi among the total cerebral cells of mock-infected or MCMV-infected fetuses at E16.5 (3 dpi) and E18.5 (5 dpi). Mean ± SEM of nine mock- and four MCMV-infected fetuses at E16.5 (3 dpi) and E18.5 (5 dpi) are shown. *P < 0.05 compared with mock-infected mice. (C) The percentages of Mo/Mφ, Mi, NK cells (CD45high/NK1.1high, NK1.1+), T cells (CD45high/CD3high, CD3+), B cells (CD45high/B220high, B220+), and neutrophils (CD45high/Gr1high, Gr1+) among the total cerebral cells of mock- or MCMV-infected fetuses at E16.5 (3 dpi). Mean ± SEM of nine mock- or four MCMV-infected fetuses. *P < 0.05 compared with mock-infected mice. MCMV, murine cytomegalovirus.
Localization of macrophages and microglia in MCMV-infected fetal cerebrum
The flow cytometric data led us to analyze the localization of brain macrophages by immunofluorescence staining of cerebral sections with mAbs against CD45 and F4/80. In the MCMV-infected cerebra, CD45- and F4/80-positive cells were found throughout the meninges (Fig. 6A–D: uninfected area and Fig. 6E–H: MCMV-infected area), while these cells were exclusively found in the infectious foci in the parenchyma (Fig. 6M–P). In both the meninges and parenchyma, the fluorescence intensity of CD45 was prominent in the infectious foci. Furthermore, CD45- and F4/80-positive cells in the infectious foci expressed the macrophage activation markers MHC class II (Fig. 6I–L and Q–T) and iNOS (Fig. 6U–X). The CD45high/CD11b+ population shown in the flow cytometry was supposed to be comprised of activated macrophages in the infectious foci and meningeal macrophages. F4/80+ microglia, localizing outside the infectious foci, increased in number but did not express the markers for macrophage activation, CD45 (Fig. 6M–P) or MHC class II (Fig. 6Q–T). These results are consistent in support of the fact that MCMV infection induces the infiltration of activated macrophages into the infectious foci and the aberrant recruitment of microglia in the uninfected parenchyma of the fetal cerebra.
Figure 6.
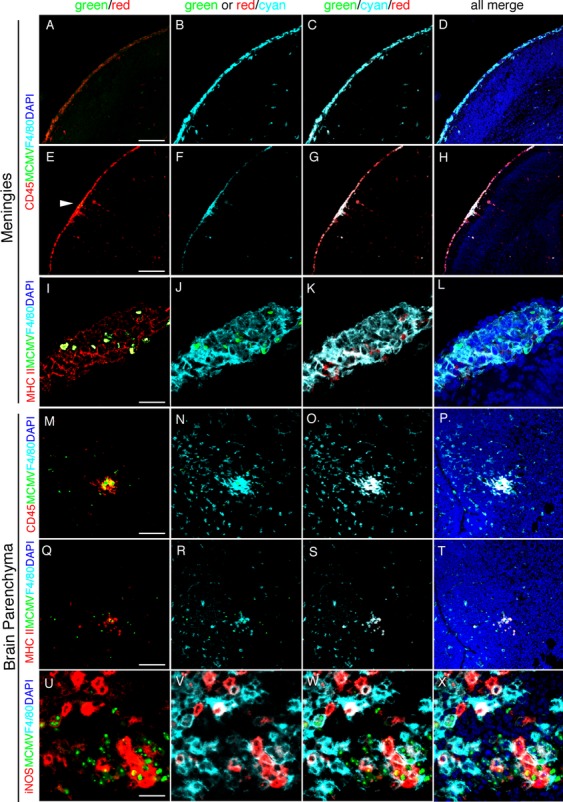
The localization of activated brain macrophages and microglia in MCMV-infected fetal cerebrum. Detection of MCMV-E1 Ag (green), F4/80 (cyan) and an indicator for macrophage activation (red), CD45, MHC class II, or iNOS, by immunofluorescence staining of sections of MCMV-infected fetal cerebrum at 5 dpi. Nuclei were stained with DAPI (blue). (A–D) The uninfected area of the meninges in the MCMV-infected brain stained with CD45 and F4/80. CD45+/F4/80+ cells exist throughout the meninges. (E–H) The area including MCMV infectious focus of the meninges stained with MCMV-E1 Ag, CD45, and F4/80. The arrowhead in (E) indicates MCMV-infected cells. CD45+/F4/80+ cells exist throughout the meninges, locating around the infectious foci. (I–L) An MCMV infectious focus of the meninges stained with MCMV-E1 Ag, MHC class II, and F4/80. MCMV-infected cells are surrounded by MHC class II+/F4/80+ activated macrophages. (M–P) An MCMV infectious focus in brain parenchyma stained with MCMV-E1 Ag, CD45, and F4/80. CD45+/F4/80+ activated macrophages infiltrate into the infectious foci. (Q–T) The serial section, adjacent to the section used for M–P, stained with MCMV-E1 Ag, MHC class II, and F4/80. MHC class II+/F4/80+ activated macrophages infiltrate into the infectious foci. F4/80+ microglia surrounding an infectious focus express neither CD45 nor MHC class II. (U–X) An MCMV infectious focus in the parenchyma stained with MCMV-E1 Ag, iNOS, and F4/80. MCMV-infected cells are surrounded by iNOS+/F4/80+ activated macrophages. Scale bar = 100 μm (A–H), 50 μm (M–T) and 20 μm (I–L and U–X). MCMV, murine cytomegalovirus; iNOS, inducible nitric oxide synthase.
