

Abstract
Drug–disease interactions involving therapeutic proteins that target cytokines and potentially impact cytochrome P450 (CYP) enzymes have been of increased interest to drug regulatory agencies and industry sponsors in recent years. This parallel-group open-label study evaluated the effects of the monoclonal antibody denosumab, an inhibitor of the cytokine RANKL, on the pharmacokinetics of the probe CYP3A4 substrate midazolam in postmenopausal women with osteoporosis. The pharmacokinetics of a 2 mg oral dose of midazolam was evaluated on days 1 and 16. Subjects in Group A received a 60 mg subcutaneous dose of denosumab on day 2, 2 weeks before the second midazolam dose, while subjects in Group B did not. For Group A (n = 17), point estimates for the ratio of least square means for midazolam exposures based on maximum observed plasma concentration (Cmax) and areas under the plasma concentration–time curve (AUCs) on day 16 versus day 1 ranged from 1.02 to 1.04 and 90% confidence intervals were within 0.80–1.25. No period effect was observed for Group B (n = 8). Midazolam and denosumab coadministration was safe and well tolerated. Inhibition of the cytokine RANKL by denosumab does not affect CYP3A4 in postmenopausal women with osteoporosis and will not alter the pharmacokinetics of drugs metabolized by this enzyme. These results are consistent with data suggesting that RANKL does not impact markers of inflammation and represent the first clinical data demonstrating a lack of effect on CYP3A4 of a therapeutic protein that is a cytokine modulator.
Keywords: CYP, cytochrome P450, cytokine modulator, denosumab, drug–drug interaction, menopause, midazolam, postmenopausal, therapeutic protein
Introduction
Denosumab is a fully human monoclonal immunoglobulin G2 (IgG2) that binds to and neutralizes RANKL activity. By blocking RANKL, denosumab inhibits osteoclast formation, function, and survival, thereby decreasing bone resorption, increasing bone mass, and reducing risk of vertebral, nonvertebral, and hip fractures in postmenopausal women with osteoporosis (Bekker et al. 2004; McClung et al. 2006; Lewiecki et al. 2007; Bone et al. 2008; Miller et al. 2008; Brown et al. 2009; Cummings et al. 2009; Miller et al. 2011). At a subcutaneous (SC) dose of 60 mg every 6 months, denosumab is approved for use in patients at high risk of fracture due to osteoporosis (men and postmenopausal women), androgen deprivation therapy for nonmetastatic prostate cancer (men), or adjuvant aromatase inhibitor therapy for breast cancer (women) (Prolia® 2012). At a SC dose of 120 mg every 4 weeks, denosumab is approved for use in the prevention of skeletal-related events in patients with bone metastases from solid tumors, and for use in the treatment of adults and skeletally mature adolescents with giant cell tumor of bone that is unresectable or where surgical resection is likely to result in severe morbidity (XGEVA® 2013).
In the years preceding and coincident to the initial approval of denosumab in osteoporosis, potential interactions between therapeutic proteins, including monoclonal antibodies, and small molecule drugs were of increased interest to drug regulatory agencies and industry sponsors (Zhou and Davis 2009; Huang et al. 2010; Lee et al. 2010), and our collective thinking has continued to develop (Girish et al. 2011; Kraynov et al. 2011; Kenny et al. 2013). Focus has been on the downregulation of some cytochrome P450 (CYP) enzymes by proinflammatory cytokines such as interleukins (IL-1, IL-6, and IL-10) and tumor necrosis factor α (TNFα), as evidenced by lower CYP expression and activity in some inflammatory disease states (Morgan 2001; Morgan et al. 2002; Aitken et al. 2006; Morgan 2009). In theory, a therapeutic protein that neutralizes or reduces the action of an inflammatory cytokine could reverse suppression of (or normalize) CYP expression, leading to faster elimination of concomitant small molecule drugs that are substrates of the affected CYPs. At present, in vitro and preclinical systems appear limited in their abilities to accurately replicate or predict the in vivo effects of a cytokine-modulating therapeutic protein on CYP activities (Dickmann et al. 2012; Evers et al. 2013; Slatter et al. 2013). For this reason, carefully conducted clinical trials investigating these potential effects are critical to inform both our evolving understanding of risk and related regulatory guidance.
It is generally recognized that in humans, CYP3A4 is involved in the elimination of roughly 50% of drugs that undergo oxidative metabolism, and CYP3A4 is downregulated by IL-6 and TNFα (Abdel-Razzak et al. 1993; Parmentier et al. 1997; Aitken and Morgan 2007; Dickmann et al. 2011). It was recently demonstrated in subjects with rheumatoid arthritis that the administration of tocilizumab, a monoclonal antibody to IL-6, caused a significant (57%) decrease in area under the plasma concentration–time curve (AUC) of the CYP3A4 probe substrate simvastatin (Schmitt et al. 2011). This result confirmed the potential for a monoclonal antibody targeting a proinflammatory cytokine to alter CYP3A4 activity in humans via a drug–disease interaction. Consistent with these data, current draft US FDA guidance (Food and Drug Administration 2012) indicates that a clinical drug–drug interaction study should be performed for a therapeutic protein that is a cytokine modulator.
