

Abstract
Multiple sclerosis (MS) is a complex disorder of the central nervous system that appears to be driven by a shift in immune functioning toward excess inflammation that results in demyelination and axonal loss. Beta interferons were the first class of disease-modifying therapies to be approved for patients with MS after treatment with this type I interferon improved the course of MS on both clinical and radiological measures in clinical trials. The mechanism of action of interferon-beta appears to be driven by influencing the immune system at many levels, including antigen-presenting cells, T cells, and B cells. One effect of these interactions is to shift cytokine networks in favor of an anti-inflammatory effect. The pleiotropic mechanism of action may be a critical factor in determining the efficacy of interferon-beta in MS. This review will focus on select immunological mechanisms that are influenced by this type I cytokine.
Introduction
Multiple sclerosis (MS) is a chronic immune-mediated disease of the central nervous system (CNS) with an unknown cause.1 In relapsing forms of MS, individuals experience inflammatory demyelination and subsequent interruption of axonal function.1 Over time, this damage to the CNS leads to significant disability and earlier death in patients with MS than those in non-MS comparators.1
A great deal of research has examined the underlying pathophysiology of the disease. Many of these efforts have focused on antigen-presenting cells (APCs), T cells (including Th1/Th2/Th17 effector cell polarization and T regulatory [Treg] cells), B cells, and cytokine networks that participate in the demyelinating process. Disease-modifying treatments (DMTs) for patients with MS must simultaneously impact multiple processes that are part of the immune system including (1) antigen presentation, (2) T-cell polarization and function, and (3) B-cell engagement in order to lead to improvements in clinical outcomes. The focus of this review will be on the immunomodulatory effects of beta interferons, the first class of DMTs to be approved for the treatment of patients with relapsing-remitting MS (RRMS).
Natural interferon-beta, type I interferon, is secreted by fibroblasts and binds to the interferon receptor, which consists of two components (IFNAR1 and IFNAR2), and activates the Janus kinase (JAK)/Signal Transducer and Activator of Transcription (STAT) pathway to phosphorylate STAT1 and STAT2.2,3 These dimerize and associate with interferon regulatory factor (IRF) 3 and then bind to interferon-stimulated response elements in the cell nucleus.4 This in turn activates interferon-stimulated genes and leads to the production of antiviral, antiproliferative, and antitumor products.4 Type II interferon (interferon-gamma) binds to IFNGR1 and IFNGR2, also activating the JAK/STAT pathway, although the resulting STAT1 homodimer complex differs from the STAT1/STAT2/IRF9 complex that is formed by type I interferons.4 Interferon gamma induces factors with weak antiviral but strong immunomodulatory effects.4
There are two commercially available formulations of recombinant interferon-beta: interferon beta-1a (intramuscular Avonex® [Biogen Idec; Cambridge, MA] and subcutaneous Rebif® [EMD Serono; Rockland, MA]), which is nearly identical to the natural interferon-beta, and interferon beta-1b (Betaferon®/Betaseron® [Bayer HealthCare Pharmaceuticals; Whippany, NJ] and the identical Extavia® [Novartis Pharmaceuticals Corporation, East Hanover, NJ]). Interferon beta-1b is expressed in a bacterial vector such as Escherichia coli and differs from interferon beta-1a in that it has one less amino acid and because it is not glycosylated.2 Interferon beta-1b also contains a serine substitution for cysteine at position 17.5 The clinical efficacy of these agents is derived from interactions with the immune system at multiple levels. Importantly, beta interferons appear to counter some pathogenic processes in MS by affecting the function of APCs, T cells, and B cells in the adaptive immune system.
The Adaptive Immune System in MS
The adaptive immune system produces antibodies and T cells that recognize and neutralize potential pathogens that enter the body.3 To accomplish this task, components of this system have both effector and regulatory functions, which are accomplished by different cell types.3 In patients with MS, the activity of these components is tipped in favor of an inflammatory response.
The exact cause of MS remains unknown. There is indirect evidence to suggest that MS is triggered by a viral infection, including the elevated levels of virus-specific antibodies in serum.6 Virus-specific oligoclonal bands and elevated immunoglobulin G have also been detected in the cerebrospinal fluid (CSF) of patients with MS.6 It is possible that some immunoglobulins may cross-react with or trigger responses to antigens within the CNS, but no clear linkage with viruses has been discovered to date. Importantly, type I interferons are expressed under natural conditions in response to viral infections.2 The role of specific viral agents responsible for the induction or persistence of the disease remains speculative and unconfirmed.
Other speculation as to the origin of MS has centered on bacteria in the gut microbiome. In animal models of demyelinating disease, changes in gut microbiota can alter outcomes.7 For example, mice genetically engineered to express a myelin basic protein peptide do not develop spontaneous experimental autoimmune encephalitis (EAE) when reared in pathogen-free environments.8 However, these mice will promptly develop EAE when colonized with commensal gut microbiota.8 In addition, germ-free mice are unable to mount an inflammatory interleukin (IL)-17-driven response in an EAE model until they are monocolonized with specific commensal bacteria such as segmented filamentous bacteria.9 Some have also speculated that MS is caused by an increase in adhesion molecules and a subsequent modification in immune reactivity and immune cell trafficking.10 Dysregulation of key components of the immune system in patients with MS suggests that the disease is caused by an overactivity and/or a loss of homeostatic balance in the immune system,10 which will be described in greater detail in the following sections.
Regardless of the cause of MS, components of the adaptive immune system are thought to become activated and respond to myelin or other targets within the CNS, leading to inflammatory demyelination and axonal loss.11 Specific CNS antigen(s) have not been isolated; however, MS is considered to be an immune-mediated disease characterized by many changes in immune regulation. This hypothesis suggests that everyone has the potential to develop MS, but the disease only manifests in certain people when the appropriate conditions are met. Much of the work that drives this speculation on disease mechanisms in MS has been derived from the EAE mouse model.3,11 Care must be taken when applying this model to human pathology because of the large differences in cellular regulation and inflammatory gene activation between humans and mice.12 In addition, EAE will incite several myelin-derived peptides, whereas in human MS, this association remains speculative.