Effects of MCMV infection on the stemness of NSPCs and cerebral corticogenesis
To investigate the impact of MCMV infection on NSPC proliferation, which reflects their ability to self-renew as a primary aspect of stemness, nuclear BrdU incorporation into SOX2+ NSPCs in the VZ/SVZ was analyzed. Infected pregnant mice were pulsed with BrdU at 3 or 5 dpi and the fetal brains were removed 3 h later. MCMV-infected NSPCs in the VZ/SVZ did not incorporate BrdU at all (Fig. 7A). Furthermore, BrdU incorporation into NSPCs aside from the infectious foci was also decreased (Fig. 7B and C). These data indicate that MCMV infection impairs NSPCs proliferation via not only direct viral cytopathicity but also indirect pathways.
Figure 7.

MCMV infection inhibits the proliferation of NSPCs via both direct and indirect mechanisms. (A–C) Mock- and MCMV-infected pregnant mice were injected with BrdU at 3 and 5 dpi, and the brains of their fetuses were removed at 3 h after the injection. (A) Detection of MCMV-IE1 Ag (green), BrdU (red), and SOX2 (magenta) by immunofluorescence staining of section of the MCMV-infected fetal cerebrum at 3 dpi. Nuclei were stained with DAPI (blue). At 3 dpi, BrdU incorporation is completely inhibited in MCMV-infected NSPCs of the VZ/SVZ between dotted lines. Scale bar = 20 μm. (B) The number of BrdU+ NSPCs adjacent to MCMV infectious focus in the MCMV-infected cerebrum (right) decreased in comparison with that in the mock-infected cerebrum (left) at 5 dpi (E18.5). Scale bar = 50 μm. (C) The number of BrdU+ cells in the uninfected areas of the VZ/SVZ in the mock- or MCMV-infected cerebrum at 3 or 5 dpi. Data are expressed as mean ± SEM of three fetuses. *P < 0.05 compared with mock-infected fetuses. (D–E) Mock- and MCMV-infected pregnant mice were injected with BrdU at 24 h after infection and then the fetuses were removed at 96 h after BrdU injection (E18.5, 5 dpi). (D) The number of BrdU+ cells in the cortical plate of the MCMV-infected cerebrum (right) decreased in comparison with the mock-infected cerebrum (left). Scale bar = 50 μm. (E) The number of BrdU+ cells in the upper layer or the lower layer of the cortical plate in the mock-infected or MCMV-infected cerebrum at 5 dpi. Data are expressed as mean ± SEM of three fetuses. *P < 0.05 compared with mock-infected fetuses. MCMV, murine cytomegalovirus; NSPC, neural stem/progenitor cells; VZ, ventricular zone; SVZ, subventricular zone.
We next examined how the impairment of the stemness of NSPCs in the VZ/SVZ influences cortical development. According to the widely accepted model of neocortical development, the upper cortical layers, unique to the mammalian brain, are derived from cortical precursor cells generated in the VZ during the period between E14 and E18.34 We injected BrdU into MCMV-infected pregnant mice at 1 dpi (E14.5) and then collected fetal brains at 5 dpi (E18.5). MCMV infection reduced the number of BrdU-labeled cells arriving at the upper layers of the CP in the E18.5 fetal brains, suggesting MCMV infection disturbs the development of the cortical upper layers (Fig. 7D and E).
To exclude the possibility that the reduction in BrdU-labeled cells was due to circulatory disturbance, we analyzed expression of the T-box brain 1 (Tbr1) transcription factor protein in the cerebral cortex, which is involved in the migration and differentiation of neurons, specifically within layer VI of developing six-layered cortex.35 There were no significant differences in the number of Tbr1+ neurons destined for layer VI between the mock- and MCMV-infected cerebra at 5 dpi (Fig. 8A and B), suggesting that MCMV infection after E13.5 produces little effect of the development of the lower layers. In contrast, the number of Brn2+ neuronal cells in the cerebral cortex, especially in the upper CP, was reduced in the MCMV-infected brain (Fig. 8A and B). Brn2, POU-homeodomain transcription factor (Pou3f2), is required for the production of the neural cells in layers II/III, as they migrate to the proper position from their departure point in the VZ.36,37 These data indicate that MCMV infection impairs the onset of Brn2 expression in NSPCs, resulting in a reduction in the number of neurons destined for the upper cortical layers. Collectively, MCMV infection impairs the stemness of NSPCs via broad and indirect mechanisms, resulting in disordered cerebral corticogenesis.
Figure 8.