During the characterization of RANKL and its receptor RANK, it was determined that they shared roughly 20-30% sequence homology with various members of the TNF ligand and receptor families and were involved in T-lymphocyte and dendritic cell interactions; RANKL was thus classified as a cytokine (Anderson et al. 1997; Wong et al. 1997; Lacey et al. 1998). However, unlike proinflammatory cytokines such as IL-6 or TNFα, to our knowledge no studies have been performed demonstrating a role of RANKL in CYP expression. Importantly, based on mRNA levels, its receptor RANK does not appear to be expressed in human or mouse liver (Su et al. 2004), in contrast to receptors for proinflammatory cytokines that impact CYP3A4 expression through their activation on hepatocyte surfaces. Thus, it appeared there was low likelihood that RANK is involved in CYP regulation in the liver or that inhibition of RANKL would have direct effects on CYP expression.
Alternatively, it was possible that inhibition of RANKL could lead to secondary effects on CYPs by altering circulating levels of proinflammatory cytokines known to impact CYP expression. However, the lifelong inhibition of RANKL in rats and mice, via over-expression of the natural RANKL inhibitor osteoprotegerin, did not alter circulating levels of IL-1, IL-6, IL-10, or TNFα (Stolina et al. 2007), and the pharmacologic inhibition of RANKL did not have significant effects on circulating levels of TNFα or IL-1β in rodent models of arthritis (Stolina et al. 2009). Moreover, in a phase 2 study in subjects with rheumatoid arthritis (Cohen et al. 2008), denosumab treatment for 1 year at 60 or 180 mg every 6 months did not alter circulating levels of C-reactive protein (Amgen Inc., Thousand Oaks, CA; R. A. Newmark, unpubl. data), which is regulated primarily by IL-6 (Eklund 2009). These preclinical and clinical data indicated that denosumab does not markedly alter levels of circulating inflammatory cytokines and was thus unlikely to indirectly impact CYPs.
However, in the context of heightened awareness of the risk of therapeutic protein-induced drug interactions involving cytokines and CYPs (Schmitt et al. 2011), and the classification of RANKL as a cytokine (Anderson et al. 1997; Wong et al. 1997; Lacey et al. 1998), this study was conducted to evaluate the effects of a 60 mg SC dose of denosumab on the pharmacokinetics of the CYP3A4 probe substrate midazolam in postmenopausal women with osteoporosis, and was a postmarketing requirement in the US FDA approval of Prolia®.
In this study, inhibition of the cytokine RANKL by denosumab did not affect CYP3A4 in postmenopausal women with osteoporosis and thus will not alter the pharmacokinetics of drugs metabolized by this enzyme. The results of this study are an important addition to the growing body of preclinical and clinical data informing decision making on whether such clinical studies are needed.
Materials and Methods
Selection of study participants
This study was conducted at two centers in the United States. Subjects could enroll if they were female, 45-75 years of age, and postmenopausal. Postmenopause was confirmed by no vaginal bleeding or spotting for at least 12 months, high follicle-stimulating hormone (≥ 50 mIU/mL), and low serum estradiol (≤ 20 pg/mL). Hormone tests were required in subjects less than 55 years of age, at the investigator's discretion in subjects 55–59 years of age, and not required in subjects ≥60 years of age. Subjects were also required to have osteoporosis, as confirmed by bone mineral density T-scores ≤–2.5 at the lumbar spine (L1–L4 or total evaluable vertebrae) or at the total hip.
Subjects were excluded if they had used any known CYP3A4 inhibitor within 14 days or five half-lives prior to study treatment, or had consumed grapefruit juice or grapefruit-containing products within 7 days prior to study treatment. Subjects were excluded if they had used any known CYP3A4 inducer or any herbal medicine with a known impact on CYP3A4 within 30 days or five half-lives prior to study treatment. Subjects were not permitted to enroll if they were currently using a prescribed medication for osteoporosis treatment or had used midazolam within 14 days prior to study treatment. Other key exclusion criteria were as follows: influenza or any other vaccination within 28 days of screening, previous exposure to denosumab, current hypocalcemia, any condition that could interfere significantly with hepatic metabolism or interpretation of study results, renal disease, Paget's disease of the bone, any unstable medical condition, or any clinically significant laboratory abnormality.
All subjects provided written informed consent to participate. The study was conducted in accordance with ethical principles of the International Conference on Harmonisation Good Clinical Practice Guidelines (ICH GCP) and the Helsinki Declaration of 1975 (as revised in 2008). An institutional review board approved the study at each center.
Study design
In this parallel-group study, all study treatment was administered open-label without masking of subjects or investigators. Prior to the first dose of study treatment, subjects were randomly assigned in a 2:1 ratio to Group A (midazolam + denosumab) or Group B (midazolam alone) using a randomization schedule that the study sponsor had generated with permuted blocks. A pharmacist at each study center used the schedule to assign subjects to a treatment group and prepared all treatments accordingly.
