

Abstract
Objective
Mitochondrial respiratory chain disorder (MRCD) is an intractable disease of infants with variable clinical symptoms. Our goal was to identify the causative mutations in MRCD patients.
Methods
The subjects were 90 children diagnosed with MRCD by enzyme assay. We analyzed whole mitochondrial DNA (mtDNA) sequences. A cybrid study was performed in two patients. Whole exome sequencing was performed for one of these two patients whose mtDNA variant was confirmed as non-pathogenic.
Results
Whole mtDNA sequences identified 29 mtDNA variants in 29 patients (13 were previously reported, the other 13 variants and three deletions were novel). The remaining 61 patients had no pathogenic mutations in their mtDNA. Of the 13 patients harboring unreported mtDNA variants, we excluded seven variants by manual curation. Of the remaining six variants, we selected two Leigh syndrome patients whose mitochondrial enzyme activity was decreased in their fibroblasts and performed a cybrid study. We confirmed that m.14439G>A (MT-ND6) was pathogenic, while m.1356A>G (mitochondrial 12S rRNA) was shown to be a non-pathogenic polymorphism. Exome sequencing and a complementation study of the latter patient identified a novel c.55C>T hemizygous missense mutation in the nuclear-encoded gene NDUFA1.
Interpretation
Our results demonstrate that it is important to perform whole mtDNA sequencing rather than only typing reported mutations. Cybrid assays are also useful to diagnose the pathogenicity of mtDNA variants, and whole exome sequencing is a powerful tool to diagnose nuclear gene mutations as molecular diagnosis can provide a lead to appropriate genetic counseling.
Introduction
The mitochondrial respiratory chain (RC) is a pathway for vital energy generation in which ATP is generated as a form of energy by the substrates generated from glycolysis and β-oxidation. The pathway is composed of five multi-enzyme complexes (complexes I–V), two electron carriers, a quinone (coenzyme Q), and a small hem-containing protein (cytochrome c) that are located in the inner mitochondrial membrane. These RC complexes are formed from subunits encoded by both mitochondrial DNA (mtDNA) and nuclear DNA (nDNA), with the exception of complex II, which is entirely encoded by nDNA.
mtDNA is a circular double-stranded DNA molecule ∼16 kb in length that encodes 37 genes comprising 13 proteins, 22 mitochondrial tRNAs, and 2 rRNAs.1,2 Defects in mitochondrial function are associated with numerous neurodegenerative diseases, such as Parkinson's disease, Alzheimer's disease, and Huntington's disease, and, in particular with mitochondrial respiratory chain disorder (MRCD). MRCD is genetically, clinically, and biochemically heterogeneous, and it can give rise to any symptoms, in any organs or tissues, at any age and with any mode of inheritance.3 One in 5000 births is a conservative realistic estimate for the minimum birth prevalence of MRCD.4 Especially in children, MRCD is an intractable disease and can be regarded as the most common group of inborn errors of metabolism.5,6
Some MRCD patients have typical clinical findings that are caused by specific point mutations or large deletions of mtDNA. Typical clinical features include mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS), myoclonus epilepsy associated with ragged-red fibers (MERRF), Leber's hereditary optic neuropathy (LHON), and chronic progressive external ophthalmoplegia (CPEO).2 Although mtDNA mutations or deletions are usually found in adults showing typical clinical findings, they account for only a minority of children with MRCD. Therefore, the diagnosis of MRCD in children by screening known mtDNA mutations is rather difficult.7 Hence, a combination of general biochemical study, histological study, and genetic analysis is essential for the diagnosis of MRCD, especially in children.6
In this study, we performed whole mtDNA sequencing for 90 children diagnosed with MRCD by RC enzyme assay with the aim of identifying causative mtDNA mutations.
Subjects, Materials, and Methods
Patients
Ninety Japanese pediatric patients diagnosed with MRCD and without characteristic clinical syndromes were studied. The primary diagnosis for these patients was definite or probable MRCD based on the criteria of Bernier et al.,8 and a mitochondrial RC residual enzyme activity of <20% in a tissue, <30% in a fibroblast cell line, or <30% in two or more tissues (Data S1). Informed consent was obtained from the patients and their families before participation in the study.
Patient summaries are shown in Tables1, 2, The details of the two patients studied in the cybrid assay are as follows: Patient (Pt) 377 is a 1-year-old girl born after a normal pregnancy to non-consanguineous parents. She has a normal brother and sister. She was hospitalized with gait difficulties at the age of 1 year. Blood lactate levels were high. Brain magnetic resonance imaging (MRI) showed bilateral and symmetrical hyperintensity foci in the basal ganglia. She developed progressive motor regression and became bedridden. Pt312 is a 5-year-old boy born after 36 weeks' gestation following a normal pregnancy to non-consanguineous parents. His birth weight was 2154 g. He has a sister who is his fraternal twin. At 5 months of age, his parents noticed hypotonia and nystagmus. At 10 months of age, he had generalized epilepsy and blood lactate and his pyruvate levels were high. A brain MRI revealed symmetrical high T2 signals in the midbrain.
Table 1.
