

Abstract
Key Clinical Message
Copy losses/gains of the Williams–Beuren syndrome (WBS) region cause neurodevelopmental disorders with variable expressivity. The WBS prenatal diagnosis cannot be easily performed by ultrasound because only few phenotypic features can be assessed. Three WBS and the first reciprocal duplication prenatal cases are described with a review of the literature.
Keywords: Array CGH, phenotype, prenatal, prenatal BACs on beads, Williams–Beuren critical region.
Introduction
Copy losses and gains of the Williams–Beuren syndrome (WBS) 7q11.23 region are responsible for neurodevelopmental disorders with multi-system involvement and variable expressivity. WBS (OMIM 194050) deletion is characterized by cardiac malformation (most frequently supravalvular aortic stenosis, SVAS), psychomotor retardation, distinctive facial appearance, and a specific cognitive and behavioral profile. The incidence of the typical presentation is 1/7500–1/10,000 live births but atypical forms, of unknown incidence, also exist [1]. The 7q11.23 reciprocal duplication includes less distinctive facial dysmorphisms than those of WBS and prominent speech delay [2].
WBS and WBS reciprocal duplication syndrome belong to a group termed “genomic disorders” caused by a rearrangement of the genome. The common recurrent 1.55 Mb microdeletion occurs by non-allelic homologous recombination (NAHR) between low copy repeats (LCRs) flanking the deleted region resulting in unequal crossing over [3–6]. A larger deletion (1.84 Mb) has been observed in about 5% of the patients [5]. Because NAHR can generate both microdeletions and microduplications, it was suspected that the reciprocal microduplication should also occur [7].
The common rearrangement-involved region includes about two dozen genes, including the elastin (ELN, OMIM 130160) and LIM kinase 1 (LIMK1, OMIM 601329) genes for which dosage-sensitive pathways may act in reciprocal fashion, resulting in converse phenotypes in deletion and duplication patients. Haploinsufficiency for the ELN gene is responsible for connective tissue and cardiovascular abnormalities in WBS patients [8], whereas LIMK1 has been implicated in their specific cognitive profile being involved in synapse formation and/or maintenance [9]. GTF2I (General Transcription Factor II-I) has been associated to the hypersociability in WBS patients [10], and at the opposite, its duplication, to separation anxiety [11]. This gene has also been reported to play a critical role in autism spectrum disorders [12]. The contributions of the remaining genes in the critical region to the various features of both syndromes remain open and studies aiming at elucidation of genotype/phenotype correlation in WBS and reciprocal duplication have been focused on the role of genes inside the deleted/duplicated interval.
While the parental transmission for the deletion in WBS is rare [13–16], there is a high frequency of parental transmission in 7q11.23 duplication patients [17].
Neither the microdeletion nor the microduplication are visible by conventional karyotyping and traditionally they have been diagnosed by fluorescence in situ hybridization (FISH) analysis. Other targeted techniques such as quantitative real-time polymerase chain reaction (PCR), segregation analysis of microsatellite, multiplex ligation-dependent-probe amplification (MLPA) and, more recently, prenatal BACs-on-Beads (BoBs) have also been shown to be reliable to detect them [18–22].
The prenatal diagnosis of the WBS cannot be easily performed because only few features of the WBS phenotype can be assessed and investigated by ultrasound (US), hence, a well-defined WBS prenatal phenotype cannot be delineated. To date, three prenatal WBS cases have been reported with a normal karyotype [20,23,24] and one with an abnormal karyotype [25]. Regarding the reciprocal duplication, prenatal cases have never been described, likely due to the milder pathological consequences that tend to arise with gene duplications compared with the reciprocal deletions.
Herein three additional prenatal cases of WBS and the first prenatal case of WBS reciprocal duplication are described.