The study design is summarized in Figure 1. Day 1 was the day on which the first dose of midazolam was administered to the subject. Subjects in both groups received oral midazolam syrup 2 mg on day 1 and day 16, after at least a 10-h fast. Midazolam administration was followed with 8 ounces of noncarbonated water, and then an additional 1-h fast. Subjects in Group A also received a single SC dose of denosumab 60 mg on day 2, administered in the abdomen; subjects in Group B did not receive denosumab. Dose administration of midazolam (day 1 and day 16) and denosumab (day 2) was scheduled at approximately the same time of the day (±1 h). All subjects were required to take supplements daily containing ≥1000 mg of elemental calcium and ≥400 IU vitamin D.
Figure 1.
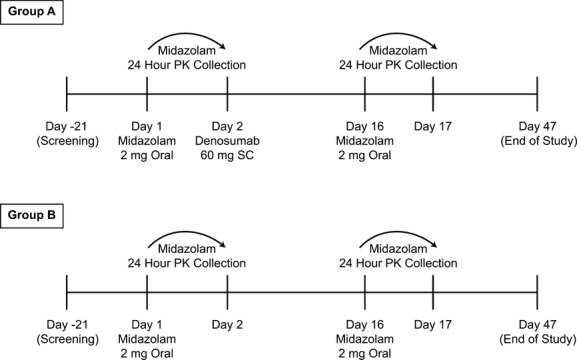
Study design and treatment schema. PK, pharmacokinetics; SC, subcutaneous.
Subjects in each group were followed up until day 47. Oral doses of midazolam on day 1 and day 16 were followed by blood sample collections for pharmacokinetics at 0.5, 1.0, 1.5, 2, 4, 8, 10, 12, and 24 h. After a 60 mg SC dose of denosumab, maximum concentration (Cmax) is observed at a median time (Tmax) of 10 days (range, 3-21 days), while maximal pharmacodynamic effects are observed by 1 week (Bekker et al. 2004). Thus, evaluating the pharmacokinetics of midazolam again on day 16 (2 weeks after administration of denosumab) was deemed appropriate, given that it was both close to Tmax for denosumab and corresponded to a time ∼1 week after maximal pharmacodynamic effects of denosumab are attained.
Additional study assessments included denosumab concentration and serum C-telopeptide (sCTX) concentration on day 2 (predose), day 16 (predose), and day 17. Serum concentrations of denosumab were measured by a validated enzyme-linked immunosorbent assay at PPD (Richmond, VA), and the lower limit of quantification (LLOQ) was 20 ng/mL. Covance (Indianapolis, IN) was responsible for sCTX testing. The LLOQ of the sCTX assay used for this study was defined as 0.2 ng/mL. Plasma concentrations of midazolam (and diltiazem and diltiazem metabolites for one subject [see results]) were measured by validated liquid chromatography tandem mass spectrometry methods at PharmaNet (Quebec, QC). The LLOQ was 20 pg/mL for midazolam and 0.500 ng/mL for diltiazem, N-desmethyl diltiazem, and desacetyl diltiazem.
Safety was assessed by adverse event reports at any time and by clinical laboratory assessments at screening (21 days before study treatment) and at day 17. Efficacy assessments were not performed.
Statistical analysis
The primary study endpoints were AUC from time 0 to t (AUC0–t), AUC from time 0 to infinity (AUC0–inf), and Cmax for midazolam in Group A. Secondary study endpoints were additional midazolam pharmacokinetics parameters (Tmax, half-life [t1/2]) and serum denosumab and sCTX concentrations in Group A; as well as AUC0–t, AUC0–inf, and Cmax for midazolam in Group B. All treated subjects were included in analyses of safety and baseline demographics. All treated subjects for whom pharmacokinetics parameters could be estimated were to be included in analyses of pharmacokinetics, but one subject in Group A used a prohibited medication during the study and was excluded from analyses of pharmacokinetics. Subjects were included in the pharmacodynamic analyses if they received denosumab and their pharmacodynamic parameters could be estimated.
Plasma midazolam concentration-time data were analyzed by noncompartmental methods using WinNonlin Enterprise v 5.1.1 (Pharsight Corporation, Mountain View, CA). Cmax and Tmax were identified by inspection of data. AUC0-t was calculated by the linear trapezoidal (linear interpolation) method. AUCt-inf was calculated by dividing the predicted concentration for the last measurable plasma concentration by the terminal rate constant (λz). λz was estimated by linear regression of the terminal log-linear portion of the plasma concentration profile using at least the last three data points with decreasing concentrations. AUC0-inf was calculated by summation of AUC0-t and AUCt-inf.