MCMV infection impairs cerebral corticogenesis in the upper layer rather than the lower layer. Detection of Tbr1 (green) and Brn2 (red), as lower and upper layer markers, respectively, by immunofluorescence staining of sections of MCMV-infected fetal cerebra at 5 dpi. Nuclei were stained with DAPI (blue). (A) The localization of Tbr1+ or Brn2+ cells in a representative cerebral section of E18.5 fetus infected with mock virus (upper panel) or MCMV (lower panel). Despite no differences in the number of Tbr1+ neurons in the lower layer between the mock- and MCMV-infected cerebra, the number of Brn2+ neurons in the upper layer in the MCMV-infected cerebrum decreased. Scale bar = 100 μm. (B) The numbers of Tbr1+ and Brn2+ neurons in the upper and lower layers in a 1.6 mm2 area of MCMV-infected cerebrum and in the corresponding area of mock-infected cerebrum. Data are expressed as mean ± SEM of three fetuses. *P < 0.05 compared with mock-infected fetuses. MCMV, murine cytomegalovirus.
Immune activation in MCMV-infected fetal cerebrum
To elucidate the mechanism involved in the reactions of brain macrophages and aberrant localization of microglia, we evaluated the immune activation status of MCMV-infected fetal cerebra. For this purpose, expression levels of iNOS, MCP-1/CCL2, and representative inflammatory cytokines in the whole cerebra including the meninges were analyzed (Fig. 9). Expressions of iNOS, TNF-α, and IL-1β, reflecting macrophage activation, increased markedly in the MCMV-infected cerebra with time after infection, but not in the mock-infected cerebra. IL-10 also showed similar mRNA expression kinetics. Interestingly, INF-α and -β, as type I interferons, contributed little to innate immunity in the MCMV-infected fetal cerebra, although they usually play an important role in the earliest innate host defense against viral infections. INF-γ and IL-6 showed a delayed increase, and MCP-1 expression peaked at 3 dpi in the MCMV-infected cerebra.
Figure 9.
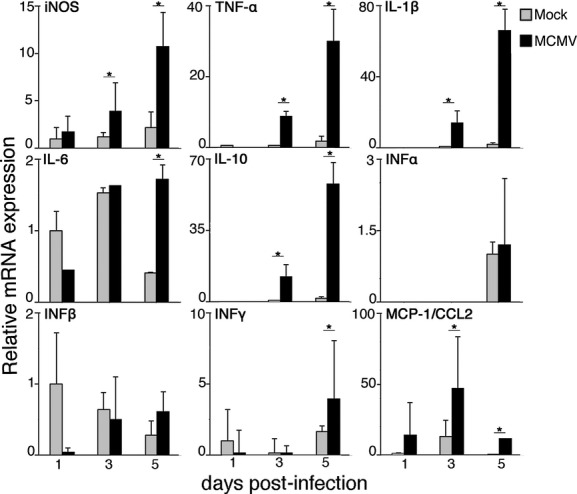
The relative mRNA expression of iNOS, cytokines, and chemokine in mock- and MCMV-infected fetal cerebrum. The mRNA expression levels of iNOS, TNF-α, IL-1β, IL-6, IL-10, INF-α, IFN-β, INF-γ, and MCP-1/CCL2 in mock-infected or MCMV-infected fetal cerebra with meninges at 1, 3, and 5 dpi were assessed by quantitative real-time PCR. The measurements were normalized against the expression of the GAPDH gene. Expression levels of samples were expressed as fold changes against the expression level in the mock-infected cerebra at 1 dpi. Since the expression levels of IL-10 at 1 dpi and INF-α at 1 and 3 dpi were undetectable, fold changes against their expression in the mock-infected cerebrum at 3 or 5 dpi are shown. Mean ± SEM of three mock- or four MCMV-infected fetuses are shown. *P < 0.05 compared with mock-infected fetuses. MCMV, murine cytomegalovirus; iNOS, inducible nitric oxide synthase.
Differential expression of iNOS, inflammatory cytokines, and interferons between activated macrophages and microglia
To determine the cellular populations responsible for the increased expression of iNOS and cytokines in the MCMV-infected fetal cerebra, four cell populations, monocyte/macrophages, microglia, CD11b− leukocytes, and other cerebral cells, were prepared by cell sorting during flow cytometry, and the expression of iNOS and cytokines in each population was analyzed (Fig. 10). The monocyte/macrophage population in the MCMV-infected cerebra was exclusively responsible for the expression of the proinflammatory cytokines, TNF-α, IL-1β, and IL-6. Expression of iNOS and INF-β were also prominent in the monocyte/macrophage population, and substantially observed in the other populations. In addition, the monocyte/macrophage population contributed to the expression of IL-10. These data appear to well explain the activated immune status of the monocyte/macrophage population in the infectious foci. In contrast, the microglia contributed little to the mRNA expression of pro- and anti-inflammatory cytokines, except for iNOS, INF-α, and -β. The expression of iNOS in the microglia of the MCMV-infected cerebra was reduced compared with that of the mock-infected cerebra. Except for the aberrant but minor expression of INF-α and -β, these results indicated a silent or suppressive immune status of microglia in the MCMV-infected fetal cerebrum. The CD11b− leukocyte population was primarily responsible for the expression of INF-γ. Although this population had been supposed to consist of CD3+/CD11b− T cells and B220+/CD11b− B cells, we found B cells were almost absent (Fig. 5C). Therefore, INF-γ, probably derived from T cells, contributed to the expression of iNOS during the late phase of MCMV infection.
Figure 10.