Distribution of mtDNA variants and clinical features.
| Characteristics | Non-pathogenic mutations | Low probability variants | New pathogenic deletions | Known variants | Total | |
|---|---|---|---|---|---|---|
| Number of subjects | 61 (100%) | 13 (100%) | 3 (100%) | 13 (100%) | 90 (100%) | |
| No consanguinity | 57 (93%) | 12 (92%) | 3 (100%) | 11 (85%) | 84 (93%) | |
| Age at onset | ≤1 y.o. | 54 (89%) | 10 (77%) | 3 (100%) | 9 (69%) | 76 (84%) |
| Status | Alive | 33 (54%) | 7 (54%) | 1 (33%) | 11 (85%) | 53 (59%) |
| Dead | 28 (46%) | 6 (46%) | 2 (67%) | 2 (15%) | 37 (41%) | |
| Sex | Female | 30 (49%) | 3 (23%) | 2 (67%) | 6 (46%) | 41 (46%) |
| Male | 31 (51%) | 10 (77%) | 1 (33%) | 7 (54%) | 49 (54%) |
y.o., years old.
Table 2.
Summary of unreported mutations and deletions.
| Patient ID | Age at onset | Clinical diagnosis | Enzyme assay (organ) | mtDNA variation | Locus | Heteroplasmy |
|---|---|---|---|---|---|---|
| 377 | 1 year | LD | 1 (Fb) | m.14439G>A | ND6 | Homo (Fb) |
| 190 | 1 year 6 months | LD | 1,4 (M) | m.11246G>A | ND4 | 73% (fb) |
| 508 | 0 days | SIDS | 1 (Hep,Car) | m.4638A>G | ND2 | 86% (Fb) 0% (Hep, Car) |
| 004 | 0 months | MC | 1 (Fb) | m.5537A>G1 | tRNATrp | 27.4% (Fb) |
| 271 | 0 months | ELBW | 1 (Hep) | m.10045T>C | tRNAGly | Homo (hep) |
| 3122 | 5 years | LD | 1 (Fb) probably | m.1356A>G | 12S rRNA | 66% (Fb) |
| 372 | 2 days | LIMD | 1 (Hep) | Deletion (3424 bp) nt12493-15916 |
65.7% (Fb) 89.9% (Hep) |
|
| 336 | 11 months | HD | 1 (Hep) | Deletion (6639 bp) nt7734-14372 |
9.2% (Fb) 92.6% (Hep) |
|
| 390 | 0 days | MC | 1,4 (M,Hep) | Deletion (5424 bp) nt8574-13997 |
44.9% (Fb) 86.4% (Hep) |
LIMD, lethal infantile mitochondrial disorder; HD, hepatic disease; LD, Leigh's disease; MC, mitochondrial cytopathy; SIDS, sudden infant death syndrome; ELBW, extremely low birth weight infant; Fb, fibroblast; Hep, liver; Car, heart; M, muscle.
Expected to be causative because of the other reported mutation on the same position.
m.1356A>G was confirmed as non-pathogenic and nDNA mutation was identified in Pt312.
Whole mtDNA sequencing and detection of variants
Genomic DNA (gDNA) was extracted from skin fibroblasts (Data S1), blood, liver, and cardiac muscle using either phenol/chloroform- or column-based extraction. Whole mtDNA was first polymerase chain reaction (PCR)-amplified as two separate large amplicons (LA1 and LA2) avoiding the nonspecific amplifications from nDNA.9 Second-round PCR was performed using 46 primer pairs (mitoSEQrTM; Applied Biosystems, Carlsbad, CA) and the LA1 and LA2 amplicon mixture from first-round PCR as a template. PCR conditions were as follows: first-round PCR was performed in a reaction mixture containing 0.2 mmol/L of each dNTP, 0.25 U of Takara Ex Taq (Takara Bio, Shiga, Japan), 1× Ex Taq Buffer, 0.3 μmol/L of each primer, and extracted gDNA in a total volume of 50 μL. Initial denaturation was performed at 94°C for 2 min, followed by 30 cycles of 94°C for 20 sec, 60°C for 20 sec, and 72°C for 5 min, with a final extension at 72°C for 11 min. Second-round PCR was performed in a reaction mixture as above except with a 10,000-fold dilution of LA1 amplicon and a 100-fold dilution of LA2 amplicon (total volume of the PCR reaction, 10 μL). Initial denaturation was performed at 96°C for 5 min, followed by 30 cycles of 94°C for 30 sec, 60°C for 45 sec, and 72°C for 45 sec, with a final extension at 72°C for 10 min.
First- and second-round PCR products were separated by 1% and 2% agarose gels, respectively, then 10 μL of second-round PCR products were incubated with 1 μL of ExoSAP-IT reagent (GE Healthcare UK Ltd., Bucks, U.K.) at 37°C for 30 min to degrade remaining primers and nucleotides. The ExoSAP-IT reagent was then inactivated by incubating at 75°C for 15 min. PCR products were sequenced using a BigDye Terminator v3.1 cycle sequencing kit (Applied Biosystems) and an ABI3130xl Genetic Analyzer (Applied Biosystems). Sequence data were compared with the revised Cambridge sequence (GenBank Accession No. NC_012920.1) and sequences present in MITOMAP (http://mitomap.org/MITOMAP) and mtSNP (http://mtsnp.tmig.or.jp/mtsnp/index_e.shtml) using SeqScape software (Applied Biosystems). Whole mtDNA sequencing of seven samples was obtained using an Ion PGM™ sequencer (Life Technologies Corporation, Carlsbad, CA).