The primary analysis was to determine the effect of denosumab on the pharmacokinetics of midazolam based on data from Group A. Log-transformed AUC0–t, AUC0–inf, and Cmax were analyzed using a mixed effect model with treatment as the fixed effect and subject as the random effect. The 90% CIs for mean differences between day 16 and day 1 were calculated. Mean differences and 90% CIs were back-transformed to produce the ratio of geometric means and their associated 90% CIs. Absence of an interaction was concluded if estimates of 90% CIs for ratios of geometric means for midazolam plus denosumab versus midazolam alone fell within the range of 0.80–1.25.
Although the same assay was used for all denosumab studies, the sCTX LLOQ determined by the central laboratory used for this study was higher (0.2 ng/mL) than the LLOQ determined by other central or bioanalytical laboratories for the majority of previous denosumab studies (0.05 ng/mL). With the 0.2 ng/mL method, all sCTX values on days 16 and 17 would have been below the LLOQ. For this reason, the analysis in this study instead used sCTX values that were below 0.2 ng/mL, but above the limit of detection of 0.05 ng/mL to avoid bias in data imputation (e.g., assigning values below the LLOQ to 0.2 ng/mL).
Intrasubject standard deviations (SD) for midazolam AUC0–inf and Cmax were assumed to be ∼18–21% based on a prior study (Padhi et al. 2008). Thus, a sample size of 18 subjects in Group A was expected to provide 90% power to achieve the 90% CI for the geometric mean ratio of AUC0–inf, AUC0–t, and Cmax for test/reference to be between 0.80 and 1.25, given the true ratio was 1. The sample size of nine subjects in Group B was based on practical considerations to allow for examination of a possible period effect on midazolam concentrations.
Results
Subject disposition
The study was conducted between December 2010 and July 2011. Thirty subjects were randomized to either Group A (midazolam + denosumab; n = 21) or Group B (midazolam alone; n = 9). Subjects in Group B were included to evaluate a potential period effect on midazolam pharmacokinetics. Of 30 subjects who were enrolled, 27 received study treatment (19 in Group A and 8 in Group B) and were evaluated for safety and baseline demographics. Four subjects discontinued: three subjects in Group A (two did not receive study treatment and one received study treatment but did not complete the study due to administrative reasons) and one subject in Group B (who did not receive treatment). Thus, 26 subjects completed the study (18 in Group A and 8 in Group B) and were evaluable for midazolam pharmacokinetics.
Baseline demographics
Baseline demographics of the 27 subjects who received study treatment are presented in Table 1. All subjects were female and postmenopausal, and most were white (23 subjects; 85%). Mean age of study participants was 64.4 years (SD, 6.16) in Group A and 66.3 years (SD, 5.34) in Group B. Ages ranged from 55 to 75 years overall and a majority of subjects (12 [63.2%] in Group A and 4 [50.0%] in Group B) were ≥65 years of age.
Table 1.
Baseline characteristics (safety population).
| Characteristic | Group A (midazolam + denosumab*) (N = 19) | Group B (Midazolam Alone†) (N = 8) |
|---|---|---|
| Gender, n (%) | ||
| Female | 19 (100.0) | 8 (100.0) |
| Race, n (%) | ||
| White | 17 (89.5) | 6 (75.0) |
| Black/African American | 1 (5.3) | 1 (12.5) |
| Asian | 1 (5.3) | 1 (12.5) |
| Age (years) | ||
| Mean (SD) | 64.4 (6.16) | 66.3 (5.34) |
| Range | 55–73 | 59–75 |
| ≥65 years, n (%) | 12 (63.2) | 4 (50.0) |
| Bone mineral density T-score, mean (SD) | ||
| Total hip | −2.07 (0.68) | −2.15 (0.65) |
| Lumbar spine | −2.96 (0.76) | −2.68 (0.92) |
SD, standard deviation.
Midazolam 2 mg orally on day 1 and day 16, and denosumab 60 mg subcutaneously on day 2.
Midazolam 2 mg orally on day 1 and day 16.
Subject exclusion
One subject in Group A was found to have an approximately fivefold higher midazolam AUC on day 16 (in the presence of denosumab) relative to day 1 (in the absence of denosumab). The investigator reported that this subject had been taking a stable dose of oral diltiazem (300 mg once daily) since 1999, but stopped taking the antihypertensive 14 days before receiving the first dose of midazolam. Diltiazem is a well-documented, moderate inhibitor of CYP3A4 and interactions with midazolam have been reported for which midazolam AUC increased approximately four- to fivefold (Zhang et al. 2009). Diltiazem and both its n-desmethyl and desacetyl metabolites were quantifiable in all plasma samples from this subject on day 16 but not on day 1, and observed plasma levels were consistent with published data for this diltiazem dose regimen (Chaudhary et al. 1993). Although the use of CYP3A4 inhibitors during the study period was proscribed, these findings clearly indicate that this subject restarted diltiazem treatment during the study and inhibition of CYP3A4 by diltiazem caused marked elevation of midazolam levels. This subject was excluded from pharmacokinetics analyses but included in all other analyses.