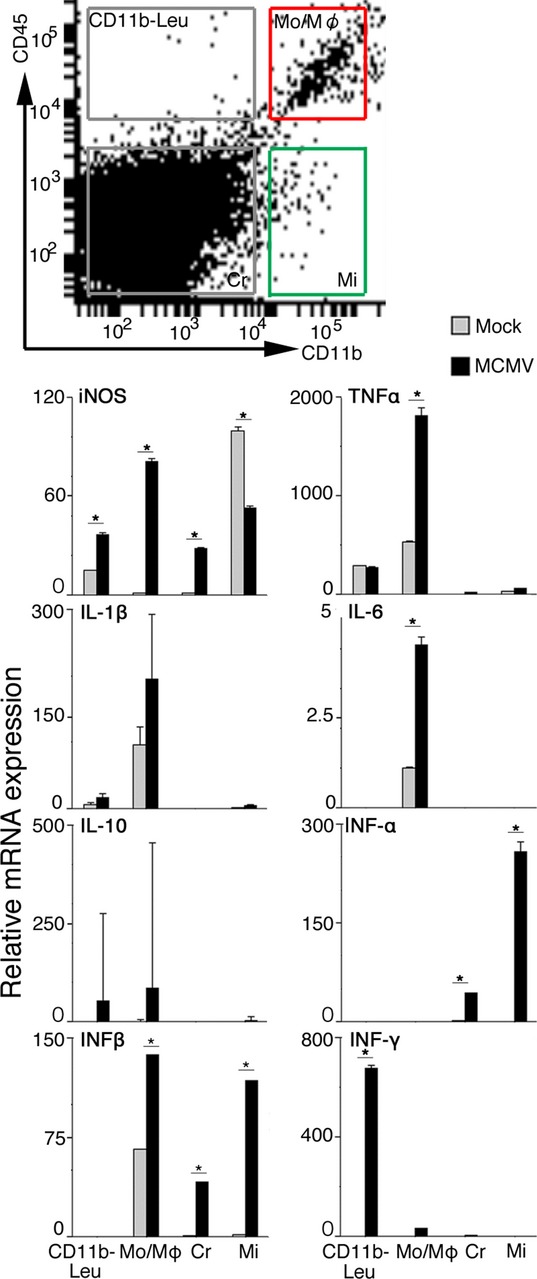
mRNA expression of iNOS and inflammatory cytokines in microglia and activated macrophages in the fetal cerebrum. Four cellular populations, monocyte/macrophage (CD45high/CD11b+, Mo/Mφ), microglia (CD45low/CD11b+, Mi), CD11b− leukocyte (CD45high/CD11b−, CD11b− Leu), and other cerebral cells (CD45low/CD11b−, Cr) were obtained from MCMV-infected fetal cerebra at 5 dpi using a flow cytometric cell sorting device as described in Materials and Methods. mRNA expression levels in each cell population were assessed by quantitative real-time PCR and normalized against the expression of GAPDH gene. Expression levels of iNOS and inflammatory cytokines in each cell population were expressed as fold changes against those of mock-infected cerebra. Mean ± SEM of three fetuses are shown. *P < 0.05 compared with mock-infected fetuses. MCMV, murine cytomegalovirus; iNOS, inducible nitric oxide synthase; PCR, polymerase chain reaction.
Discussion
The present study, using the mouse model of intraplacental MCMV infection, focused on the effects of MCMV infection on late corticogenesis from E14.5 to E18.5. This time frame corresponds to the period from 11 to 15 weeks during the first to second trimesters of human pregnancy.8 During this period, the upper cortical layers II and III are generated both in mice and humans, and disturbances in these layers in humans have been implicated in the etiologies of neurodevelopmental disorders, such as schizophrenia.38 The neonatal mice models with intracranial,30,39 or intraperitoneal MCMV inoculation40 instead of MCMV intraplacental infection, have offered important of the neuropathogenesis of congenital CMV infection. However, in these models, it was difficult to investigate the effects of MCMV infection on cerebral corticogenesis in terms of the route of infection and the stage of CNS development. In this study, we have established an improved model system of congenital MCMV infection using a highly accurate and less invasive method for intraplacental MCMV inoculation based on the use of a fine glass micropipette. This less invasive method improved the survival rate of fetuses infected with MCMV intraplacentally. Furthermore, it minimized the influence of immune response incidentally induced by surgical procedures.
The main findings of the present study are as follows: (1) intraplacentally inoculated MCMV invades the cerebrum via the bloodstream from the placenta in the same way as congenital HCMV infection; (2) brain macrophages are not only the major cells infected in the MCMV-infected fetal brain, but also crucial cells for primitive innate immune response; (3) MCMV directly infects NSPCs and impairs their stemness. Furthermore, we found that MCMV infection impairs their stemness by global and indirect mechanisms during the late cerebral corticogenesis. As the global and indirect mechanisms for the impairment of the stemness of NSPCs in MCMV-infected fetal brain, we would like to propose the disturbances caused by soluble mediators from activated macrophages and the alteration of the brain microenvironment due to the improper distribution of microglia together with an aberrant increase in number.