Characterization of mtDNA deletions
We searched for mtDNA deletions by focusing on the size of first-round PCR products in agarose electrophoresis. If PCR products were smaller than controls, we suspected mtDNA deletion and performed further analysis. The smaller PCR products were recovered from the gel and amplified by second-round PCR, as described above, and analyzed for an mtDNA deletion. Second-round PCR was performed using fewer (25–26) PCR cycles to avoid un-targeted DNA amplification. To identify the location of the deletion, we first compared the density of bands and screened the faint bands with agarose electrophoresis. The precise deletion boundaries were confirmed by sequencing analysis with primers used for second-round PCR that were close to the probable deletion region.
Results
Patient characteristics and their mtDNA mutations
A total of 90 patients (49 were men and 41 were women) with MRCD were subjected to whole mtDNA sequencing analysis (Table1). Eighty-four subjects (93%) were non-consanguineous. Seventy-six subjects (84%) were aged 1 year or younger. We identified 13 previously reported mtDNA mutations, 13 unreported variants, and three novel deletions (Fig.1). The remaining 61 subjects had normal polymorphisms in their mtDNA (Fig.1).
Figure 1.
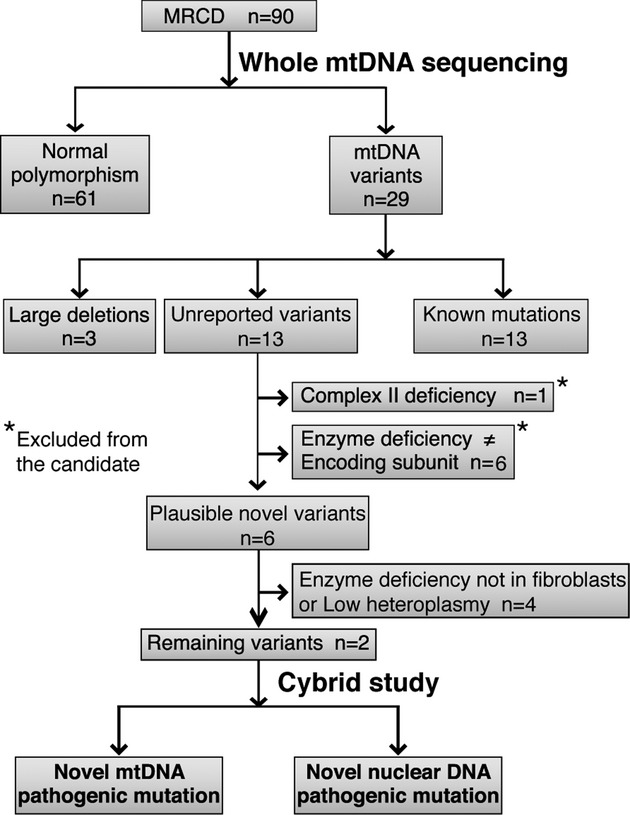
Flow diagram of study analysis. Ninety MRCD patients were analyzed in this study. Sixty-one patients had normal polymorphisms and 29 had mtDNA variants. Of these variants, 13 patients had MRCD causative mutations that had been previously described. We identified three novel large deletions and 13 unreported variants. Of the unreported variants, one patient with complex II deficiency was excluded because complex II is not encoded by mtDNA. Six patients were excluded because their enzyme deficiency pattern did not coincide with the variants found in mtDNA. Four patients were excluded because of the lack of fibroblast enzyme deficiency or low heteroplasmy. The remaining two cases were analyzed by cybrid study.
Large mtDNA deletions were identified in three patients
Agarose gel electrophoresis of first-round PCR from fibroblast and liver mtDNA clearly showed the presence of mtDNA deletions in Pt336, 390, and 372 (Fig.2A). The precise deletion sites were confirmed by sequencing analysis. The expected size of the first-round PCR LA2 product in wild-type mtDNA from an MRCD patient was 11.2 kb, which enabled us to estimate the deletion sizes of Pt336, 390, and 372 as 6639, 5424, and 3424 bp, respectively (Fig.2A and B). In Pt336, the 6639-bp deletion was located between nucleotides 7734 and 14,372 and was flanked by 5-bp perfect direct repeats. This deletion results in the loss of 15 genes (CO2, ATP8, ATP6, CO3, ND3, ND4L, ND4, ND5, ND6, and six tRNA genes). The heteroplasmy ratio of this deletion was 9.2% in the fibroblasts (Fb) and 92.6% in the liver (Hep) (Table2 and Data S1). In Pt390, the 5424-bp deletion was located between nucleotide positions 8574 and 13,997 and was flanked by 11-bp imperfect direct repeats. This deletion results in the loss of 11 genes (ATP6, CO3, ND3, ND4L, ND4, ND5, and five tRNA genes). The heteroplasmy ratio of this deletion was 44.9% (Fb) and 86.4% (Hep) (Table2). In Pt372, the 3424-bp deletion was located between nucleotides 12,493 and 15,916 and was flanked by 6-bp imperfect direct repeats. This deletion results in the loss of five genes (ND5, ND6, CYB, and two tRNA genes). The heteroplasmy ratio of this deletion was 65.7% (Fb), and 89.9% (Hep) (Table2).
Figure 2.