Midazolam pharmacokinetics
Mean plasma midazolam concentration–time profiles for Group A in the absence (day 1) and presence (day 16) of denosumab in postmenopausal women were essentially superimposable (Fig. 2). Point estimates for the ratio of the least square means for AUC0–t, AUC0–inf, and Cmax were 1.02, 1.02, and 1.04, respectively, and all 90% CIs were within the range of 0.80–1.25 (Table 2 and Fig. 3), demonstrating that a single 60 mg SC dose of denosumab had no effect on the pharmacokinetics of midazolam in postmenopausal women with osteoporosis. Mean Tmax was 0.5 h on both day 1 and day 16, and mean t1/2 on these days was 6.34 h and 6.69 h, respectively (Table 3). Mean pharmacokinetics parameters for midazolam were similar between Group A and Group B (Table 3).
Figure 2.
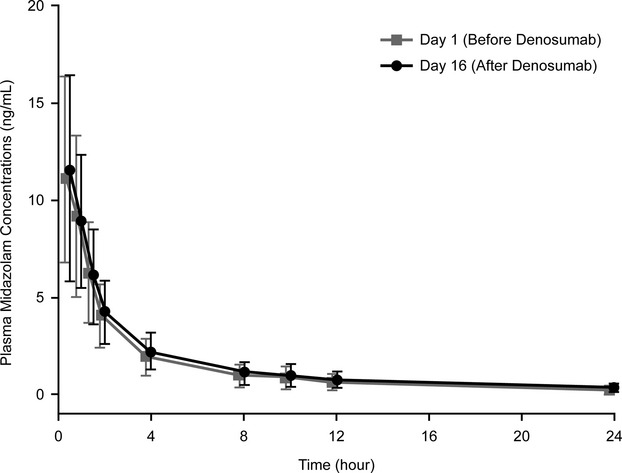
Mean (SD) Plasma midazolam concentration–time profiles following two midazolam oral doses (2 mg) in the absence (day 1) and presence (day 16) of denosumab in postmenopausal women (Group A; n = 17*). * Of the 18 subjects who completed study treatment in Group A, 17 were included in pharmacokinetics parameter estimates and 1 was excluded because of prohibited medication use (diltiazem; see text).
Table 2.
Midazolam pharmacokinetics parameter estimates on day 1 and day 16 and ratio of day 16 to day 1 (pharmacokinetics population; Group A: midazolam + denosumab*).
| Day 16 (test) (N = 17†) | Day 1 (reference) (N = 17†) | Ratio of day 16/day 1 | ||
|---|---|---|---|---|
| Parameter (units) | LS mean | LS mean | LS mean | (90% CI) |
| AUC0–t (ng·h/mL) | 30.96 | 30.45 | 1.02 | (0.95, 1.09) |
| AUC0–inf (ng·h/mL) | 32.80 | 32.05 | 1.02 | (0.96, 1.09) |
| Cmax (ng/mL) | 11.08 | 10.61 | 1.04 | (0.93, 1.17) |
AUC, area under the concentration-time curve; CI, confidence interval; Cmax, maximum concentration; LS mean, least squares geometric mean.
Midazolam 2 mg orally on day 1 and day 16, and denosumab 60 mg subcutaneously on day 2.
Of the 18 subjects who completed study treatment in Group A, 17 were included in pharmacokinetics parameter estimates and 1 was excluded because of prohibited medication use (diltiazem; see text).
Figure 3.
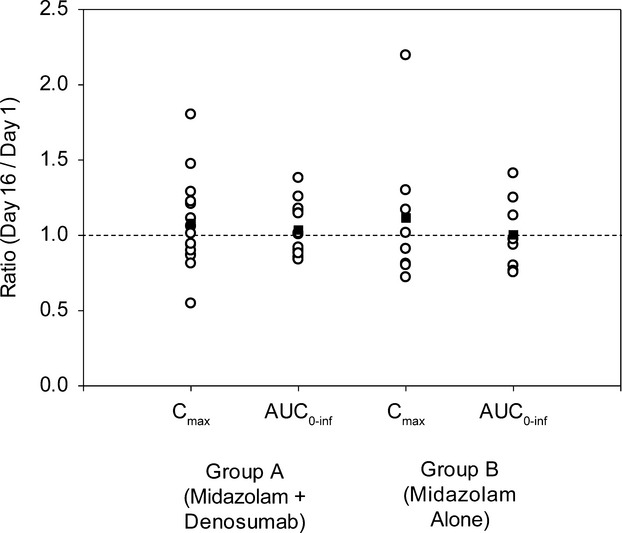
Individual (Circles) and mean (Squares) Cmax and AUC0-inf ratios for midazolam in Groups A (midazolam + denosumab) and B (midazolam alone).
Table 3.