In our mouse model, multifocal and disseminated MCMV infectious foci were formed through the whole fetal body, including the brain, as shown in congenital HCMV infection.41 In the cerebrum of our mouse model, MCMV infectious foci were distributed along blood vessels throughout the brain parenchyma and meninges, with the periventricular area as a preferred site of MCMV infection. The infectious foci mostly consisted of infiltrated macrophages, and only a part of them were infected with MCMV. Since immature myelomonocytic leukocytes have been shown to participate in MCMV dissemination via the bloodstream,42 it is supposed that these immature monocyte/macrophage lineage cells may contribute to transvascular invasion of blood-borne MCMV into the fetal cerebrum.
In the present study, it is worthy of note that focal MCMV infection of the fetal cerebrum globally induced a dramatic and aberrant increase in brain macrophages, including microglia, and this increase was dependent on the recruitment of precursor cells from other sites rather than in situ proliferation. Activated macrophages were few in number compared with the other brain macrophage populations, but exclusively expressed MHC class II and iNOS, indicating macrophage activation. These activated macrophages in the infectious foci were the primary source of iNOS and proinflammatory cytokines, TNF-α and IL-1β, which have been reported to suppress MCMV replication.43,44 Considering the delayed expression of INF-γ, we speculate that the expression of these mediators may be first induced by the direct interaction between MCMV and macrophages, perhaps via mechanisms involving the activation of pattern recognition receptors.45 This macrophage activation was restricted to the infectious foci with active MCMV replication, as reported previously for the MCMV infection of the postnatal brain,30 and possibly prevented the spread of MCMV infection. It is likely that both the activated macrophages in the infectious foci and the recruited meningeal macrophages derive from the fetal liver where the major fetal hematopoiesis occurs after E12, instead of from the yolk sac in mice.46 In contrast to the activated macrophages, microglia showed no potent activation but improperly appeared in the CP associated with an aberrant increase in their number, suggesting this improper and excessive distribution of microglia could adversely affect cerebral development. However, the source of the increase in microglia, whether primitive macrophages in the yolk sac or fetal liver monocytes, remains to be clarified.47
With regard to the neuropathogenesis of congenital CMV infection, great attention has been paid to NSPCs,19,20 as HCMV preferentially infects the VZ/SVZ in the brains of congenitally infected patients.48 A loss of stemness in NSPCs caused by direct CMV cytopathicity has been demonstrated in previous in vitro studies.11–13 Our intraplacental MCMV infection model simulated transplacental HCHV infection, and demonstrated that intraplacentally inoculated MCMV preferentially infects NSPCs in the developing fetal cerebrum and directly impairs their stemness. At the same time, global and indirect impairment of NSPCs was observed in the MCMV-infected fetal cerebra. Postnatal mouse models infected by intraventricular MCMV injection showed delayed cerebellar development with decreases in granular neuron proliferation and migration40 and improved development of MCMV-infected cerebellum in response to suppression of inflammation by glucocorticoid treatment.49 These reports are highly suggestive of an impact by focal MCMV infection on CNS development via broad inflammatory responses. The dysregulation of immune mediators has been suggested to adversely affect neurodevelopment.50 In this study, the induction of iNOS, TNF-α, IL-1β, and IL-10 from activated macrophages infiltrating infections foci was the earliest primitive innate immune response against MCMV infection in the fetal cerebrum. These immune mediators, TNF-α, IL-1β, and IL-10, are known to adversely influence the stemness of NSPCs and are associated with neurodevelopmental disorders, such as schizophrenia and epilepsy.51–55 In addition to the activated macrophage response, MCMV infection in the developing fetal cerebrum induced improper and excess distribution of microglia, which physiologically regulate NSPCs and supports cerebral cortical development.56,57 iNOS expression was suppressed in these aberrantly recruited microglia during MCMV infection. NO produced by iNOS is an important player in neuronal development.58 These aberrantly recruited microglia may disturb normal brain development via alterations in the microenvironment not only due to their improper and excessive distribution in the fetal brain, but also due to their adverse immune response.
Recent studies have proposed an association between aberrant microglial status and impairment of corticogenesis in neurodevelopmental diseases, such as schizophrenia.59 However, the origin of aberrant microglia and the specific causal relationships between microglial status and diseases remain unclear. Our present study implies that inflammatory responses during embryogenesis can trigger microglial dysregulation and that such dysregulation may result in certain kinds of neuropsychiatric diseases. It would be interesting to see the relationship between neurophysiological and neuroethological outcomes and aberrant microglia status in the brains of adult mice that survive congenital MCMV infection in our model.
In conclusion, we have established an improved mouse model that can closely reflect congenital CMV infection in humans. The model indicated that immune responses to viral infection, including aberrant recruitment and activation of brain macrophages, compromise the stemness of NSPCs, thus resulting in neurodevelopmental disorders.
Acknowledgments
The authors thank Ms. Kawashima, Ms. Suzuki, Mr. Kaneta (Department of Regenerative and Infectious Pathology, Hamamatsu University School of Medicine), Mr. Furukawa (Department of Neurophysiology, Hamamatsu University School of Medicine), and Mr. Shibata (Research Equipment Center, Hamamatsu University School of Medicine) for their excellent technical assistance.
Authors' Contributions
M. S. and I. K. conceived and designed the study. M. S., H. K., T. A., S. M., and I. K. contributed to the data acquisition. M. S., N. I., and I. K. analyzed and interpreted the data. M. S., H. M., T. I., Y. T., N. I., and I. K. wrote and reviewed the manuscript. All authors read and approved the final manuscript.