APC Function in the Innate Immune System
Under normal physiological conditions, APCs process and present non-self-molecules to T cells, which are primed through interaction with costimulatory molecules on the surface of the APCs.3 In patients with an autoimmune disease, APCs process and present proteins that are part of the patient's own body.3 In the case of MS, APCs may/are believed to present myelin antigens in the CNS,3 although evidence supporting this hypothesis is weak. Among the different types of APCs (i.e., dendritic cells, monocytes, macrophages, and B cells), dendritic cells are the most potent type and can be further differentiated into myeloid or plasmacytoid cells.3 B cells will be discussed in a separate section below.
T cells are transformed into active phenotypes through a process of polarization that requires two signals, the presence of costimulatory proteins, and secreted cytokines from APCs and lymphocytes (including IL-4, IL-5, IL-6, IL-12, IL-13, IL-23, and TGF-β), which control the balance between effectors and regulators in the immune system.3,13 The first signal in this process occurs when the peptide-loaded human leukocyte antigen on the APC binds to the T-cell receptor.3 Once bound, a second costimulatory protein on APCs activates the T cell and triggers the innate immune response.3
Dendritic cells are particularly important in this process and are activated by toll-like receptors (TLRs) and cytokines to provide all of the necessary signals for polarization of naïve T cells.3 TLRs are pattern recognition molecules that are constitutively expressed, primarily on APCs and, to a lesser extent, on lymphocytes.3 A subtype of dendritic cells, myeloid dendritic cells, expresses TLR2, which responds to microbial products and induces dendritic cells to secrete IL-12p70 to stimulate T cells.13,14 Plasmacytoid dendritic cells express TLR7 and TLR9 (which respond to microbial nucleic acids) and secrete type I interferon when activated.13,14 Elevated type I interferon induces Treg phenotypes and Th1 cells.13,15
Although a myelin-specific antigen has not been identified in patients with MS, such an “MS antigen” would presumably have some structural similarities with myelin. However, evidence to back up this assumption is limited. Support for this hypothesis is derived from experiments involving the administration of altered peptide ligands in mice after induction of EAE.16 To date, trials using tissue from human subjects have not found evidence of myelin-specific antigens.
In patients with MS, dendritic cell function is shifted in favor of inflammatory activity (Table 1). For example, patients with MS have a high number of plasmacytoid dendritic cells in CSF, which increases further during relapses.13 These cells express excessive levels of costimulatory molecules and Th1-promoting interferon-alpha in patients with RRMS.13,17 In addition, decreased secretion of interferon-alpha compared with healthy controls when the plasmacytoid dendritic cells of patients with MS are stimulated suggests an impairment in immune regulation in that plasmacytoid cells have a decreased capacity to induce mature cell phenotypes.18
Table 1.
Overview of the immunomodulatory effects of interferon-beta
| Dysfunction in MS | Effect of interferon-beta | |
|---|---|---|
| APCs | • May respond to myelin in the CNS | • ↓ Antigen presentation and T-cell stimulation |
| • ↓ Secretion of Th1-promoting interferon-alpha | • ↓ Dendritic cell concentration in peripheral blood | |
| • ↓ Suppression of dendritic cell activity | • ↓ TLR9-mediated interferon-alpha secretion | |
| • ↑ Expression of pro-inflammatory costimulatory markers and cytokines | • ↓ Pro-inflammatory cytokine production | |
| T cells | • CD8+ cells attack oligodendrocytes | • ↑ Apoptosis of pro-inflammatory CD4+ and CD8+ Th17 cells |
| • ↑ Percentage of Th17 cells | • ↓ Pro-inflammatory cytokine production | |
| • ↓ CD8+ regulatory cells in blood and CSF | • Channel T cells into lymphoid tissues and thereby reduce T-cell activation | |
| • ↑ Production of Treg | ||
| • ↓T-cell adhesion to blood–brain barrier | ||
| B cells | • ↑ MHC II | • ↓ MHC II expression |
| • ↑ CD80+ cells, CD80+/CD86+ ratio | • ↓ CD80+ cells | |
| • ↑ Plasma BAFF | • ↑ IL-10 and TGFβ secretion | |
| Cytokine networks | • ↑ IL-17 secretion | • ↓ IL-17 secretion by Th17 cells |
| • ↑ IL-22 secretion leads to increased development of lymphoid cells | • ↑ Secretion of Th2-promoting cytokines (IL-4, IL-5, IL-13) | |
| • ↑ IL-23 secretion leads to increased T-cell polarization | • ↑ IL-27 secretion increases the induction of Treg | |
| Other effects | • ↑ Secretion of MMPs, allowing activated macrophages to enter the CNS | • ↓ MMP-9, restore MMP-9/TIMP-1 ratio |
| • Expression of BDNF receptor on neurons and glia near lesions | • ↑ BDNF secretion, which theoretically could lead to axonal repair |
MS, multiple sclerosis; APCs, antigen-presenting cells; CNS, central nervous system; CSF, cerebrospinal fluid; MHC, major histocompatibility complex; IL, interleukin; TGF, transforming growth factor; BAFF, B-cell activating factor of the TNF family; MMPs, matrix metalloproteinases; BDNF, brain-derived neurotrophic factor; Treg, regulatory T cell.
Myeloid dendritic cell phenotypes also change with disease form in patients with MS.13 When the disease reaches the secondary progressive stage, myeloid dendritic cells upregulate costimulatory markers that are indicative of a pro-inflammatory phenotype.13 In contrast, patients with RRMS typically have myeloid dendritic cells with an immature phenotype.13 Overall, increased dendritic cell activity appears to be a key feature in the pathogenesis of MS.