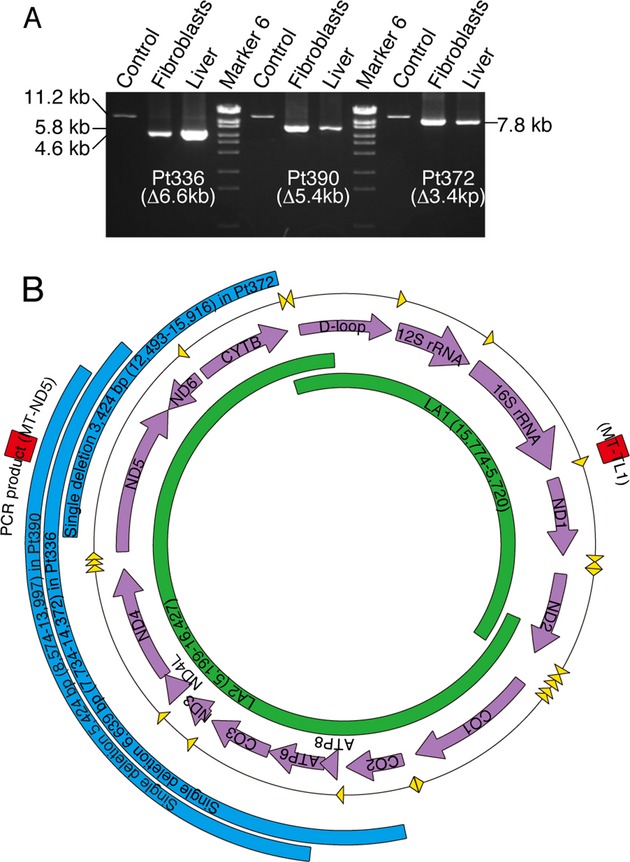
Identification of three large deletions. (A) Characterization of the three novel mtDNA deletions using agarose electrophoresis. First-round PCR products amplified from patient fibroblast and liver DNA clearly showed the presence of mtDNA deletions in Pt336, 390, and 372. Normal mtDNA from an MRCD patient was used as a positive control. (B) Positions of the novel mtDNA deletions are shown in blue. LA1 and LA2 amplification is shown in green. Two red squares represent real-time PCR amplicons MT-ND5 and MT-TL1.
Unreported variants of mtDNA detected in 13 patients
We identified 13 unreported mtDNA variants. Of these, seven were excluded by manual curation (Fig.1). One of these was excluded because the enzyme deficiency was specific to complex II, which is not encoded by mtDNA. The other six were excluded because their enzyme deficiency pattern did not coincide with the variants found in mtDNA. From the remaining six plausible mtDNA variants, we determined whether they were causative using the following inclusion criteria for further analysis: (1) cells were viable for further assay, (2) mtDNA variants corresponded to the enzyme assay data in the RC subunit, (3) enzyme deficiency was observed in the fibroblasts, and (4) variants had high heteroplasmy ratios (Fig.1 and Table2). On the basis of these criteria, we selected two patients whose mtDNA variants (m.14439G>A in MT-ND6 and m.1356A>G in 12S rRNA) were suitable for further analysis as shown in Figure1. The other four patients were excluded because they did not show enzyme deficiency in their fibroblasts or because of low heteroplasmy ratios (Table2).
m.14439G>A (MT-ND6), but not m.1356A>G (12S rRNA), is a causative mutation
The m.14439G>A (MT-ND6) variant was observed in fibroblasts from Pt377 (Fig.3A). PCR- restriction fragment length polymorphism (RFLP) analysis with the Hpy188I restriction enzyme found Pt377 fibroblasts to be homoplasmic, and the m.14439G>A variant was not detected in the blood of the patient's parents (Fig.3A and B). This mutation changes the proline to a serine at amino acid position 79, which is highly conserved among vertebrates (Fig.3C). ND6 is one of the mtDNA-encoded complex I subunits and alignment of the ND6 protein in different species revealed conservation of amino acids. The activity level of the RC complex I was coincidentally reduced in the patient's fibroblasts (Fig.4A). To further confirm whether this mutation was causative of mitochondrial dysfunction, we performed cybrid analysis (Data S1). The cybrids showed a reduction in the complex I activity level consistent with the respiratory enzyme assay in the patient's fibroblasts (Fig.4B). These data strongly support the idea that the m.14439G>A (ND6) mutation detected in Pt377 is responsible for the complex I deficiency.
Figure 3.