Methods
The study has been approved by the TOMA laboratory Institutional Review Board (IRB) (IRB project #0000008; 22 December 2011). After obtaining parental informed consent, karyotype analysis was performed in all cases of amniotic fluid (AF, cases 1 and 2) and of chorionic villous samples (CVS, cases 3 previously included in Gruchy et al., 2012 and case 4) in agreement with the European guidelines. Prenatal BoBs™ CE-IVD analysis (PerkinElmer LAS, Wallac, Turku, Finland) was also performed as a rapid diagnostic or confirmatory test in three pregnancies (cases 1, 2, and 4) to provide the dosage of chromosomes 13, 18, 21, X/Y, and of critical regions associated with nine well-characterized microdeletion syndromes with significant newborn morbidity and mortality but without specific echographic findings by 24–26 weeks of gestation (wg) [26]. Confirmatory FISH analysis on the fetuses and parents were performed with ELN-specific probes (in case 1 probe by Cytocell, Newmarket Road, Cambridge, UK; in cases 2 and 4 probe by Vysis, Inc., Downers Grove, IL; in case 3 probe by Oncor, Inc., Gaithersburg, MD). Because chromosome 7 is imprinted and more complex underlying rearrangements cannot be excluded, uniparental disomy testing of microsatellite markers located along the involved chromosome was performed on parental and AF DNAs of the first pregnancy. In this case, the parental origin of the microdeletion was also assessed by segregation analysis of the microsatellite makers for the parents and fetus located in the WBS critical region (D7S489, D7S2479, D7S2476, D7S2523, and D7S3015). In fetus 3, bacterial artificial chromosome (BAC) array comparative genomic hybridization (aCGH) by a CytoChip Focus slide (BlueGnome®; Illumina, Cambridge, UK) was performed on cell-free fetal (Cff) DNA [27] to further investigate the cause of fetal abnormalities. In the fetus with WBS-CR duplication genome-wide BAC aCGH (Genome-wide BAC platform ConstitutionalChip4.0; PerkinElmer LAS, Wallac, Turku, Finland) analysis was conducted on DNA extracted from a fragment of the native CVS.
Results
Case 1 (WBS-CR deletion)
The first case with WBS microdeletion was a 36-years-old patient, gravida 2 para 1, that was referred because of intrauterine growth restriction (IUGR) at 20 + 2 week of gestation (wg). The US examination confirmed that the fetus was small for gestational age. At 22 wg the anatomical survey did not reveal abnormalities, but the femur was disproportionately shortened (<−3 SD from the mean) compared with head and trunk size, and the bowel appeared echogenic. Utero-placental insufficiency was considered unlikely because of the past obstetric history (the patient had previously delivered a good size infant after an uneventful pregnancy).
The possibility of a complex congenital anomaly was discussed with the couple and an amniocentesis was performed to obtain a karyotype. Prenatal BoBs analysis showed the presence of the deletion of all BACs spanning the WBS critical region (Fig.1A). The abnormality was confirmed by FISH on metaphase chromosomes. Uniparental disomy of chromosome 7 was excluded on DNA from AF. Parental studies by karyotype and FISH analyses showed normal results indicating that the microdeletion was de novo in the fetus. The karyotype was therefore: 46,XX.rsa 7q11.23(ELN)x1.ish del(7)(q11.23q11.23)(ELN-,LIMK1-,D7S613-)dn (Fig.1B). The segregation analysis of the parents and fetus of the microsatellite makers located in the WBS critical region showed that the microdeletion arose on the paternal allele (Fig.1C). After the genetic counseling, the parents decided to terminate the pregnancy and the autopsy at 23 wg showed left kidney hypoplasia and the ectasis of the IV ventricle.
Figure 1.
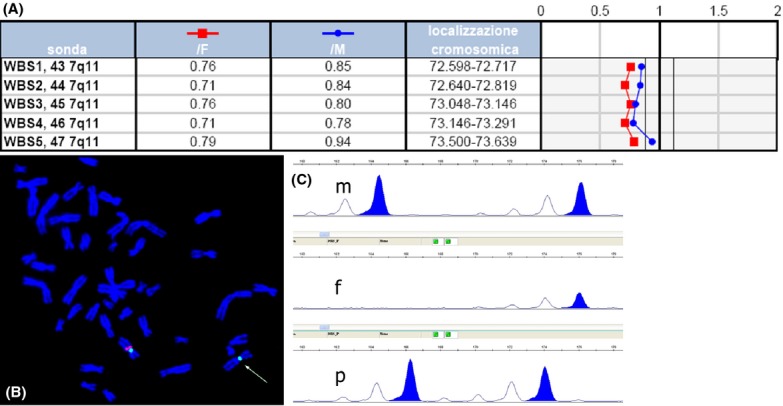
Genetic results of the first case with WBS microdeletion. (A) Prenatal BoBs profile showing copy loss for all beads mapping in the WBS-CR; (B) WBS microdeletion confirmed by fluorescence in situ hybridization (FISH) on metaphases with a specific probe for the elastin (ELN) gene 46,XX. ish del(7)(q11.23q11.23)(ELN-,LIMK1-,D7S613-); (C) segregation analysis from parents (m and p = maternal and paternal alleles) to fetus (f = fetal alleles) of an informative microsatellite maker located in the WBS critical region showing that the microdeletion involved the paternal allele.