APCs, including dendritic cells, can activate Th1 cells, which release pro-inflammatory cytokines that enable leukocytes to cross the blood–brain barrier into the CNS.11 The chemokine receptor CCR7 facilitates the entry of T cells into peripheral lymph nodes and away from the CNS.11 In patients with MS, there is decreased expression of this receptor on CD8+ effector T cells, which should increase trafficking of T cells into the CNS.19
Effects of Interferon-Beta on Antigen Presentation
Interferon-beta reduces myeloid dendritic concentrations in peripheral blood. It also alters the function of dendritic cells and other APCs to downregulate antigen presentation and the ability of APCs to stimulate T-cell responses.20 Beta interferons nonetheless help to maintain the number of type I interferon-secreting plasmacytoid dendritic cells.13 However, they downregulate TLR9, which decreases TLR9-mediated secretion of Th1-promoting interferon-alpha from the plasmacytoid dendritic cells.17 In addition, on plasmacytoid dendritic cells, interferon-beta upregulates the expression of TLR3, TLR7, and MyD88, the TLR adaptor molecule that is hypothesized to increase immune regulation and decrease the likelihood of virus-mediated MS relapses (as well as other anti-inflammatory effects that have not yet been fully explained).21 In another mechanism, increased CCR7 expression with interferon-beta may also facilitate the channeling of T cells away from the CNS and toward peripheral lymph nodes.22 In the CNS, interferon-beta may also alter antigen presentation by monocytes and microglial cells to reduce presentation to T cells.20,23 These combined effects would potentially reduce the presentation of myelin-specific antigens.
T-Cell (CD4+ and CD8+) Function
CD4+ and CD8+ T cells recognize antigens through major histocompatibility complex (MHC) molecules bound to APCs.3 Once activated, CD4+ T cells become polarized to differentiate into Th1 cells (also known as antiviral and antitumor effector T cells, which are increased in MS) or Th2 cells (which counter Th1 cells and promote antibody secretion and allergies).3 Both cell types are involved in different immune-mediated disease states. CD8+ T cells have two effector phenotypes, with CD8+ CD28+ cytotoxic lymphocytes functioning as autoreactive effectors.24 The CD8+ CD28− suppressor cells regulate immune function and are critical in MS.24 Effector T cells are critical for eliminating pathogens, while regulatory T cells are essential for suppressing immune activation.3 Patients with MS show evidence of disordered activity of both aspects of T-cell activity. In MS, Th1 and Th17 differentiation by activated dendritic cells may be the primary source of disease pathogenesis.3 Th2 cells, which can also have an effector phenotype (depending on the specific disease condition), serve a more regulatory function in CNS demyelinating disease and inhibit Th1 cells.3
Once activated, T cells secrete pro-inflammatory cytokines that enable them to bind to the endothelial cells that make up the blood–brain barrier.11 These bound T cells secrete matrix metalloproteinases (MMPs) that compromise the extracellular matrix proteins of the subendothelial basement membrane of the blood–brain barrier, thereby allowing activated macrophages to enter the CNS and demyelinate axons.11 MMP-9 is found in high levels in the serum and CSF of patients with MS and correlates with disease severity.25,26 Patients with MS also have a higher ratio of MMP-9 to tissue inhibitor of metalloproteinase-1 (TIMP-1), which is the main inhibitor of MMP-9.27 Within the CNS, macrophages and CD8+ T cells attack oligodendrocytes, transect axons, and promote vascular permeability.3,11 This disruption of the blood–brain barrier is thought to be a critical step in the formation of demyelinated lesions. These lesions are presumably the main site of damage to the axon; neurons and glia near these lesions express receptors for brain-derived neurotrophic factor (BDNF), a trophic factor related to neuronal growth and repair.28 Sites of local inflammation in the CNS in patients with MS also show decreased Treg activity, suggesting a loss of control over the inflammatory response.29,30
MS is marked by an increase in the percentage of T cells that secrete pro-inflammatory IL-17, known as Th17 cells (Fig. 1A).30 Th17 cells can induce disease, and neutralization of IL-17 will reduce the symptoms of EAE.31 In humans, Th17 concentration in peripheral blood mononuclear cells is generally low in patients with stable MS as well as in healthy controls but increases significantly during active MS.30 IL-17 gene expression is also high in the CSF of patients with MS, as well as within MS lesions.32
Figure 1.
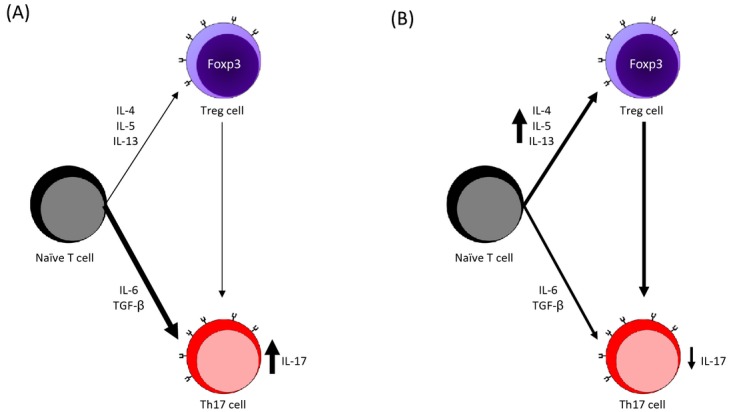
T-cell polarization (A) in patients with untreated MS and (B) under the effects of interferon-beta. Effector and regulatory T-cell polarization is driven by cytokines that influence the phenotype of the polarized cell. Patients with MS have an increased number of Th17 cells, which are polarized by IL-6 and TGF-β, leading to increased secretion of pro-inflammatory IL-17.3,30 Interferon-beta inhibits Th17 differentiation, leading to a decrease in IL-17 secretion.41 In addition to this effector cell phenotype, patients with MS also have a dysregulated Treg response by Foxp3+ cells that is unable to suppress Th17 cell activity.3 Treatment with interferon-beta increases the number of Foxp3+ cells by increasing production of IL-4, IL-5, and IL-13, thereby helping to return the secretion of IL-17 by Th17 cells to a level similar to healthy patients.3,45 MS, multiple sclerosis; IL, interleukin; TGF, transforming growth factor.
Levels of IL-17 and the related cytokine IL-22 correlate with disease activity.30 Furthermore, stimulation with myelin antigen-specific cells increases the number of IL-17-secreting cells, which in turn leads to sustained tissue inflammation.30 IL-22, which is a member of the IL-10 cytokine family and is produced by IL-17-secreting T cells, may be of particular interest with regard to pathogenesis in MS as it plays a critical role in the inflammatory cascade of various autoimmune diseases.33 This cytokine stimulates the development of innate lymphoid cells that are gut derived.33 Furthermore, the IL-22RA2 gene has been identified as a risk factor for the development of MS.34 All of these findings suggest that Th17 cells and their related cytokines play a critical role in MS exacerbations.