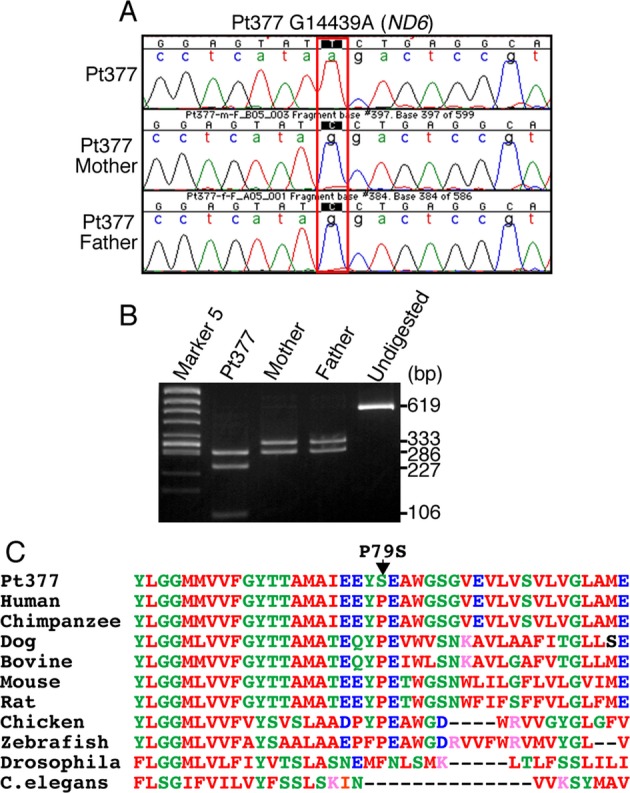
Novel mutation m.14439G>A in Pt377 mtDNA. (A) Trio-sequencing analysis of m.14439G>A (MT-ND6 p.P79S) change in Pt377 family. Sequence chromatograms show that the m.14439G>A is detectable only in Pt377. (B) PCR-RFLPanalysis using fibroblast mtDNA from Pt377 and blood from both parents. A 619-bp PCR fragment was digested with Hpy188I. Wild-type mtDNA was cleaved into two fragments of 333 and 286 bp as shown in “Mother” and “Father”, whereas the PCR product containing the m.14439G>A mutation was cleaved into three fragments: 286, 227, and 106 bp (“Pt377”). Undigested = undigested PCR product. (C) Alignment of MT-ND6 protein between different species shows the conservation of amino acid Proline 79. Amino acid sequences of MT-ND6 gene products were aligned by ClustalW program (http://www.ebi.ac.uk/Tools/msa/clustalw2/) and NCBI/homologene (http://www.ncbi.nlm.nih.gov/homologene).
Figure 4.
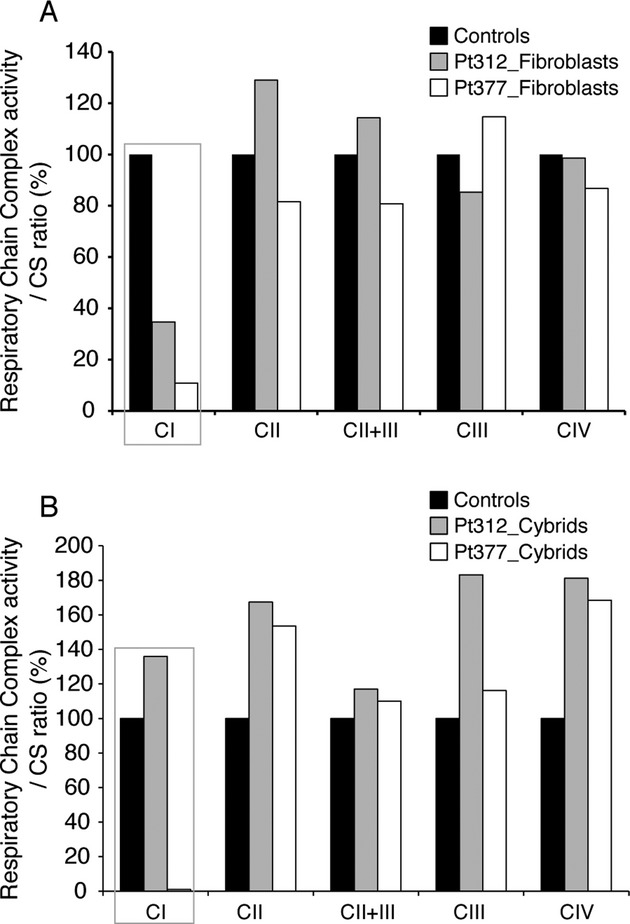
Biochemical assay for respiratory chain enzyme activity in fibroblasts and cybrid cells from Pt377 and Pt312. (A) Respiratory chain complex enzyme activity for CI, CII, CII + III, and CIV in skin fibroblast mitochondria from Pt312 and Pt377 compared with normal controls. The activity of each complex was calculated as a ratio relative to citrate synthase (CS). CI showed a reduction in enzyme activity in Pt312 and 377 fibroblasts. (B) Respiratory chain complex enzyme activity of cybrids established from Pt312 and Pt377 fibroblasts. Cybrids were established from rho0-HeLa cell and Pt312 or Pt377 fibroblasts. The activity of each complex in these cybrids was calculated as a ratio relative to that of citrate synthase (CS).
The m.1356A>G (12S rRNA) variant was observed in fibroblasts from Pt312, which showed reduced activity levels of RC complex I (Fig.4A). By mismatch PCR-RFLPanalysis using the StyI restriction enzyme, this variant was determined at a heteroplasmy ratio of 66% in the patient's fibroblasts (Table2). The cybrids harboring this variant showed a recovery of complex I enzyme activity compared with the original patient's fibroblasts (Fig.4B). These data suggest that reduced complex I enzyme activity was rescued by nuclear DNA and that this mtDNA variation is not causative. This further indicates that the nuclear gene mutation is the cause of MRCD in this patient.