Case 2 (WBS-CR deletion)
The second case was a 31-years-old propositus, gravida 1 para 1, that showed at 32 wg IUGR (fetal biometric data <3rd centile). Karyotype of AF was 46,XX and Prenatal BoBs™ analysis, requested in parallel, disclosed the presence of the WBS critical region microdeletion. This microdeletion was confirmed by FISH. Parental studies showed a normal FISH signal pattern and a normal karyotype. The result was therefore: 46,XX.rsa 7q11.23(ELN)x1.ish del(7)(q11.23q11.23)(ELN-)dn.
The couple requested to terminate the pregnancy at 34 wg and the autopsy showed dolichocephaly, fifth finger clinodactyly, peculiar facial dysmorphisms (malar hypoplasia, bulbous nose, wide mouth with thick lower lip, pointed chin, and large earlobes) (Fig.2A), aortic stenosis (base of the aorta Ao 4.5 mm; base of the pulmonary artery 7 mm), and long aneurismal ductus arteriosus as observed in Marfan syndrome. The histology examination showed abnormal large arteries (Fig.2B) and abnormal sclera (sparse, with irregular fibers).
Figure 2.
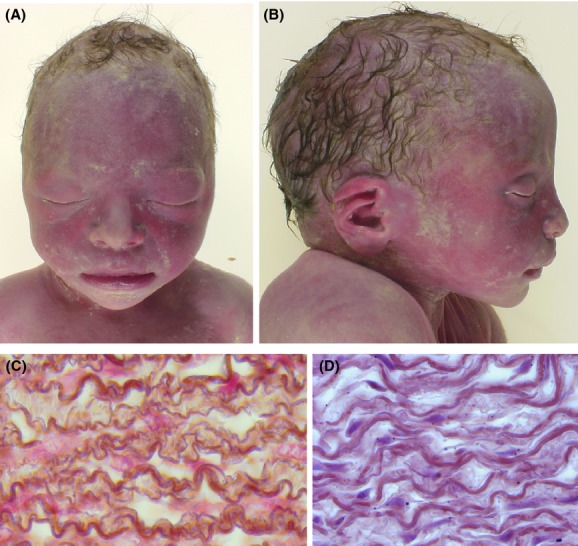
Fetopathological examination of the second case with WBS microdeletion. (A and B) dolichocephaly, peculiar facial dysmorphisms (malar hypoplasia, bulbous nose, wide mouth with thick lower lip, pointed chin, and large earlobes) are visible; (C and D) histology examination of descending aorta showed abnormal large arteries with abnormal sclera (sparse, with irregular fibers) (C) compared with the control (D).
Case 3 (WBS-CR deletion)
In the third case, a 30-year-old patient was referred because of omphalocele at 13 wg. Fetal karyotype from CVS was normal (46,XY). At 18 wg, IUGR was detected and at this time termination of pregnancy occurred and AF was obtained for cytogenetic analysis. ArrayCGH conducted on Cff DNA showed the presence of WBS critical region microdeletion arr[hg18] 7q11.23 (72,171,274-74,159,511)x1. FISH analysis confirmed the aCGH result on the fetus. Parental studies showed a normal FISH signal pattern and a normal karyotype. The result was therefore: 46,XY. arr[hg18] 7q11.23 (72,171,274-74,159,511)x1.ish del(7)(q11.23q11.23)(ELN-)dn.
The autopsy revealed facial dysmorphism (anteverted nares, large mouth, micrognathia, low-set ears), fifth finger clinodactyly, lung abnormal lobulation, gallbladder hypoplasia, and rib number anomalies (13 ribs on left side) (Fig.3).
Figure 3.
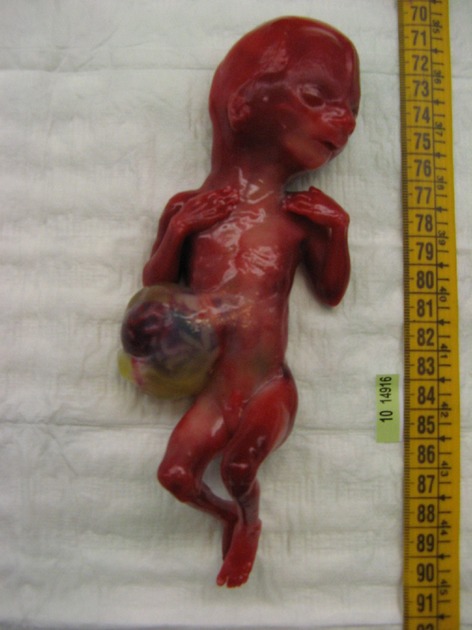
Fetopathological examination of the third case with WBS microdeletion: in utero growth retardation, peculiar facial dysmorphisms (anteverted nares, large mouth, micrognathia, low-set ears), clinodactyly V, large omphalocele, lung abnormal lobulation (three lobes on left lung), and rib number anomalies (13 ribs on left side) were detected.