In MS, regulation of the inflammatory response falters. CD8+ regulatory T cells secrete IL-10, inhibit the maturation of dendritic cells, and secrete transforming growth factor (TGF)-β, which blocks the activation of lymphocytes and normally should reduce the proliferation of myelin-specific T cells.29 The percentage and function of the CD8+ CD28− suppressor/regulatory cells in peripheral blood are reduced in patients with MS relative to healthy controls, and there is a significant reduction in these cells during disease exacerbations.35,36 In addition to a reduction in the number of CD8+ suppressor cells, patients with active MS also show reduced expression of CD8 protein on T cells,36 perhaps interfering with thymic education or regulatory cell effector function. CD8+ CD25+ FOXP3+ regulatory T cells are also reduced in the CSF of patients with RRMS relative to those with inactive disease or healthy controls, likely due to excess inflammation in the CNS.29
Concentrations of CD4+ CD25+ FOXP3+ Treg cells are low in MS lesions; however, their numbers in peripheral blood and CSF are increased.37 This finding suggests that patients with MS do not have an insufficient number of Treg cells; rather, this regulatory cell population is dysfunctional.37 The dysfunctional CD4+ CD25+ FOXP3+ cell activity in the CNS may also be related to insufficient trafficking of these cells into the CNS,37 thereby limiting the ability of this population of Treg cells to curb the inflammation associated with MS. A picture emerges in which patients with MS have an increased inflammatory response without an adequate regulatory response.
Effects of Interferon-Beta on T-Cell-Mediated Immunity
Consistent with the hypothesis that a broad spectrum of immunological activity should be maximally effective in the treatment of patients with MS, interferon-beta exerts a number of effects on T cells. Through the previously described mechanisms mediated by APCs, interferon-beta may indirectly help to control the inflammatory response in patients with MS by reducing APCs' ability to activate T cells.11 Direct effects on T cells also prevent adhesion to and crossing of the blood–brain barrier.11 Interferon-beta also facilitates the induction of regulatory T cells, including CD4+ CD25+ FOXP3+ Treg cells and CD8+ regulatory cells (Fig. 1B).11,38 Through regulation of chemokine receptors, interferon-beta can also channel autoreactive T cells into lymphoid tissues and away from the CNS.11 While the decrease in MMP-9 appears to be transient, the increase in TIMP-1 seems to a more long-lasting response.39 Other effects at the site of MS lesions can be seen in the upregulation of the neurotrophic factor BDNF, which could theoretically lead to neuronal repair.40
Type I interferons also inhibit Th17 cell differentiation, thereby reducing the secretion of IL-17 (Fig. 1b).41 In vitro experiments using peripheral blood collected from patients with active MS showed that interferon-beta causes apoptosis in proinflammatory CD4+ and CD8+ Th17 cells in a dose-dependent manner.27,39 Production of the regulatory protein IL-27 by macrophages is increased, suppressing the inflammatory response.41 Importantly, IL-27 can significantly inhibit development of EAE when administered to mice, perhaps by its effect on the induction of T regulatory cells.41,42 Interferon-beta also decreases concentrations of TNF in patients with MS; TNF limits maturation of T cells and should have a negative effect on T-cell activation in mouse models, but this effect may not be present in humans.41,43,44 Overall, treatment with interferon-beta is thought to shift cytokine production in favor of Treg-promoting cytokines, including IL-4, IL-5, and IL-13,45 although murine studies46 and data from the Reder laboratory47 find that there is also a shift to some Th1-promoting cytokines.
B-Cell Function
B cells produce antibodies but also have the capacity to present antigens and utilize costimulatory proteins to activate T cells.48 B cells can be APCs48 and are quite potent when antigen concentrations are low because the B-cell receptor is antigen specific.49
Through the expression of MHC class II, B cells facilitate antigen presentation to CD4+ T cells.49 Once antigen is bound to the B-cell receptor and is presented to T cells, the interaction between CD28 on T cells and CD80/CD86 on B cells leads to T-cell activation.50 The costimulatory proteins CD80 and CD86 that are expressed on B cells also facilitate T-cell differentiation into effector or regulatory cells via their interaction with CD40 on B cells and its T-cell ligand CD154.49 This is followed by CD80+ cell activation of Th1 cells for a pro-inflammatory effect and CD86+ cell activation of Th2 cells for a regulatory effect via their interaction with the specific T-cell costimulatory molecules CD28 and CTLA-4.49 Interferon-beta reduces CD80 on B cells, likely interrupting this highly specific B-cell arm of immunity.49,51 B cells also produce the critical anti-inflammatory cytokine, IL-10, that has profound anti-inflammatory effects by modulation of the migration of dendritic cells, activation of macrophages, and regulation of B- and T-cell functioning.49,51
In MS, recent clinical trials utilizing B-cell depletion (via ocrelizumab52 or rituximab53) also suggest that B cells would be critical for the presentation of the neuro- and other antigens that are hypothesized to exist in MS and also for the secretion of cytokines to enhance T-cell function. B cells may also be responsible for the production of autoimmune antibodies to these antigens. Evidence for dysregulated B-cell functioning can be seen in the increased number of CD80+ cells in the blood of patients with active MS.48 This increased inflammatory effect does not appear to be countered by a regulatory effect, as the ratio of CD80+ to CD86+ cells is also higher in patients with active MS than in healthy controls.48 Patients with MS also have an elevated number of CF80 (B7.1)-expressing B cells.48,50 B cells of patients with MS also have a reduced capacity to secrete IL-10, suggesting a lack of regulatory modulation (up or down) of dendritic cell activity.49,51 Furthermore, within the meningeal spaces of the CNS (the germinal center-like areas), B cells produce inflammatory mediators that stimulate plasma cells to produce immunoglobulins, which appear as oligoclonal bands in CSF.49 B-cell activating factor of the TNF family (BAFF) is elevated in the blood of patients with MS.54 This crucial factor for maintenance of B cells is induced at sites of inflammation and can exacerbate local inflammation by stimulating the survival of B cells.54 BAFF is also expressed in MS lesions.55
Effects of Interferon-Beta on B-Cell Function