Identification of the c.55C>T (NDUFA1) mutation in Pt312 by whole exome sequencing
To search for the causative nuclear gene mutation in Pt312, we performed whole exome sequencing (Data S1). This identified a single hemizygous mutation (c.55C>T) in exon 1 of the NDUFA1 gene, which altered the amino acid residue at position 19 from proline to serine (p. P19S). The mutation was confirmed by Sanger sequencing (Fig.5A). This conserved proline residue lies within the hydrophobic N-terminal side constituting a functional domain that is involved in mitochondrial targeting, import, and orientation of NDUFA1.10,11 SIFT and PolyPhen, which predict the function of non-synonymous variants (http://genetics.bwh.harvard.edu/pph/), also revealed that the p.P19S mutation “probably” damages the function of the NDUFA1 protein (damaging score, 0.956). Alignment of the NDUFA1 protein between different species revealed the conservation of three amino acids, including the proline at position 19, which is highly conserved among vertebrates (Fig.5B). To further confirm if the complex I deficiency in Pt312 occurred because of the mutation in NDUFA1, we overexpressed NDUFA1 cDNA to determine if the enzyme deficiency could be recovered (Data S1). Lentiviral transfection of NDUFA1 resulted in a significant increase in complex I assembly level as determined by blue native polyacrylamide gel electrophoresis. By contrast, lentiviral transfection of control mtTurboRFP did not rescue the phenotype (Fig.5C). These data indicate that the c.55C>T mutation in NDUFA1 is responsible for the complex I deficiency in Pt312.
Figure 5.
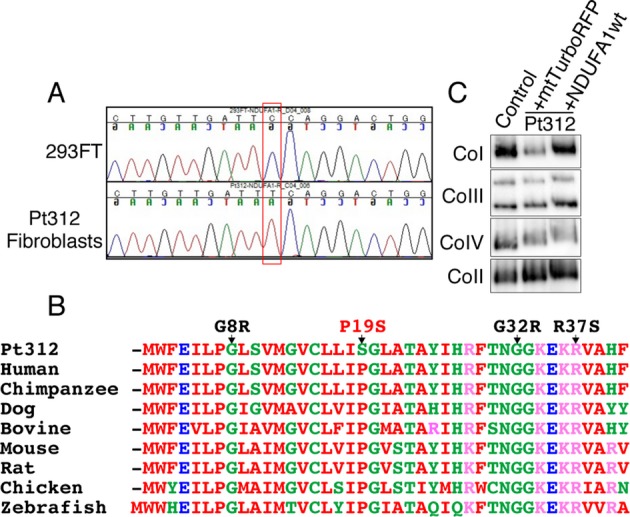
The novel nDNA mutation c.55C>T in NDUFA1. (A) Sequence chromatograms showing the c.55C>T (NDUFA1 p.P19S) mutation in Pt312 and 293FT genomic DNA as a wild-type control. (B) Alignment of amino acid sequences of NDUFA1 subunit between different species shows the high conservation of amino acid Proline 19. G8R, G32R, and R37S show reported pathogenic mutations in NDUFA1. (C) Blue native polyacrylamide gel electrophoresis for CI, CII, CIII, and CIV following lentiviral transductions. Transduction of wild-type NDUFA1 cDNA into Pt312 fibroblasts using recombinant lentivirus rescued complex I assembly levels of the fibroblasts, similar to the transduction of mtTurboRFP into normal fibroblasts (fHDF). As control gene of candidate genes, mtTurboRFP was used which inserted mitochondrial targeting signal sequence to N terminal of TurboRFP protein. By contrast, lentiviral transduction of control mtTurboRFP into Pt312 fibroblasts decreased the assembly level of complex I.
Discussion
MRCD is particularly difficult to diagnose in pediatric cases as the clinical features are highly variable. We, therefore, propose a systematic approach for diagnosing MRCD that starts with a biochemical enzyme assay and is followed by whole mtDNA sequencing. In this study, we performed whole mtDNA sequencing for 90 children with MRCD, and identified 29 mtDNA variants. Of these, we identified 13 known causative mutations, three large deletions, and further confirmed that m.14439G>A (MT-ND6) and c.55C>T (NDUFA1) are new causative mutations for MRCD from the results of a cybrid assay, whole exome sequencing, and a complementation study. The diagnosis of MRCD was then confirmed as definite by molecular analysis in these 18 cases.
Whole mitochondrial DNA sequencing identified 13 cases (14%) harboring known causative mtDNA mutations. mt. 10191T>C (ND3) and mt. 8993T>C or G (ATP6) mutations were detected in three and two patients, respectively (data not shown). Both are common causative mutations of infantile Leigh syndrome. Previous reports found that most common MRCD causative mutations are primarily responsible for adult-onset disease, whereas few are responsible for childhood-onset MRCD;12,13 only 14% of our cases were attributed to known mtDNA mutations.
Most patients in this study were 1-year old or younger at the onset of disease, with no family history. We used the RC complex enzyme assay to diagnose pediatric patients who had not been diagnosed with MRCD in a clinical setting. Several MRCD cases in children were previously reported to be difficult to diagnose with nonspecific clinical presentations in contrast to the characteristic clinical syndromes such as MELAS and MERRF caused by common mtDNA mutations.6,12
We identified three novel deletions that we concluded were causative because they include several genes that could explain the deficiency of the RC enzymes. Generally, most mtDNA deletions share similar structural characteristics, are located in the major arc between two proposed origins of replication (OH and OL; Mitomap), and are predominantly (∼85%) flanked by short direct repeats.14,15 Single mtDNA deletions are reported to be the common causes of sporadic MRCD such as Kearns-Sayre syndrome (KSS), CPEO, and Pearson's syndrome. In this study, all three deletions were located in the major arc and were flanked by repeat sequences, similar to previous studies. Although Pt390 was diagnosed with Pearson's syndrome, the other two patients (Pt336 and Pt372) did not show a common phenotype caused by a single deletion such as KSS, CPEO, or Pearson's syndrome. Therefore, screening by mtDNA size differences is important even in those patients not clinically suspected to have mtDNA deletions.