Conflict of Interest
None declared.
References
- 1.Kenneson A, Cannon MJ. Review and meta-analysis of the epidemiology of congenital cytomegalovirus (CMV) infection. Rev Med Virol. 2007;17:253–276. doi: 10.1002/rmv.535. [DOI] [PubMed] [Google Scholar]
- 2.Manicklal S, Emery VC, Lazzarotto T, et al. The “silent” global burden of congenital cytomegalovirus. Clin Microbiol Rev. 2013;26:86–102. doi: 10.1128/CMR.00062-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cannon MJ, Davis KF. Washing our hands of the congenital cytomegalovirus disease epidemic. BMC Public Health. 2005;5:70. doi: 10.1186/1471-2458-5-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iannetti P, Nigro G, Spalice A, et al. Cytomegalovirus infection and schizencephaly: case reports. Ann Neurol. 1998;43:123–127. doi: 10.1002/ana.410430122. [DOI] [PubMed] [Google Scholar]
- 5.Malinger G, Lev D, Zahalka N, et al. Fetal cytomegalovirus infection of the brain: the spectrum of sonographic findings. AJNR Am J Neuroradiol. 2003;24:28–32. [PMC free article] [PubMed] [Google Scholar]
- 6.Engman ML, Lewensohn-Fuchs I, Mosskin M, Malm G. Congenital cytomegalovirus infection: the impact of cerebral cortical malformations. Acta Paediatr. 2010;99:1344–1349. doi: 10.1111/j.1651-2227.2010.01852.x. [DOI] [PubMed] [Google Scholar]
- 7.Teissier N, Fallet-Bianco C, Delezoide AL, et al. Cytomegalovirus-induced brain malformations in fetuses. J Neuropathol Exp Neurol. 2014;73:143–158. doi: 10.1097/NEN.0000000000000038. [DOI] [PubMed] [Google Scholar]
- 8.Clancy B, Darlington RB, Finlay BL. Translating developmental time across mammalian species. Neuroscience. 2001;105:7–17. doi: 10.1016/s0306-4522(01)00171-3. [DOI] [PubMed] [Google Scholar]
- 9.Bosnjak VM, Daković I, Duranović V, et al. Malformations of cortical development in children with congenital cytomegalovirus infection – A study of nine children with proven congenital cytomegalovirus infection. Coll Antropol. 2011;35(Suppl. 1):229–234. [PubMed] [Google Scholar]
- 10.Ramalho-Santos M, Yoon S, Matsuzaki Y, et al. “Stemness”: transcriptional profiling of embryonic and adult stem cells. Science. 2002;298:597–600. doi: 10.1126/science.1072530. [DOI] [PubMed] [Google Scholar]
- 11.Kosugi I, Shinmura Y, Kawasaki H, et al. Cytomegalovirus infection of the central nervous system stem cells from mouse embryo: a model for developmental brain disorders induced by cytomegalovirus. Lab Invest. 2000;80:1373–1383. doi: 10.1038/labinvest.3780145. [DOI] [PubMed] [Google Scholar]
- 12.Odeberg J, Wolmer N, Falci S, et al. Human cytomegalovirus inhibits neuronal differentiation and induces apoptosis in human neural precursor cells. J Virol. 2006;80:8929–8939. doi: 10.1128/JVI.00676-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luo MH, Hannemann H, Kulkarni AS, et al. Human cytomegalovirus infection causes premature and abnormal differentiation of human neural progenitor cells. J Virol. 2010;84:3528–3541. doi: 10.1128/JVI.02161-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boksa P. Effects of prenatal infection on brain development and behavior: a review of findings from animal models. Brain Behav Immun. 2010;24:881–897. doi: 10.1016/j.bbi.2010.03.005. [DOI] [PubMed] [Google Scholar]
- 15.Soumiya H, Fukumitsu H, Furukawa S. Prenatal immune challenge compromises the normal course of neurogenesis during development of the mouse cerebral cortex. J Neurosci Res. 2011;89:1575–1585. doi: 10.1002/jnr.22704. [DOI] [PubMed] [Google Scholar]
- 16.Hagberg H, Gressens P, Mallard C. Inflammation during fetal and neonatal life: implications for neurologic and neuropsychiatric disease in children and adults. Ann Neurol. 2012;71:444–457. doi: 10.1002/ana.22620. [DOI] [PubMed] [Google Scholar]
- 17.Harvey L, Boksa P. Prenatal and postnatal animal models of immune activation: relevance to a range of neurodevelopmental disorders. Dev Neurobiol. 2012;72:1335–1348. doi: 10.1002/dneu.22043. [DOI] [PubMed] [Google Scholar]
- 18.Shi L, Tu N, Patterson PH. Maternal influenza infection is likely to alter fetal brain development indirectly: the virus is not detected in the fetus. Int J Dev Neurosci. 2005;23:299–305. doi: 10.1016/j.ijdevneu.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 19.Tsutsui Y, Kosugi I, Kawasaki H. Neuropathogenesis in cytomegalovirus infection: indication of the mechanisms using mouse models. Rev Med Virol. 2005;15:327–345. doi: 10.1002/rmv.475. [DOI] [PubMed] [Google Scholar]