Midazolam pharmacokinetics parameter estimates following midazolam 2 mg doses on day 1 and day 16.
| Group A (midazolam + denosumab*) (N = 17†) | Group B (midazolam alone‡) (N = 8) | |||||||
|---|---|---|---|---|---|---|---|---|
| Day | Tmax (h) | Cmax (ng/mL) | AUCinf (ng·h/mL) | t1/2 (h) | Tmax (hr) | Cmax (ng/mL) | AUCinf (ng·h/mL) | t1/2 (h) |
| 1 | 0.5 (0.5–1.0) | 11.6 (5.1) | 35.6 (16.4) | 6.34 (1.83) | 0.5 (0.5–1.0) | 11.3 (5.8) | 31.0 (21.1) | 6.20 (2.01) |
| 16 | 0.5 (0.5–1.0) | 12.0 (4.6) | 36.3 (16.3) | 6.69 (1.67) | 0.5 (0.5–1.0) | 10.9 (3.7) | 29.3 (18.9) | 6.22 (2.83) |
Data are reported as mean (standard deviation) except for Tmax, which is reported as median (range). Note: Study treatment in Group A was midazolam 2 mg orally on day 1 and day 16, and denosumab 60 mg subcutaneously on day 2. AUC, area under the concentration-time curve; Cmax, maximum concentration; t1/2, half-life; Tmax, time of Cmax.
Midazolam 2 mg orally on day 1 and day 16, and denosumab 60 mg subcutaneously on day 2.
Of the 18 subjects who completed study treatment in Group A, 17 were included in pharmacokinetics parameter estimates and 1 was excluded because of prohibited medication use (diltiazem; see text).
Midazolam 2 mg orally on day 1 and day 16.
In Group B, no substantial period effect was observed; point estimates for the ratio of the least square means for both AUC0–t and AUC0–inf were 0.98, with 90% CIs within the range of 0.80–1.25, and the point estimate for the ratio of the least square means for Cmax was 1.05, with 90% CI from 0.82 to 1.33 (Fig. 3).
Denosumab exposures and pharmacodynamics
Mean (SD) denosumab concentration following a 60 mg SC dose on day 16 (14 days after denosumab administration) was 5790 (1800) ng/mL. The mean (SD) baseline concentration of sCTX on day 2 was 0.493 (0.296) ng/mL and the mean percent change from baseline on day 16 was –81.2% (Table 4).
Table 4.
Percent change in sCTX from baseline to day 16 (pharmacodynamic population; Group A: midazolam + denosumab*)
| Mean | SD | Minimum | Quartile 1 | Median | Quartile 3 | Maximum | |
|---|---|---|---|---|---|---|---|
| % Change in sCTX† | –81.22 | 16.33 | –95.4 | –91.45 | –87.52 | –80.01 | –43.4 |
sCTX, serum C-telopeptide.
Midazolam 2 mg orally on day 1 and day 16, and denosumab 60 mg subcutaneously on day 2.
Including values below the lower limit of quantification (0.2 ng/mL) but above the limit of detection (0.05 ng/mL).
Safety
Of the 27 subjects who received treatment, 12 of 19 subjects (63%) in Group A and 3 of 8 subjects (38%) in Group B had at least one adverse event. Somnolence was the most frequently reported adverse event in both Groups A and B; in Group A, somnolence was reported in 7 of 19 subjects (37%) on day 1 (prior to administration of denosumab), in no subjects from days 2 to 15, and in 7 of 18 subjects (39%) on day 16 (after the second midazolam dose). In Group B, somnolence was reported in 2 of 8 subjects (25%) on day 1, in no subjects from days 2 to 15, and in no subjects on day 16. Other commonly reported adverse events (≥10%) were as follows: Group A – headache (16%), nausea (16%), dizziness (11%), and injection site pain (11%); Group B – constipation (13%), headache (13%), and dizziness (13%). No deaths, withdrawal due to adverse events, or serious adverse events were reported in this study. Changes in clinical laboratory values from baseline were unremarkable.
Discussion
A single, SC dose of 60 mg denosumab had no effect on CYP3A4 activity in postmenopausal women with osteoporosis based on unaltered midazolam pharmacokinetics. No new safety findings were apparent, and there were no fatal adverse events, adverse events leading to study discontinuation, or serious adverse events. There appeared to be a higher incidence of adverse events overall in Group A than in Group B (e.g., incidences of somnolence on day 1 in Groups A and B were 37% and 25%, respectively), but this was potentially a chance finding due to small group sizes. For Group B, although the upper bound of the 90% CI for Cmax (of 1.33) slightly exceeded 1.25, the 90% CI for both AUC measures were within 0.8–1.25. Thus, a lack of notable period effect is concluded. This suggests that a marked period effect did not confound the results for Group A.
In many subjects, Tmax was at the time of the first post dose sample collection (0.5 h), and thus Cmax potentially was not captured. However, all subjects in a recent study (Winter et al. 2013) with a 2 mg oral midazolam dose and sample collection at 0.25 h displayed Tmax of 0.5 or 1 h and the mean Cmax (11.9 ng/mL) reported was similar to those observed in the present work. It is, therefore, likely that Cmax was adequately characterized in this study.