Manual curation identified six plausible mtDNA variants that had not previously been reported (Fig.1). We attempted to carry out a functional assay of the two patients whose fibroblasts are enzyme deficient, although it was difficult to apply this strategy to those fibroblasts with normal enzyme activity. In this sense, it is important to collect patients with similar phenotypes and carrying the same mtDNA variants to accurately diagnose the causal mutation. Thus, this study of patients harboring unreported mtDNA variants will be useful in a clinical situation. Of these, the m.14439G>A (MT-ND6) variant was experimentally confirmed to be a novel causative mtDNA mutation, while 1356A>G (12S rRNA) was confirmed to be non-pathogenic by a cybrid assay. The remaining four novel variants have yet to be experimentally elucidated, but m.5537A>G (mt-tRNA trp) in Pt004 is likely to be causative because m.5537AinsT was reported to be disease causing.16
ND6 is an mtDNA-encoded complex I subunit that is essential for the assembly of complex I and the maintenance of its structure.17–19 ND6 mutations were previously found to be associated with Leigh syndrome20 and MELAS,21 and this gene region is also reported to be a hot spot for LHON mutations.22 Mitochondrial 12S rRNA is a hot spot for mutations associated with aminoglycoside ototoxicity and non syndromic hearing loss, although mutations in this gene have not been reported to cause syndromic mitochondrial disorders.23 We found that the m.14439G>A mutation altered an evolutionarily conserved proline to a serine in the hydrophilic inner membrane space of the ND6 protein22 (Fig.3C). As this mutation was homoplasmic in the patient's fibroblasts and absent from the blood of unaffected parents (Fig.3A and B), this suggests that it developed de novo.
Exome sequencing in this study identified a single hemizygous change (c.55C>T, p.P19S) in exon 1 of the X-linked NDUFA1 gene. To date, three missense mutations (G8R,10 G32R,24 and R37S10) have been reported in NDUFA1 that are associated with neurological symptoms. NDUFA1 was shown to interact with the subunits encoded by mtDNA during the complex I assembly process.11
Cybrid study is a powerful tool for detecting pathogenicity of either mtDNA or nDNA origin, although patients' cells showing RC enzyme deficiency are inevitable. Nevertheless, a major limitation of this technique is the length of time to establish transmitochondrial cybrids. We would, therefore, propose a systematic approach for diagnosing MRCD that starts with a biochemical enzyme assay and is followed by whole mtDNA sequencing. For patients with no apparent putative mtDNA mutations, whole exome sequencing is a powerful tool to diagnose nuclear gene mutations especially in cases when molecular diagnosis leads to appropriate genetic counseling.
Acknowledgments
We thank T. Hirata and Y. Yatsuka for their technical assistance. This study was supported in part by a grant from the Research Program of Innovative Cell Biology by Innovative Technology (Cell Innovation), a Grant-in-Aid for the Development of New Technology from The Promotion and Mutual Aid Corporation for Private Schools of Japan from MEXT (to Y. O.), a Grant-in-Aid research grants for Scientific Research (A-22240072, B-21390459, A-25242062) from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT) of Japan to M. T., and a Grant-in-Aids (H23-016, H23-119, and H24-005) for the Research on Intractable Diseases (Mitochondrial Disease) from the Ministry of Health, Labour and Welfare (MHLW) of Japan to M. T. and A. O., and a Grant-in-Aids (H23-001, H24-017, H24-071) for the Research on Intractable Diseases from the Ministry of Health, Labour and Welfare (MHLW) of Japan to A. O.
Conflict of Interest
None declared.
Supporting Information
Supplementary methods.