Case 4 (WBS-CR duplication)
In the case of WBS duplication, a 34-year-old patient was referred because of increased nuchal translucency (NT, 4.8 mm), absence of the nasal bone, and inversion of the “a” wave of the ductus venosus at 11 + 4 wg. The couple had five previous pregnancies: two of them (the first and the fifth) ended in a spontaneous abortion in the first trimester and the remaining three delivered at term one male and two females, one of them with speech delay. The karyotype on CV was normal (46,XY). US investigations detected choroid plexus cysts and bilateral renal pyelectasis at 16 wg; head circumference at 90th centile with a femur/head circumference between −1 and −2 SD from the mean and a borderline unilateral ventriculomegaly at 19 + 4 wg (left 11.1 mm, right 9.0 mm); increased NT, ventriculomegaly, bilateral renal pyelectasis. Choroid plexus cysts were still visible at 20 + 3 wg. Array CGH detected a submicroscopic copy number gain involving the WBS critical region arr[hg18] 7q11.23 (72,390,001-73,910,001)x3 that was also confirmed by Prenatal BoBs (Fig.4) and, after the termination of pregnancy, by FISH analysis on nuclei from skin fibroblasts [nuc ish (ELNx3)] (data not shown). The result was therefore: 46,XY. arr[hg18] 7q11.23 (72,390,001-73,910,001)x3.rsa 7q11.23(ELN)x3.nuc ish (ELNx3). Parental karyotype and FISH analyses were normal.
Figure 4.
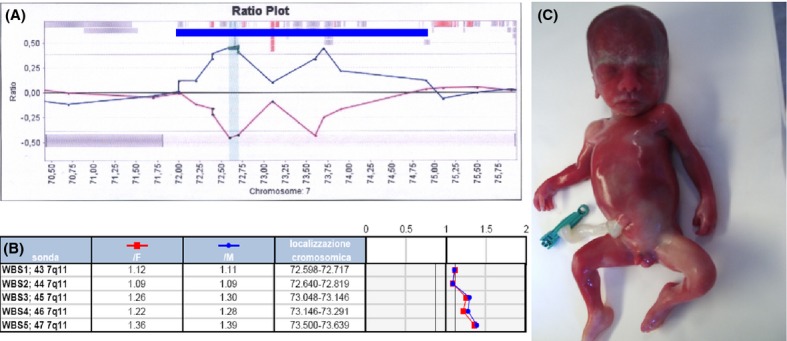
Fetus with WBS microduplication. (A) BAC array comparative genomic hybridization profile of the WBS region: the blue bar indicates the maximal extension of the duplicated segment; (B) confirmatory Prenatal BoBs profile showing copy gain of WBS-CR; (C) fetus at 21 wg with WBS duplication. Craniofacial dysmorphic features include prevalence of neurocranium compared to visceral cranium, hypertelorism, periorbital edema, mild malar hypoplasia, high nasal bridge, broad nose, and mild micrognathia
Magnetic resonance investigation at 21 wg showed enlarged lateral ventricles (atrium width 13 mm left and 11 mm right) and underdevelopment of sylvian, hippocampal, and parietooccipital fissures (lissencephaly-type abnormality). After termination of pregnancy, external physical examination of the fetus showed the prevalence of neurocranium compared to visceral cranium, facial dysmorphisms (hypertelorism, periorbital edema, mild malar hypoplasia, high nasal bridge, broad nose, and mild micrognathia), and mild nuchal edema. The autopsy revealed an increased weight for gestational age (603 g; >2.0 SD from the mean; reference 518 g), increased Crown-rump length (20 cm >2.0 SD; reference 16.0 cm), abdominal circumference 16 cm (50–95th centile) [28,29], and confirmed the presence of a mild bilateral ventriculomegaly, bilateral renal pyelectasis and choroid plexus cysts.
A seventh pregnancy is currently ongoing and fetal karyotype and FISH analyses performed at 16 wg on 100 nuclei showed normal results [46,XX.nuc ish(ELNx2)].