Interferon-beta therapy induces changes in B-cell functioning that alter antigen presentation. Expression of MHC II on B cells is decreased after treatment with interferon-beta through reduction in costimulatory CD80, which should attenuate antigen presentation to CD8+ T cells.20,48 Also, the number of CD80+ B cells is decreased with interferon-beta treatment, bringing the ratio of CD80+ to CD86+ cells closer to that of healthy controls.48,50 The decrease in CD80-expressing cells may result from an interferon-beta-mediated decrease in the production of interferon gamma or other cytokines that induce CD80+ B cells.48 In addition, treatment with interferon-beta increases the number of CD86 (B7.2)-expressing B cells.50 This increase in CD86+ cells should contribute to the downregulation of Th1 cell responses.50 BAFF levels in blood leukocytes and serum are also increased with interferon-beta treatment, suggesting a consequential increase in B-cell functioning.54 With the right combination of stimuli, this could lead to increased B-cell secretion of the anti-inflammatory cytokines IL-10 and TGF-β, suggesting that interferon-beta could induce CD4 and CD8 Treg as well as regulatory B cells.56 The benefits of the induction of regulatory B cells could depend on the characteristics of the patient, as B-cell induction could have undesirable consequences for patients with B-cell-mediated disease.54
Conclusions
Type 1 interferons have a wide range of effects on the immune system that can be associated with shifts in specific immune-mediated pathways that are involved in MS disease pathogenesis. Through various mechanisms, interferon-beta affects antigen presentation, potentially shifts Th1/Th2/Th17 polarization to a more anti-inflammatory state, increases regulatory T-cell and B-cell activity, and reduces the ability of B cells to present antigens.
Attempts have been made to identify biomarkers for treatment response. For example, nonresponders to interferon-beta treatment (based on magnetic resonance imaging criteria) show decreased IL-10 levels during the course of treatment.57 Treatment nonresponders may also have a lower IL-10/interferon-gamma and IL-4/interferon-gamma ratio than responders.40 The same study found that nonresponders had lower BDNF levels at baseline than responders.40 Induction of TIMP-1, the main inhibitor of MMP-9, may also be a marker of response to interferon-beta.39 Differences in molecular signatures include upregulation of many genes related to immune regulation, but also into protective and antioxidant profiles in most treated patients.58 In parallel, an exaggerated molecular response to interferon-beta in a subset of patients with MS has been associated with a poor response to treatment.59 While these and other findings suggest that biomarkers for treatment response may exist, more research is needed before these can be put into clinical practice.
Interestingly, current data would suggest that effective therapy for patients with MS requires intervention in some combination of immune compartments. For example, T-cell-specific therapies such as ustekinumab, an anti-IL-12/IL-23 antibody, failed to reduce the cumulative number of new lesions in a phase 2 trial.60 Also, the anti-CD4 antibody, cM-T412, produced long-lasting reductions in circulating CD4+ T cells, but this alone did not affect the number of active lesions in patients with MS.61 In a third approach, T-cell vaccination did not produce clinical or radiological improvements in previously treated patients with RRMS.62 In addition to these T-cell-targeted therapies, the fusion protein atacicept, which limits BAFF binding and B-cell maturation, actually increased inflammation in a phase 2 trial.63 In contrast, the B-cell-specific anti-CD20 antibody, rituximab, which destroys B cells and may also impact the capacity of B cells to present antigens to T cells, was very effective at reducing MS relapses.63,64 These studies suggest that some therapeutic approaches to treatment of patients with MS that target only one immunologic pathway or process may differ in effectiveness relative to those that affect the immune system at multiple levels. Notably, the very late antigen-4 antagonist natalizumab remains the exception to this hypothesis as it has a very focused mechanism of action but has clear efficacy in patients with RRMS.65 Interferon-beta, which has pleiotropic effects on immunity and brain cells, may be considered a broad spectrum therapeutic.
Acknowledgments
We would like to thank Mark J. Rametta, DO (Bayer HealthCare Pharmaceuticals, Whippany, NJ) for his assistance with drafting this manuscript. Additionally, we thank Robert C. Ristuccia, Ph.D. (Precept Medical Communications, Warren, NJ) for medical writing assistance that was funded by Bayer HealthCare Pharmaceuticals in the preparation of the manuscript.
Conflict of Interest
L. H. Kasper has been a consultant to and has received honoraria from Bayer HealthCare, EMD Serono, Teva Neuroscience, Genzyme, ONO Pharmaceutical, Novartis, Centocor, Genentech, Biogen/Idec, National Institutes of Health, and the National MS Society. He has served on speakers' bureaus for Bayer HealthCare, EMD Serono, Genzyme, and Teva Neuroscience. A. T. Reder has received compensation or has been on the advisory board/a consultant for Abbott Laboratories; American Medical Association; Astra Merck; Athena Neurosciences; Aventis Pharma; Bayer HealthCare Pharmaceuticals; Berlex Laboratories; BioMS Medical Corp.; Biogen and Biogen Idec; Blue Cross Blue Shield; Boehringer Ingelheim Pharmaceuticals, Inc.; Caremark Rx; Centocor, Inc.; Cephalon, Inc.; Connectics/Connective Therapeutics; CroMedica Global, Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; Genentech; Genzyme Corporation; GlaxoSmithKline; Hoechst Marion Roussel Canada Research, Inc.; Hoffmann-La Roche; Idec; Immunex; Institute for Health Care Quality; Johnson & Johnson; Pharmaceutical Research & Development; Medlink Neurology electronic journal (editor and author); NARCOMS; National MS Societies of Italy, Turkey, and the United States; NMSS & Paralyzed Veterans of America; Neurocrine Biosciences; Novartis Corporation; Parke-Davis; Pfizer, Inc.; Pharmacia & Upjohn; Protein Design Labs, Inc.; Quantum Biotechnologies, Inc.; Questcor; Quintiles, Inc.; RENEW study (postmarketing study of Novantrone in MS); Sandoz (now Novartis) and Novartis; Sention, Inc.; Schering; Serono; SmithKline Beecham; Specialized Therapeutics, a division of Berlipharm, Inc.; Takeda Pharmaceuticals; and Teva-Marion. He has also received research support from Bayer; Serono; Teva; the US National MS Society; and the Chinese, Egyptian, Illinois, Turkish, and United States governments.