- 20.Cheeran MC, Lokensgard JR, Schleiss MR. Neuropathogenesis of congenital cytomegalovirus infection: disease mechanisms and prospects for intervention. Clin Microbiol Rev. 2009;22:99–126. doi: 10.1128/CMR.00023-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li RY, Tsutsui Y. Growth retardation and microcephaly induced in mice by placental infection with murine cytomegalovirus. Teratology. 2000;62:79–85. doi: 10.1002/1096-9926(200008)62:2<79::AID-TERA3>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 22.Arai Y, Ishiwata M, Baba S, et al. Neuron-specific activation of murine cytomegalovirus early gene e1 promoter in transgenic mice. Am J Pathol. 2003;163:643–652. doi: 10.1016/S0002-9440(10)63691-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yamada S, Kosugi I, Katano H, et al. In vivo imaging assay for the convenient evaluation of antiviral compounds against cytomegalovirus in mice. Antiviral Res. 2010;88:45–52. doi: 10.1016/j.antiviral.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 24.Wentworth BB, French L. Plaque assay of cytomegalovirus strains of human origin. Proc Soc Exp Biol Med. 1970;135:253–258. doi: 10.3181/00379727-135-35031. [DOI] [PubMed] [Google Scholar]
- 25.Bonnin A, Goeden N, Chen K, et al. A transient placental source of serotonin for the fetal forebrain. Nature. 2011;472:347–350. doi: 10.1038/nature09972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu XF, Yan S, Abecassis M, Hummel M. Biphasic recruitment of transcriptional repressors to the murine cytomegalovirus major immediate-early promoter during the course of infection in vivo. J Virol. 2010;84:3631–3643. doi: 10.1128/JVI.02380-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wagner M, Jonjic S, Koszinowski UH, Messerle M. Systematic excision of vector sequences from the BAC-cloned herpesvirus genome during virus reconstitution. J Virol. 1999;73:7056–7060. doi: 10.1128/jvi.73.8.7056-7060.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trgovcich J, Stimac D, Polić B, et al. Immune responses and cytokine induction in the development of severe hepatitis during acute infections with murine cytomegalovirus. Arch Virol. 2000;145:2601–2618. doi: 10.1007/s007050070010. [DOI] [PubMed] [Google Scholar]
- 29.Tsutsui Y, Naruse I. Murine cytomegalovirus infection of cultured mouse embryos. Am J Pathol. 1987;127:262–270. [PMC free article] [PubMed] [Google Scholar]
- 30.Kosugi I, Kawasaki H, Arai Y, Tsutsui Y. Innate immune responses to cytomegalovirus infection in the developing mouse brain and their evasion by virus-infected neurons. Am J Pathol. 2002;161:919–928. doi: 10.1016/S0002-9440(10)64252-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pont-Lezica L, Béchade C, Belarif-Cantaut Y, et al. Physiological roles of microglia during development. J Neurochem. 2011;119:901–908. doi: 10.1111/j.1471-4159.2011.07504.x. [DOI] [PubMed] [Google Scholar]
- 32.Sedgwick JD, Schwender S, Imrich H, et al. Isolation and direct characterization of resident microglial cells from the normal and inflamed central nervous system. Proc Natl Acad Sci USA. 1991;88:7438–7442. doi: 10.1073/pnas.88.16.7438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ajami B, Bennett JL, Krieger C, et al. Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat Neurosci. 2011;14:1142–1149. doi: 10.1038/nn.2887. [DOI] [PubMed] [Google Scholar]
- 34.Dehay C, Kennedy H. Cell-cycle control and cortical development. Nat Rev Neurosci. 2007;8:438–550. doi: 10.1038/nrn2097. [DOI] [PubMed] [Google Scholar]
- 35.Hevner RF, Shi L, Justice N, et al. Tbr1 regulates differentiation of the preplate and layer 6. Neuron. 2001;29:353–366. doi: 10.1016/s0896-6273(01)00211-2. [DOI] [PubMed] [Google Scholar]
- 36.McEvilly RJ, de Diaz MO, Schonemann MD, et al. Transcriptional regulation of cortical neuron migration by POU domain factors. Science. 2002;295:1528–1532. doi: 10.1126/science.1067132. [DOI] [PubMed] [Google Scholar]
- 37.Dominguez MH, Ayoub AE, Rakic P. POU-III transcription factors (Brn1, Brn2, and Oct6) influence neurogenesis, molecular identity, and migratory destination of upper-layer cells of the cerebral cortex. Cereb Cortex. 2013;23:2632–2643. doi: 10.1093/cercor/bhs252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Garey L. When cortical development goes wrong: schizophrenia as a neurodevelopmental disease of microcircuits. J Anat. 2010;217:324–333. doi: 10.1111/j.1469-7580.2010.01231.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shinmura Y, Kosugi I, Aiba-Masago S, et al. Disordered migration and loss of virus-infected neuronal cells in developing mouse brains infected with murine cytomegalovirus. Acta Neuropathol. 1997;93:551–557. doi: 10.1007/s004010050651. [DOI] [PubMed] [Google Scholar]