Discussion
In this study, three prenatal cases with WBS and one with WBS reciprocal duplication have been described. Four additional WBS prenatal cases are present in literature [20,25–27]. Regarding the reciprocal duplication, to the best of our knowledge, this is the first reported case detected by prenatal diagnosis. Table1 summarizes clinical findings in our group of fetuses and in the reported cases. In the total series of fetuses (deleted and duplicated, current group + reported cases) growth defects were present as reciprocal phenotypes; IUGR in fetuses with WBS-CR microdeletion and increased weight and length for gestational age in the fetus with the complementary microduplication. This supports the hypothesis that dosage-sensitive genes with putative effect on growth are present in the WBS-CR (GTF2I family) [30] and that they also act in prenatal period. In case 3, the presence of omphalocele in early gestation could have influenced the early occurrence of IUGR condition at 18 wg. In the rest of the cohort, isolated IUGR was detectable as early as 20 weeks. IUGR has also been reported in WBS adult patients, the mean decrease in adult height compared to target height is nearly 10 cm and head circumference can be also reduced [31,32]. Recently, growth defects and impaired cognitive-behavioral abilities have been observed in Eif4h null mice, whose human homolog maps in WBSCR1 region [33]. We hypothesize that IUGR can be an early indication for the suspicion of WBS that should be prenatally investigated in addition to the other chromosomal causes (e.g.,: 4p16.3, 6q24-q25, and 15q26-qter microdeletions and uniparental disomy of chromosome 7) after a normal karyotype. In nearly all deleted prenatal cases, cardiac defects were present, and in case by Kontos et al. [20], a small ventricular septal defect was detected which is not among the common cardiac features of WBS. Minor SVAS together with the characteristic facial dysmorphisms were identified only after a targeted US examination at 30 wg prompted by the cytogenetic findings [24]. Haploinsufficiency of the ELN gene produces the cardiovascular pathology of WBS; arteriopathy with vascular stenoses were well documented in case 2 of this study.
Table 1.
Phenotypic features of fetuses with deletion or duplication of 7q11.23 region (present study + literature)
| WBS deletion fetus | Karyotype | Referral reason and week of gestation | Growth features | Cardiac defects | Other findings |
|---|---|---|---|---|---|
| 1-present cohort | 46,XX.rsa 7q11.23(ELN)x1.ish del(7)(q11.23q11.23)(ELN-,LIMK1-,D7S613-)dn | IUGR, 20 + 2 wg | IUGR (<5th centile) at 20 + 2 wg | Not visible | At 20 + 2 wg echogenic bowel, short femur; at 23 wg left kidney hypoplasia, ectasis of the IV right ventricle |
| 2-present cohort | 46,XX.rsa 7q11.23(ELN)x1.ish del(7)(q11.23q11.23)(ELN-) | IUGR, 32 wg | IUGR (<3rd centile) at 32 wg | Aortic stenosis, ductus arteriosus | Dolichocephaly, clinodactyly V, peculiar facial dysmorphisms |
| 3-present cohort | 46,XY. arr[hg18] 7q11.23 (72,171,274-74,159,511)x1.ish del(7)(q11.23q11.23)(ELN-)dn | Omphalocele, 13 wg | IUGR at 18 wg | Not visible | Clinodactyly V, lung abnormal lobulation, gallbladder hypoplasia, rib number anomalies and peculiar dysmorphisms (anteverted nares, large mouth, micrognathia, low set ears) |
| [25] | 46,XX, t(6;7)(q27;q11.23).ish t(6;7)(ELNdim;ELN+) | Hydrops fetalis, polyhydramnios, absence of diastolic umbilical blood flow, and biophysical profile score of 2/8 at 30 wg | NA | Severe arteriopathy supravalvular aortic and pulmonary stenosis | Large anterior fontanel, high forehead, frontal bossing, hypoplastic supraorbital ridges, downslanting palpebral fissures, long philtrum, retrognathia, and low-set prominent and folded ears |
| [20] | 46,XX. rsa(13,18,21,X)x2. rsa7q11.23(P064B)x1. nuc ish (ELNx1) | Ventricular septal defect (VSD), 23 wg | NA | Small VSD | No further abnormalities |