References
- 1.Compston A, Coles A. Multiple sclerosis. Lancet. 2008;372:1502–1517. doi: 10.1016/S0140-6736(08)61620-7. [DOI] [PubMed] [Google Scholar]
- 2.Dhib-Jalbut S. Mechanisms of action of interferons and glatiramer acetate in multiple sclerosis. Neurology. 2002;58:S3–S9. doi: 10.1212/wnl.58.8_suppl_4.s3. [DOI] [PubMed] [Google Scholar]
- 3.Kasper LH, Shoemaker J. Multiple sclerosis immunology: the healthy immune system vs the MS immune system. Neurology. 2010;74(suppl 1):S2–S8. doi: 10.1212/WNL.0b013e3181c97c8f. [DOI] [PubMed] [Google Scholar]
- 4.Bekisz J, Sato Y, Johnson C, et al. Immunomodulatory effects of interferons in malignancies. J Interferon Cytokine Res. 2013;33:154–161. doi: 10.1089/jir.2012.0167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Runkel L, Meier W, Pepinsky RB, et al. Structural and functional differences between glycosylated and non-glycosylated forms of human interferon-beta (IFN-beta) Pharm Res. 1998;15:641–649. doi: 10.1023/a:1011974512425. [DOI] [PubMed] [Google Scholar]
- 6.Owens GP, Gilden D, Burgoon MP, et al. Viruses and multiple sclerosis. Neuroscientist. 2011;17:659–676. doi: 10.1177/1073858411386615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ochoa-Reparaz J, Mielcarz DW, Begum-Haque S, et al. Gut, bugs, and brain: role of commensal bacteria in the control of central nervous system disease. Ann Neurol. 2011;69:240–247. doi: 10.1002/ana.22344. [DOI] [PubMed] [Google Scholar]
- 8.Berer K, Mues M, Koutrolos M, et al. Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature. 2011;479:538–541. doi: 10.1038/nature10554. [DOI] [PubMed] [Google Scholar]
- 9.Lee YK, Menezes JS, Umesaki Y, et al. Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA. 2011;108(suppl 1):4615–4622. doi: 10.1073/pnas.1000082107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Engelhardt B. Immune cell entry into the central nervous system: involvement of adhesion molecules and chemokines. J Neurol Sci. 2008;274:23–26. doi: 10.1016/j.jns.2008.05.019. [DOI] [PubMed] [Google Scholar]
- 11.Dhib-Jalbut S, Marks S. Interferon-beta mechanisms of action in multiple sclerosis. Neurology. 2010;74(suppl 1):S17–S24. doi: 10.1212/WNL.0b013e3181c97d99. [DOI] [PubMed] [Google Scholar]
- 12.Seok J, Warren HS, Cuenca AG, et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci USA. 2013;110:3507–3512. doi: 10.1073/pnas.1222878110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nuyts A, Lee W, Bashir-Dar R, et al. Dendritic cells in multiple sclerosis: key players in the immunopathogenesis, key players for new cellular immunotherapies? Mult Scler. 2013;19:995–1002. doi: 10.1177/1352458512473189. [DOI] [PubMed] [Google Scholar]
- 14.Kaisho T. Pathogen sensors and chemokine receptors in dendritic cell subsets. Vaccine. 2012;30:7652–7657. doi: 10.1016/j.vaccine.2012.10.043. [DOI] [PubMed] [Google Scholar]
- 15.Bayas A, Stasiolek M, Kruse N, et al. Altered innate immune response of plasmacytoid dendritic cells in multiple sclerosis. Clin Exp Immunol. 2009;157:332–342. doi: 10.1111/j.1365-2249.2009.03964.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zamvil S, Nelson P, Trotter J, et al. T-cell clones specific for myelin basic protein induce chronic relapsing paralysis and demyelination. Nature. 1985;317:355–358. doi: 10.1038/317355a0. [DOI] [PubMed] [Google Scholar]
- 17.Balashov KE, Aung LL, Vaknin-Dembinsky A, et al. Interferon-beta inhibits toll-like receptor 9 processing in multiple sclerosis. Ann Neurol. 2010;68:899–906. doi: 10.1002/ana.22136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stasiolek M, Bayas A, Kruse N, et al. Impaired maturation and altered regulatory function of plasmacytoid dendritic cells in multiple sclerosis. Brain. 2006;129:1293–1305. doi: 10.1093/brain/awl043. [DOI] [PubMed] [Google Scholar]
- 19.Ifergan I, Kebir H, Alvarez JI, et al. Central nervous system recruitment of effector memory CD8+ T lymphocytes during neuroinflammation is dependent on alpha4 integrin. Brain. 2011;134:3560–3577. doi: 10.1093/brain/awr268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang H, Milo R, Swoveland P, et al. Interferon beta-1b reduces interferon gamma-induced antigen-presenting capacity of human glial and B cells. J Neuroimmunol. 1995;61:17–25. doi: 10.1016/0165-5728(95)00072-a. [DOI] [PubMed] [Google Scholar]
- 21.Derkow K, Bauer JM, Hecker M, et al. Multiple sclerosis: modulation of toll-like receptor (TLR) expression by interferon-beta includes upregulation of TLR7 in plasmacytoid dendritic cells. PLoS One. 2013;8:e70626. doi: 10.1371/journal.pone.0070626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vallittu AM, Saraste M, Airas L. CCR7 expression on peripheral blood lymphocytes is up-regulated following treatment of multiple sclerosis with interferon-beta. Neurol Res. 2007;29:763–766. doi: 10.1179/016164107X228633. [DOI] [PubMed] [Google Scholar]
- 23.Li Q, Milo R, Panitch H, et al. Effect of propranolol and IFN-beta on the induction of MHC class II expression and cytokine production by IFN-gamma IN THP-1 human monocytic cells. Immunopharmacol Immunotoxicol. 1998;20:39–61. doi: 10.3109/08923979809034808. [DOI] [PubMed] [Google Scholar]