- 40.Koontz T, Bralic M, Tomac J, et al. Altered development of the brain after focal herpesvirus infection of the central nervous system. J Exp Med. 2008;205:423–435. doi: 10.1084/jem.20071489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abdel-Latif Mel A, Sugo E. Images in clinical medicine. Congenital cytomegalovirus infection. N Engl J Med. 2010;362:833. doi: 10.1056/NEJMicm0804100. [DOI] [PubMed] [Google Scholar]
- 42.Noda S, Aguirre SA, Bitmansour A, et al. Cytomegalovirus MCK-2 controls mobilization and recruitment of myeloid progenitor cells to facilitate dissemination. Blood. 2006;107:30–38. doi: 10.1182/blood-2005-05-1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cheeran MC, Hu S, Gekker G, Lokensgard JR. Decreased cytomegalovirus expression following proinflammatory cytokine treatment of primary human astrocytes. J Immunol. 2000;164:926–933. doi: 10.4049/jimmunol.164.2.926. [DOI] [PubMed] [Google Scholar]
- 44.Noda S, Tanaka K, Sawamura S, et al. Role of nitric oxide synthase type 2 in acute infection with murine cytomegalovirus. J Immunol. 2001;166:3533–3541. doi: 10.4049/jimmunol.166.5.3533. [DOI] [PubMed] [Google Scholar]
- 45.Tabeta K, Georgel P, Janssen E, et al. Toll-like receptors 9 and 3 as essential components of innate immune defense against mouse cytomegalovirus infection. Proc Natl Acad Sci USA. 2004;101:3516–3521. doi: 10.1073/pnas.0400525101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Naito M, Takahashi K, Nishikawa S. Development, differentiation, and maturation of macrophages in the fetal mouse liver. J Leukoc Biol. 1990;48:27–37. doi: 10.1002/jlb.48.1.27. [DOI] [PubMed] [Google Scholar]
- 47.Ginhoux F, Lim S, Hoeffel G, et al. Origin and differentiation of microglia. Front Cell Neurosci. 2013;7:45. doi: 10.3389/fncel.2013.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Perlman JM, Argyle C. Lethal cytomegalovirus infection in preterm infants: clinical, radiological, and neuropathological findings. Ann Neurol. 1992;31:64–68. doi: 10.1002/ana.410310112. [DOI] [PubMed] [Google Scholar]
- 49.Kosmac K, Bantug GR, Pugel EP, et al. Glucocorticoid treatment of MCMV infected newborn mice attenuates CNS inflammation and limits deficits in cerebellar development. PLoS Pathog. 2013;9:e1003200. doi: 10.1371/journal.ppat.1003200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kokaia Z, Martino G, Schwartz M, Lindvall O. Cross-talk between neural stem cells and immune cells: the key to better brain repair? Nat Neurosci. 2012;15:1078–1087. doi: 10.1038/nn.3163. [DOI] [PubMed] [Google Scholar]
- 51.Ben-Hur T, Ben-Menachem O, Furer V, et al. Effects of proinflammatory cytokines on the growth, fate, and motility of multipotential neural precursor cells. Mol Cell Neurosci. 2003;24:623–631. doi: 10.1016/s1044-7431(03)00218-5. [DOI] [PubMed] [Google Scholar]
- 52.Liu YP, Lin HI, Tzeng SF. Tumor necrosis factor-alpha and interleukin-18 modulate neuronal cell fate in embryonic neural progenitor culture. Brain Res. 2005;1054:152–158. doi: 10.1016/j.brainres.2005.06.085. [DOI] [PubMed] [Google Scholar]
- 53.Meyer U, Murray PJ, Urwyler A, et al. Adult behavioral and pharmacological dysfunctions following disruption of the fetal brain balance between pro-inflammatory and IL-10-mediated anti-inflammatory signaling. Mol Psychiatry. 2008;13:208–221. doi: 10.1038/sj.mp.4002042. [DOI] [PubMed] [Google Scholar]
- 54.Crampton SJ, Collins LM, Toulouse A, et al. Exposure of foetal neural progenitor cells to IL-1β impairs their proliferation and alters their differentiation – a role for maternal inflammation? J Neurochem. 2012;120:964–973. doi: 10.1111/j.1471-4159.2011.07634.x. [DOI] [PubMed] [Google Scholar]
- 55.Pineda E, Shin D, You SJ, et al. Maternal immune activation promotes hippocampal kindling epileptogenesis in mice. Ann Neurol. 2013;74:11–19. doi: 10.1002/ana.23898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cunningham CL, Martínez-Cerdeño V, Noctor SC. Microglia regulate the number of neural precursor cells in the developing cerebral cortex. J Neurosci. 2013;33:4216–4233. doi: 10.1523/JNEUROSCI.3441-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ueno M, Fujita Y, Tanaka T, et al. Layer V cortical neurons require microglial support for survival during postnatal development. Nat Neurosci. 2013;16:543–551. doi: 10.1038/nn.3358. [DOI] [PubMed] [Google Scholar]
- 58.Béchade C, Pascual O, Triller A, Bessis A. Nitric oxide regulates astrocyte maturation in the hippocampus: involvement of NOS2. Mol Cell Neurosci. 2011;46:762–769. doi: 10.1016/j.mcn.2011.02.009. [DOI] [PubMed] [Google Scholar]
- 59.Blank T, Prinz M. Microglia as modulators of cognition and neuropsychiatric disorders. Glia. 2013;61:62–70. doi: 10.1002/glia.22372. [DOI] [PubMed] [Google Scholar]