References
- Calvo S, Jain M, Xie X, et al. Systematic identification of human mitochondrial disease genes through integrative genomics. Nat Genet. 2006;38:576–582. doi: 10.1038/ng1776. [DOI] [PubMed] [Google Scholar]
- Zeviani M, Di Donato S. Mitochondrial disorders. Brain. 2004;127(Pt 10):2153–2172. doi: 10.1093/brain/awh259. [DOI] [PubMed] [Google Scholar]
- Munnich A, Rötig A, Chretien D, et al. Clinical presentation of mitochondrial disorders in childhood. J Inherit Metab Dis. 1996;19:521–527. doi: 10.1007/BF01799112. [DOI] [PubMed] [Google Scholar]
- Skladal D, Halliday J, Thorburn DR. Minimum birth prevalence of mitochondrial respiratory chain disorders in children. Brain. 2003;126(Pt 8):1905–1912. doi: 10.1093/brain/awg170. [DOI] [PubMed] [Google Scholar]
- Kisler JE, Whittaker RG, McFarland R. Mitochondrial diseases in childhood: a clinical approach to investigation and management. Dev Med Child Neurol. 2010;52:422–433. doi: 10.1111/j.1469-8749.2009.03605.x. [DOI] [PubMed] [Google Scholar]
- Thorburn DR. Mitochondrial disorders: prevalence, myths and advances. J Inherit Metab Dis. 2004;27:349–362. doi: 10.1023/B:BOLI.0000031098.41409.55. [DOI] [PubMed] [Google Scholar]
- Zeviani M, Bertagnolio B, Uziel G. Neurological presentations of mitochondrial diseases. J Inherit Metab Dis. 1996;19:504–520. doi: 10.1007/BF01799111. [DOI] [PubMed] [Google Scholar]
- Bernier FP, Boneh A, Dennett X, et al. Diagnostic criteria for respiratory chain disorders in adults and children. Neurology. 2002;59:1406–1411. doi: 10.1212/01.wnl.0000033795.17156.00. [DOI] [PubMed] [Google Scholar]
- Akanuma J, Muraki K, Komaki H, et al. Two pathogenic point mutations exist in the authentic mitochondrial genome, not in the nuclear pseudogene. J Hum Genet. 2000;45:337–341. doi: 10.1007/s100380070004. [DOI] [PubMed] [Google Scholar]
- Potluri P, Davila A, Ruiz-Pesini E, et al. A novel NDUFA1 mutation leads to a progressive mitochondrial complex I-specific neurodegenerative disease. Mol Genet Metab. 2009;96:189–195. doi: 10.1016/j.ymgme.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadava N, Houchens T, Potluri P, Scheffler IE. Development and characterization of a conditional mitochondrial complex I assembly system. J Biol Chem. 2004;279:12406–12413. doi: 10.1074/jbc.M313588200. [DOI] [PubMed] [Google Scholar]
- Elliott HR, Samuels DC, Eden JA, et al. Pathogenic mitochondrial DNA mutations are common in the general population. Am J Hum Genet. 2008;83:254–260. doi: 10.1016/j.ajhg.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoffner JM. Oxidative phosphorylation disease diagnosis. Ann N Y Acad Sci. 1999;893:42–60. doi: 10.1111/j.1749-6632.1999.tb07817.x. [DOI] [PubMed] [Google Scholar]
- Bua E, Johnson J, Herbst A, et al. Mitochondrial DNA-deletion mutations accumulate intracellularly to detrimental levels in aged human skeletal muscle fibers. Am J Hum Genet. 2006;79:469–480. doi: 10.1086/507132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan KJ, Reeve AK, Samuels DC, et al. What causes mitochondrial DNA deletions in human cells? Nat Genet. 2008;40:275–279. doi: 10.1038/ng.f.94. [DOI] [PubMed] [Google Scholar]
- Tulinius M, Moslemi AR, Darin N, et al. Leigh syndrome with cytochrome-c oxidase deficiency and a single T insertion nt 5537 in the mitochondrial tRNATrp gene. Neuropediatrics. 2003;34:87–91. doi: 10.1055/s-2003-39607. [DOI] [PubMed] [Google Scholar]
- Bai Y, Attardi G. The mtDNA-encoded ND6 subunit of mitochondrial NADH dehydrogenase is essential for the assembly of the membrane arm and the respiratory function of the enzyme. EMBO J. 1998;17:4848–4858. doi: 10.1093/emboj/17.16.4848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardol P, Matagne RF, Remacle C. Impact of mutations affecting ND mitochondria-encoded subunits on the activity and assembly of complex I in Chlamydomonas. Implication for the structural organization of the enzyme. J Mol Biol. 2002;319:1211–1221. doi: 10.1016/S0022-2836(02)00407-2. [DOI] [PubMed] [Google Scholar]
- Ugalde C, Triepels RH, Coenen MJ, et al. Impaired complex I assembly in a Leigh syndrome patient with a novel missense mutation in the ND6 gene. Ann Neurol. 2003;54:665–669. doi: 10.1002/ana.10734. [DOI] [PubMed] [Google Scholar]
- Kirby DM, Kahler SG, Freckmann ML, et al. Leigh disease caused by the mitochondrial DNA G14459A mutation in unrelated families. Ann Neurol. 2000;48:102–104. [PubMed] [Google Scholar]
- Ravn K, Wibrand F, Hansen FJ, et al. An mtDNA mutation, 14453G–>A, in the NADH dehydrogenase subunit 6 associated with severe MELAS syndrome. Eur J Hum Genet. 2001;9:805–809. doi: 10.1038/sj.ejhg.5200712. [DOI] [PubMed] [Google Scholar]
- Chinnery PF, Brown DT, Andrews RM, et al. The mitochondrial ND6 gene is a hot spot for mutations that cause Leber's hereditary optic neuropathy. Brain. 2001;124(Pt 1):209–218. doi: 10.1093/brain/124.1.209. [DOI] [PubMed] [Google Scholar]
- Prezant TR, Agapian JV, Bohlman MC, et al. Mitochondrial ribosomal RNA mutation associated with both antibiotic-induced and non-syndromic deafness. Nat Genet. 1993;4:289–294. doi: 10.1038/ng0793-289. [DOI] [PubMed] [Google Scholar]
- Fernandez-Moreira D, Ugalde C, Smeets R, et al. X-linked NDUFA1 gene mutations associated with mitochondrial encephalomyopathy. Ann Neurol. 2007;61:73–83. doi: 10.1002/ana.21036. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary methods.