- 24.Gravano DM, Hoyer KK. Promotion and prevention of autoimmune disease by CD8+ T cells. J Autoimmun. 2013;45:68–79. doi: 10.1016/j.jaut.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 25.Lee MA, Palace J, Stabler G, et al. Serum gelatinase B, TIMP-1 and TIMP-2 levels in multiple sclerosis. A longitudinal clinical and MRI study. Brain. 1999;122(Pt 2):191–197. doi: 10.1093/brain/122.2.191. [DOI] [PubMed] [Google Scholar]
- 26.Waubant E, Goodkin D, Bostrom A, et al. IFNbeta lowers MMP-9/TIMP-1 ratio, which predicts new enhancing lesions in patients with SPMS. Neurology. 2003;60:52–57. doi: 10.1212/wnl.60.1.52. [DOI] [PubMed] [Google Scholar]
- 27.Boz C, Ozmenoglu M, Velioglu S, et al. Matrix metalloproteinase-9 (MMP-9) and tissue inhibitor of matrix metalloproteinase (TIMP-1) in patients with relapsing-remitting multiple sclerosis treated with interferon beta. Clin Neurol Neurosurg. 2006;108:124–128. doi: 10.1016/j.clineuro.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 28.Stadelmann C, Kerschensteiner M, Misgeld T, et al. BDNF and gp145trkB in multiple sclerosis brain lesions: neuroprotective interactions between immune and neuronal cells? Brain. 2002;125:75–85. doi: 10.1093/brain/awf015. [DOI] [PubMed] [Google Scholar]
- 29.Correale J, Villa A. Role of CD8+ CD25+ Foxp3+ regulatory T cells in multiple sclerosis. Ann Neurol. 2010;67:625–638. doi: 10.1002/ana.21944. [DOI] [PubMed] [Google Scholar]
- 30.Durelli L, Conti L, Clerico M, et al. T-helper 17 cells expand in multiple sclerosis and are inhibited by interferon-beta. Ann Neurol. 2009;65:499–509. doi: 10.1002/ana.21652. [DOI] [PubMed] [Google Scholar]
- 31.Baeten DL, Kuchroo VK. How cytokine networks fuel inflammation: interleukin-17 and a tale of two autoimmune diseases. Nat Med. 2013;19:824–825. doi: 10.1038/nm.3268. [DOI] [PubMed] [Google Scholar]
- 32.Wang X, Ma C, Wu J, et al. Roles of T helper 17 cells and interleukin-17 in neuroautoimmune diseases with emphasis on multiple sclerosis and Guillain-Barre syndrome as well as their animal models. J Neurosci Res. 2013;91:871–881. doi: 10.1002/jnr.23233. [DOI] [PubMed] [Google Scholar]
- 33.Pan HF, Li XP, Zheng SG, et al. Emerging role of interleukin-22 in autoimmune diseases. Cytokine Growth Factor Rev. 2013;24:51–57. doi: 10.1016/j.cytogfr.2012.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beyeen AD, Adzemovic MZ, Ockinger J, et al. IL-22RA2 associates with multiple sclerosis and macrophage effector mechanisms in experimental neuroinflammation. J Immunol. 2010;185:6883–6890. doi: 10.4049/jimmunol.1001392. [DOI] [PubMed] [Google Scholar]
- 35.Antel J, Arnason BG. Pathogenesis of multiple sclerosis. Eur J Clin Invest. 1977;7:461–463. doi: 10.1111/j.1365-2362.1977.tb01636.x. [DOI] [PubMed] [Google Scholar]
- 36.Reder AT, Antel JP, Oger JJ, et al. Low T8 antigen density on lymphocytes in active multiple sclerosis. Ann Neurol. 1984;16:242–249. doi: 10.1002/ana.410160214. [DOI] [PubMed] [Google Scholar]
- 37.Fritzsching B, Haas J, Konig F, et al. Intracerebral human regulatory T cells: analysis of CD4+ CD25+ FOXP3+ T cells in brain lesions and cerebrospinal fluid of multiple sclerosis patients. PLoS One. 2011;6:e17988. doi: 10.1371/journal.pone.0017988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Noronha A, Toscas A, Jensen MA. Interferon beta augments suppressor cell function in multiple sclerosis. Ann Neurol. 1990;27:207–210. doi: 10.1002/ana.410270219. [DOI] [PubMed] [Google Scholar]
- 39.Comabella M, Rio J, Espejo C, et al. Changes in matrix metalloproteinases and their inhibitors during interferon-beta treatment in multiple sclerosis. Clin Immunol. 2009;130:145–150. doi: 10.1016/j.clim.2008.09.010. [DOI] [PubMed] [Google Scholar]
- 40.Dhib-Jalbut S, Sumandeep S, Valenzuela R, et al. Immune response during interferon beta-1b treatment in patients with multiple sclerosis who experienced relapses and those who were relapse-free in the START study. J Neuroimmunol. 2013;254:131–140. doi: 10.1016/j.jneuroim.2012.08.012. [DOI] [PubMed] [Google Scholar]
- 41.Guo B, Chang EY, Cheng G. The type I IFN induction pathway constrains Th17-mediated autoimmune inflammation in mice. J Clin Invest. 2008;118:1680–1690. doi: 10.1172/JCI33342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mascanfroni ID, Yeste A, Vieira SM, et al. IL-27 acts on DCs to suppress the T cell response and autoimmunity by inducing expression of the immunoregulatory molecule CD39. Nat Immunol. 2013;14:1054–1063. doi: 10.1038/ni.2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Caminero A, Comabella M, Montalban X. Tumor necrosis factor alpha (TNF-alpha), anti-TNF-alpha and demyelination revisited: an ongoing story. J Neuroimmunol. 2011;234:1–6. doi: 10.1016/j.jneuroim.2011.03.004. [DOI] [PubMed] [Google Scholar]
- 44.Cope AP, Liblau RS, Yang XD, et al. Chronic tumor necrosis factor alters T cell responses by attenuating T cell receptor signaling. J Exp Med. 1997;185:1573–1584. doi: 10.1084/jem.185.9.1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yong VW, Chabot S, Stuve O, et al. Interferon beta in the treatment of multiple sclerosis: mechanisms of action. Neurology. 1998;51:682–689. doi: 10.1212/wnl.51.3.682. [DOI] [PubMed] [Google Scholar]