The C-terminal telopeptide of type I collagen in bone is a widely accepted bone turnover marker and higher levels in serum (sCTX) indicate higher levels of bone turnover. sCTX concentrations on day 16 in this study were, on average, ∼81% lower than those observed on day 2, indicating robust pharmacodynamic effects of denosumab at inhibiting RANKL and suppressing bone turnover in this study. Denosumab exposures and the level of pharmacodynamic effects in this study were comparable to those observed in previous denosumab studies in healthy subjects and postmenopausal women (Bekker et al. 2004; Eastell et al. 2011; Sutjandra et al. 2011).
Given the clear lack of effects of concomitant denosumab administration on CYP3A4 activity in this study, the results can be extrapolated to other CYP3A4 substrate drugs in this patient population. Over 90 cytokines have been identified to date (Tato and Cua 2008a,b,c,d); most are not proinflammatory and, to our knowledge, the majority has not been shown to influence CYP expression or activity in vitro, preclinically, or in humans. As noted in the Introduction, the current draft US FDA guidance (Food and Drug Administration 2012) indicates that a clinical drug–drug interaction study should be performed for any therapeutic protein that is a cytokine modulator. Results from this study with denosumab indicate that such broad recommendations may warrant further consideration. It may also be premature for such guidance to recommend clinical studies only for drugs targeting cytokines that have been demonstrated to affect CYPs. In vitro and preclinical systems are presently insufficient for demonstrating such effects, while for agents such as denosumab that are directed at novel targets, their very nature as novel agents would likely preclude the existence of clinical data demonstrating an effect (or lack of effect) on CYPs.
This study was conducted in postmenopausal women with osteoporosis, which is one of the patient populations for denosumab 60 mg every 6 months. Age and menopause do not affect CYP3A4 activity in women based on studies with midazolam (Gorski et al. 2003), triazolam (Greenblatt et al. 2004), or erythromycin (Harris et al. 1996), and osteoporosis does not appear to be associated with increased levels of proinflammatory cytokines (Özmen et al. 2007). Thus, available data suggest that in postmenopausal women with osteoporosis, CYP3A4 levels and activity do not differ markedly from healthy (younger) adults, which is the typical subject population for drug–drug interaction studies. In cases where CYPs are not different in the target population, there is no CYP suppression to normalize, and thus the magnitude of a potential drug interaction is likely small. The potential importance of this factor is illustrated by the tocilizumab study (Schmitt et al. 2011) and a recent drug–drug interaction assessment with tofacitinib (Gupta et al. 2012). The former study, as noted previously, illustrated a notable (∼57%) reduction in simvastatin AUC in the presence of IL-6 inhibition in subjects with rheumatoid arthritis. The latter investigation evaluated the effects of the disease-modifying antirheumatic Janus kinase inhibitor tofacitinib on CYP3A4 activity (midazolam pharmacokinetics) in healthy volunteers, demonstrating a lack of effects in this study population. However, in three phase 2 trials of tofacitinib in subjects with rheumatoid arthritis, the drug consistently caused 60–80% decreases in C-reactive protein across the numerous dose levels evaluated (Tanaka et al. 2011; Fleischmann et al. 2012; Kremer et al. 2012). These decreases are on par with those observed for tocilizumab, suggesting that a drug–drug interaction evaluation for tofacitinib in subjects with rheumatoid arthritis may yield different results than those observed in healthy adults. Moreover, because tofacitinib is a small molecule, the example suggests that current focus on therapeutic proteins as potential perpetrators of cytokine-related drug–disease interactions should be broadened and thus independent of drug modality, with focus instead on the magnitude of demonstrated disease-state effects on CYPs. The apparent increase in simvastatin exposures in rheumatoid arthritis, based on the level of “de-suppression” observed in the tocilizumab trial (Schmitt et al. 2011), suggests a disease-state effect of rheumatoid arthritis similar to roughly three- to fourfold increases in simvastatin exposure with diltiazem or grapefruit juice administration (Mousa et al. 2000; Lilja et al. 2004). Yet neither current US prescribing information for simvastatin (ZOCOR 2012) nor, to our knowledge, that for any CYP3A4 substrate, conveys this potentially important information.
When combined with information on the expression of RANK and RANKL and the lack of denosumab effects on C-reactive protein in humans, these results suggest that assessment of drug–drug interaction risk involving therapeutic proteins may be informed by:
Expression of the target and related proteins in the liver, as relevant to potential direct effects
In vitro and preclinical data, such as the lack of treatment effects on circulating inflammatory cytokines
Lack of treatment-related effects on C-reactive protein levels in relevant patient populations
The magnitude of demonstrated disease-state effects on CYP levels or activity
The magnitude of potential normalization of CYP activity in the context of inherent inter-subject variability in pharmacokinetics and the therapeutic window of the concomitant medication.
Despite current focus on therapeutic proteins as perpetrators of cytokine and CYP-related drug–drug interactions, equal focus should be placed on better characterizing disease-state effects on CYPs in disease populations and the impact of disease-modifying treatments, regardless of drug modality, on those effects.