| [23] | 46,XY.ish del(7)(q11.23)(ELN-) | Symmetrical IUGR, 20 wg | IUGR (5th centile) at 20 wg | Narrowing of the ascending aorta (SVAS at 29 wg autopsy) | At 22 wg echogenic bowel; at 29 wg (autopsy) low-set ears, small flat nose; large mouth, long philtrum, everted lower lip |
| [24] | 46,XX.rsa[hg18] 7q11.23(72,639,987-73,639,000)x1. ish del(7)(q11.23)(ELN-) | IUGR, 25 wg | IUGR at 25 wg | Small SVAS visible at 30 wg | At 30 wg bulbous nasal tip, long philtrum, wide mouth with an everted full lower lip |
| WBS duplication fetus | Karyotype | Referral reason and week of gestation | Growth features | Brain MRI findings | Other findings |
| 4-present cohort | 46,XY. arr[hg18] 7q11.23 (72,390,001-73,910,001)x3.rsa 7q11.23(ELN)x3.nuc ish (ELNx3) | Increased NT, absence of nasal bone, and inversion of the “a” wave of the ductus venosus, 11 + 4 wg | Increased length and weight (>2 SD) for gestational age at 21 wg | Mild bilateral ventriculomegaly, Lissencephaly-type abnormality | Bilateral renal pyelectasis, choroid plexus cysts, prevalence of neurocranium compared to visceral cranium, facial dysmorphisms (hypertelorism, periorbital edema, mild malar hypoplasia, high nasal bridge, broad nose and, mild micrognathia) |
Phenotypic features in patients with 7q11.23 duplication include normal growth in the majority of cases, severe language impairment, and mild intellectual disability, hypotonia is a feature in more than 50% of cases and autism is present in probably less than 50%. Epilepsy is present in less than 25% of the patients. Brain magnetic resonance imaging (MRI) is abnormal in the majority of cases including ventricular dilatation, hypoplasia of the corpus callosum and, in one case, a simplified gyral pattern but no consistent brain abnormalities were obvious [17,34,35]. Paradoxically, in the present case, the prenatal phenotype of the duplication of 7q11.23 seems to be more severe than the reciprocal deletion and brain MRI detected a mild bilateral ventriculomegaly with Lissencephaly-type abnormalities at 21 wg. Ventriculomegaly could be a relatively early sign of a neuronal migration defect and could represent an indication to prenatally investigate the presence of the 7q11.23 duplication in addition to the other chromosomal causes (e.g.: 17p13.3 microdeletion) after a normal karyotype.
Microarray or Prenatal BoBs™ analyses on extensive prenatal cohorts will clarify the incidence of WBS-CR microdeletion and the reciprocal microduplication in fetuses with IUGR and ventriculomegaly, respectively, with a normal karyotype.
Conflict of Interests
Federico Maggi is the President, Founder and CEO of TOMA laboratory. Giuseppe Simoni is the Scientific Director of TOMA laboratory. Federico Maggi and Giuseppe Simoni are the holders of TOMA Advanced Biomedical Assays S.p.A.. Livia Marcato, Simona De Toffol and Francesca R. Grati are full-time employees of TOMA Advanced Biomedical Assays S.p.A. and declare no conflict of interest. The remaining authors have no conflicts of interest to declare.
References
- 1.Strømme P, Bjørnstad PG, Ramstad K. Prevalence estimation of Williams syndrome. J. Child Neurol. 2002;17:269–271. doi: 10.1177/088307380201700406. [DOI] [PubMed] [Google Scholar]
- 2.Merla G, Brunetti-Pierri N, Micale L, Fusco C. Copy number variants at Williams-Beuren syndrome 7q11.23 region. Hum. Genet. 2010;128:3–26. doi: 10.1007/s00439-010-0827-2. [DOI] [PubMed] [Google Scholar]
- 3.Stankiewicz P, Lupski JR. Genome architecture, rearrangements and genomic disorders. Trends Genet. 2002;18:74–82. doi: 10.1016/s0168-9525(02)02592-1. [DOI] [PubMed] [Google Scholar]
- 4.Urbán Z, Helms C, Fekete G, Csiszár K, Bonnet D, Munnich A, et al. 7q11.23 deletions in Williams syndrome arise as a consequence of unequal meiotic crossover. Am. J. Hum. Genet. 1996;59:958–962. [PMC free article] [PubMed] [Google Scholar]