- 46.Lowther DE, Chong DL, Ascough S, et al. Th1 not Th17 cells drive spontaneous MS-like disease despite a functional regulatory T cell response. Acta Neuropathol. 2013;126:501–515. doi: 10.1007/s00401-013-1159-9. [DOI] [PubMed] [Google Scholar]
- 47.Feng X, Yau D, Holbrook C, et al. Type I interferons inhibit interleukin-10 production in activated human monocytes and stimulate IL-10 in T cells: implications for Th1-mediated diseases. J Interferon Cytokine Res. 2002;22:311–319. doi: 10.1089/107999002753675730. [DOI] [PubMed] [Google Scholar]
- 48.Genc K, Dona DL, Reder AT. Increased CD80(+) B cells in active multiple sclerosis and reversal by interferon beta-1b therapy. J Clin Invest. 1997;99:2664–2671. doi: 10.1172/JCI119455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dalakas MC. B cells as therapeutic targets in autoimmune neurological disorders. Nat Clin Pract Neurol. 2008;4:557–567. doi: 10.1038/ncpneuro0901. [DOI] [PubMed] [Google Scholar]
- 50.Huang H, Ito K, Dangond F, et al. Effect of interferon beta-1a on B7.1 and B7.2 B-cell expression and its impact on T-cell proliferation. J Neuroimmunol. 2013;258:27–31. doi: 10.1016/j.jneuroim.2013.02.010. [DOI] [PubMed] [Google Scholar]
- 51.Ireland S, Monson N. Potential impact of B cells on T cell function in multiple sclerosis. Mult Scler Int. 2011;2011:423971. doi: 10.1155/2011/423971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kappos L, Li D, Calabresi PA, et al. Ocrelizumab in relapsing-remitting multiple sclerosis: a phase 2, randomised, placebo-controlled, multicentre trial. Lancet. 2011;378:1779–1787. doi: 10.1016/S0140-6736(11)61649-8. [DOI] [PubMed] [Google Scholar]
- 53.Hawker K, O'Connor P, Freedman MS, et al. Rituximab in patients with primary progressive multiple sclerosis: results of a randomized double-blind placebo-controlled multicenter trial. Ann Neurol. 2009;66:460–471. doi: 10.1002/ana.21867. [DOI] [PubMed] [Google Scholar]
- 54.Krumbholz M, Faber H, Steinmeyer F, et al. Interferon-beta increases BAFF levels in multiple sclerosis: implications for B cell autoimmunity. Brain. 2008;131:1455–1463. doi: 10.1093/brain/awn077. [DOI] [PubMed] [Google Scholar]
- 55.Krumbholz M, Theil D, Derfuss T, et al. BAFF is produced by astrocytes and up-regulated in multiple sclerosis lesions and primary central nervous system lymphoma. J Exp Med. 2005;201:195–200. doi: 10.1084/jem.20041674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Meinl E, Krumbholz M, Hohlfeld R. B lineage cells in the inflammatory central nervous system environment: migration, maintenance, local antibody production, and therapeutic modulation. Ann Neurol. 2006;59:880–892. doi: 10.1002/ana.20890. [DOI] [PubMed] [Google Scholar]
- 57.Graber JJ, Ford D, Zhan M, et al. Cytokine changes during interferon-beta therapy in multiple sclerosis: correlations with interferon dose and MRI response. J Neuroimmunol. 2007;185:168–174. doi: 10.1016/j.jneuroim.2007.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Croze E, Yamaguchi KD, Knappertz V, et al. Interferon-beta-1b-induced short- and long-term signatures of treatment activity in multiple sclerosis. Pharmacogenomics J. 2013;13:443–451. doi: 10.1038/tpj.2012.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rudick RA, Rani MR, Xu Y, et al. Excessive biologic response to IFNbeta is associated with poor treatment response in patients with multiple sclerosis. PLoS One. 2011;6:e19262. doi: 10.1371/journal.pone.0019262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Segal BM, Constantinescu CS, Raychaudhuri A, et al. Repeated subcutaneous injections of IL12/23 p40 neutralising antibody, ustekinumab, in patients with relapsing-remitting multiple sclerosis: a phase II, double-blind, placebo-controlled, randomised, dose-ranging study. Lancet Neurol. 2008;7:796–804. doi: 10.1016/S1474-4422(08)70173-X. [DOI] [PubMed] [Google Scholar]
- 61.van Oosten BW, Lai M, Hodgkinson S, et al. Treatment of multiple sclerosis with the monoclonal anti-CD4 antibody cM-T412: results of a randomized, double-blind, placebo-controlled, MR-monitored phase II trial. Neurology. 1997;49:351–357. doi: 10.1212/wnl.49.2.351. [DOI] [PubMed] [Google Scholar]
- 62.Fox E, Wynn D, Cohan S, et al. A randomized clinical trial of autologous T-cell therapy in multiple sclerosis: subset analysis and implications for trial design. Mult Scler. 2012;18:843–852. doi: 10.1177/1352458511428462. [DOI] [PubMed] [Google Scholar]
- 63.Hartung HP, Kieseier BC. Atacicept: targeting B cells in multiple sclerosis. Ther Adv Neurol Disord. 2010;3:205–216. doi: 10.1177/1756285610371146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Castillo-Trivino T, Braithwaite D, Bacchetti P, et al. Rituximab in relapsing and progressive forms of multiple sclerosis: a systematic review. PLoS One. 2013;8:e66308. doi: 10.1371/journal.pone.0066308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Miller DH, Khan OA, Sheremata WA, et al. A controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2003;348:15–23. doi: 10.1056/NEJMoa020696. [DOI] [PubMed] [Google Scholar]