- 5.Bayés M, Magano LF, Rivera N, Flores R, Pérez Jurado LA. Mutational mechanisms of Williams-Beuren syndrome deletions. Am. J. Hum. Genet. 2003;73:131–151. doi: 10.1086/376565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hillier LW, Fulton RS, Fulton LA, Graves TA, Pepin KH, Wagner-McPherson C, et al. The DNA sequence of human chromosome 7. Nature. 2003;424:157–164. doi: 10.1038/nature01782. [DOI] [PubMed] [Google Scholar]
- 7.Lupski JR. Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 1998;14:417–422. doi: 10.1016/s0168-9525(98)01555-8. [DOI] [PubMed] [Google Scholar]
- 8.Li DY, Faury G, Taylor DG, Davis EC, Boyle WA, Mecham RP, et al. Novel arterial pathology in mice and humans hemizygous for elastin. J. Clin. Invest. 1998;102:1783–1787. doi: 10.1172/JCI4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scott RW, Olson MF. LIM kinases: function, regulation and association with human disease. J. Mol. Med. 2007;85:555–568. doi: 10.1007/s00109-007-0165-6. [DOI] [PubMed] [Google Scholar]
- 10.Sakurai T, Dorr NP, Takahashi N, McInnes LA, Elder GA, Buxbaum JD. Haploinsufficiency of Gtf2i, a gene deleted in Williams Syndrome, leads to increases in social interactions. Autism Res. 2011;4:28–39. doi: 10.1002/aur.169. [DOI] [PubMed] [Google Scholar]
- 11.Mervis CB, Dida J, Lam E, Crawford-Zelli NA, Young EJ, Henderson DR, et al. Duplication of GTF2I results in separation anxiety in mice and humans. Am. J. Hum. Genet. 2012;90:1064–1070. doi: 10.1016/j.ajhg.2012.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Malenfant P, Liu X, Hudson ML, Qiao Y, Hrynchak M, Riendeau N, et al. Association of GTF2i in the Williams-Beuren syndrome critical region with autism spectrum disorders. J. Autism Dev. Disord. 2012;42:1459–1469. doi: 10.1007/s10803-011-1389-4. [DOI] [PubMed] [Google Scholar]
- 13.Morris CA, Thomas IT, Greenberg F. Williams syndrome: autosomal dominant inheritance. Am. J. Med. Genet. 1993;47:478–481. doi: 10.1002/ajmg.1320470409. [DOI] [PubMed] [Google Scholar]
- 14.Ounap K, Laidre P, Bartsch O, Rein R, Lipping-Sitska M. Familial Williams-Beuren syndrome. Am. J. Med. Genet. 1998;80:491–493. doi: 10.1002/(sici)1096-8628(19981228)80:5<491::aid-ajmg10>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 15.Pankau R, Siebert R, Kautza M, Schneppenheim R, Gosch A, Wessel A, et al. Familial Williams-Beuren syndrome showing varying clinical expression. Am. J. Med. Genet. 2001;98:324–329. doi: 10.1002/1096-8628(20010201)98:4<324::aid-ajmg1103>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 16.Sadler LS, Robinson LK, Verdaasdonk KR, Gingell R. The Williams syndrome: evidence for possible autosomal dominant inheritance. Am. J. Med. Genet. 1993;47:468–470. doi: 10.1002/ajmg.1320470406. [DOI] [PubMed] [Google Scholar]
- 17.Van der Aa N, Rooms L, Vandeweyer G, van den Ende J, Reyniers E, Fichera M, et al. Fourteen new cases contribute to the characterization of the 7q11.23 microduplication syndrome. Eur. J. Med. Genet. 2009;52:94–100. doi: 10.1016/j.ejmg.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 18.Vialard F, Simoni G, Gomes DM, Abourra A, De Toffol S, Bru F, et al. Prenatal BACs-on-Beads™: the prospective experience of five prenatal diagnosis laboratories. Prenat. Diagn. 2012;32:329–335. doi: 10.1002/pd.2934. [DOI] [PubMed] [Google Scholar]
- 19.Dutra RL, Pieri Pde C, Teixeira AC, Honjo RS, Bertola DR, Kim CA. Detection of deletions at 7q11.23 in Williams-Beuren syndrome by polymorphic markers. Clinics. 2011;66:959–964. doi: 10.1590/S1807-59322011000600007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kontos H, Manolakos E, Malligiannis P, Plachouras N, Ploumis N, Mihalatos M, et al. Prenatal diagnosis of a fetus with 7q11.23 deletion detected by multiplex ligation-dependent probe amplification (MLPA) screening. Prenat. Diagn. 2008;28:556–558. doi: 10.1002/pd.2020. [DOI] [PubMed] [Google Scholar]
- 21.Howald C, Merla G, Digilio MC, Amenta S, Lyle R, Deutsch S, et al. Two high throughput technologies to detect segmental aneuploidies identify new Williams-Beuren syndrome patients with atypical deletions. J. Med. Genet. 2006;43:266–273. doi: 10.1136/jmg.2005.034009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schubert C, Laccone F. Williams-Beuren syndrome: determination of deletion size using quantitative real-time PCR. Int. J. Mol. Med. 2006;18:799–806. [PubMed] [Google Scholar]
- 23.Krzeminska D, Steinfeld C, Cloez JL, Vibert M, Chery M, Menzies D, et al. Prenatal diagnosis of Williams syndrome based on ultrasound signs. Prenat. Diagn. 2009;29:710–712. doi: 10.1002/pd.2263. [DOI] [PubMed] [Google Scholar]
- 24.Popowski T, Vialard F, Leroy B, Bault JP, Molina-Gomes D. Williams-Beuren syndrome: the prenatal phenotype. Am. J. Obstet. Gynecol. 2011;205:e6–e8. doi: 10.1016/j.ajog.2011.09.017. [DOI] [PubMed] [Google Scholar]
- 25.von Dadelszen P, Chitayat D, Winsor EJ, Cohen H, MacDonald C, Taylor G, et al. De novo 46,XX,t(6;7)(q27;q11;23) associated with severe cardiovascular manifestations characteristic of supravalvular aortic stenosis and Williams syndrome. Am. J. Med. Genet. 2000;90:270–275. doi: 10.1002/(sici)1096-8628(20000214)90:4<270::aid-ajmg2>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 26.Vialard F, Simoni G, Aboura A, De Toffol S, Molina Gomes D, Marcato L, et al. Prenatal BACs-on-Beads™: a new technology for rapid detection of aneuploidies and microdeletions in prenatal diagnosis. Prenat. Diagn. 2011;31:500–508. doi: 10.1002/pd.2727. [DOI] [PubMed] [Google Scholar]
- 27.Gruchy N, Decamp M, Richard N, Jeanne-Pasquier C, Benoist G, Mittre H, et al. Array CGH analysis in high-risk pregnancies: comparing DNA from cultured cells and cell-free fetal DNA. Prenat. Diagn. 2012;32:383–388. doi: 10.1002/pd.2861. [DOI] [PubMed] [Google Scholar]
- 28.Hall JG, Froster-Iskenius UG, Allanson JE. Handbook of normal physical measurements. New York: Oxford medical publications. Oxford Univ. Press; 1989. [Google Scholar]
- 29.Salomon LJ, Bernard JP, Ville Y. Estimation of fetal weight: reference range at 20–36 weeks' gestation and comparison with actual birth-weight reference range. Ultrasound Obstet. Gynecol. 2007;29:550–555. doi: 10.1002/uog.4019. [DOI] [PubMed] [Google Scholar]
- 30.Pober BR. Williams-Beuren syndrome. N. Engl. J. Med. 2010;362:239–252. doi: 10.1056/NEJMra0903074. [DOI] [PubMed] [Google Scholar]
- 31.Pankau R, Partsch CJ, Gosch A, Oppermann HC, Wessel A. Statural growth in Williams-Beuren syndrome. Eur. J. Pediatr. 1992;151:751–755. doi: 10.1007/BF01959084. [DOI] [PubMed] [Google Scholar]
- 32.Pankau R, Partsch CJ, Neblung A, Gosch A, Wessel A. Head circumference of children with Williams-Beuren syndrome. Am. J. Med. Genet. 1994;52:285–290. doi: 10.1002/ajmg.1320520307. [DOI] [PubMed] [Google Scholar]
- 33.Capossela S, Muzio L, Bertolo A, Bianchi V, Dati G, Chaabane L, et al. Growth defects and impaired cognitive-behavioral abilities in mice with knockout for Eif4h, a gene located in the mouse homolog of the Williams-Beuren syndrome critical region. Am. J. Pathol. 2012;180:1121–1135. doi: 10.1016/j.ajpath.2011.12.008. [DOI] [PubMed] [Google Scholar]
- 34.Torniero C, Dalla Bernardina B, Novara F, Cerini R, Bonaglia C, Pramparo T, et al. Dysmorphic features, simplified gyral pattern and 7q11.23 duplication reciprocal to the Williams-Beuren deletion. Eur. J. Hum. Genet. 2008;16:880–887. doi: 10.1038/ejhg.2008.42. [DOI] [PubMed] [Google Scholar]
- 35.Orellana C, Bernabeu J, Monfort S, Roselló M, Oltra S, Ferrer I, et al. Duplication of the Williams-Beuren critical region: case report and further delineation of the phenotypic spectrum. J. Med. Genet. 2008;45:187–189. doi: 10.1136/jmg.2007.054064. [DOI] [PubMed] [Google Scholar]