Acknowledgments
This study was supported by Amgen Inc., which participated in the study design, the analysis and interpretation of data, the writing of the report, and the decision to submit the manuscript for publication. All authors are employees and stockholders of Amgen Inc. Jonathan Latham of PharmaScribe, LLC (on behalf of Amgen Inc.) and Michelle N. Bradley of Amgen Inc. provided assistance with drafting and submitting the manuscript. The authors thank J. Greg Slatter, Ph.D. (Clinical Pharmacology, Amgen Inc.) for critical review of and insightful comments on the manuscript. The authors also thank Jolene K. Berg, M.D. (Cetero Research during the study; now at DaVita Clinical Research) and Mark P. Christiansen, M.D. (Diablo Clinical Research) for their contributions as study investigators.
Glossary
- AUC
area under the plasma concentration-time curve
- Cmax
maximum concentration
- CYP
cytochrome P450
- ICH GCP
International Conference on Harmonisation Good Clinical Practice Guidelines
- IgG2
immunoglobulin G2
- IL
interleukin
- LLOQ
lower limit of quantification
- SC
subcutaneous
- sCTX
serum C-telopeptide
- SD
standard deviation
- t1/2
half-life
- Tmax
time to maximum concentration
- TNFα
tumor necrosis factor α
- λz
terminal rate constant
Disclosure
None declared.
References
- Abdel-Razzak Z, Loyer P, Fautrel A, Gautier JC, Corcos L, Turlin B, et al. Cytokines down-regulate expression of major cytochrome P-450 enzymes in adult human hepatocytes in primary culture. Mol Pharmacol. 1993;44:707–715. [PubMed] [Google Scholar]
- Aitken AE, Morgan ET. Gene-specific effects of inflammatory cytokines on cytochrome P450 2C, 2B6 and 3A4 mRNA levels in human hepatocytes. Drug Metab Dispos. 2007;35:1687–1693. doi: 10.1124/dmd.107.015511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aitken AE, Richardson TA, Morgan ET. Regulation of drug-metabolizing enzymes and transporters in inflammation. Annu Rev Pharmacol Toxicol. 2006;46:123–149. doi: 10.1146/annurev.pharmtox.46.120604.141059. [DOI] [PubMed] [Google Scholar]
- Anderson DM, Maraskovsky E, Billingsley WL, Dougall WC, Tometsko ME, Roux ER, et al. A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature. 1997;390:175–179. doi: 10.1038/36593. [DOI] [PubMed] [Google Scholar]
- Bekker PJ, Holloway DL, Rasmussen AS, Murphy R, Martin SW, Leese PT, et al. A single-dose placebo-controlled study of AMG 162, a fully human monoclonal antibody to RANKL, in postmenopausal women. J Bone Miner Res. 2004;19:1059–1066. doi: 10.1359/JBMR.040305. [DOI] [PubMed] [Google Scholar]
- Bone HG, Bolognese MA, Yuen CK, Kendler DL, Wang H, Liu Y, et al. Effects of denosumab on bone mineral density and bone turnover in postmenopausal women. J Clin Endocrinol Metab. 2008;93:2149–2157. doi: 10.1210/jc.2007-2814. [DOI] [PubMed] [Google Scholar]
- Brown JP, Prince RL, Deal C, Recker RR, Kiel DP, de Gregorio LH, et al. Comparison of the effect of denosumab and alendronate on BMD and biochemical markers of bone turnover in postmenopausal women with low bone mass: a randomized, blinded, phase 3 trial. J Bone Miner Res. 2009;24:153–161. doi: 10.1359/jbmr.0809010. [DOI] [PubMed] [Google Scholar]
- Chaudhary RS, Gangwal SS, Avachat MK, Shah YN, Jindal KC. Determination of diltiazem hydrochloride in human serum by high-performance liquid chromatography. J Chromatogr. 1993;614:261–266. doi: 10.1016/0378-4347(93)80317-w. [DOI] [PubMed] [Google Scholar]
- Cohen SB, Dore RK, Lane NE, Ory PA, Peterfy CG, Sharp JT, et al. Denosumab treatment effects on structural damage, bone mineral density, and bone turnover in rheumatoid arthritis: a twelve-month, multicenter, randomized, double-blind, placebo-controlled, phase II clinical trial. Arthritis Rheum. 2008;58:1299–1309. doi: 10.1002/art.23417. [DOI] [PubMed] [Google Scholar]
- Cummings SR, San Martin J, McClung MR, Siris ES, Eastell R, Reid IR, et al. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med. 2009;361:756–765. doi: 10.1056/NEJMoa0809493. [DOI] [PubMed] [Google Scholar]
- Dickmann LJ, Patel SK, Rock DA, Wienkers LC, Slatter JG. Effects of interleukin-6 (IL-6) and an anti-IL-6 monoclonal antibody on drug-metabolizing enzymes in human hepatocyte culture. Drug Metab Dispos. 2011;39:1415–1422. doi: 10.1124/dmd.111.038679. [DOI] [PubMed] [Google Scholar]
- Dickmann LJ, Patel SK, Wienkers LC, Slatter JG. Effects of interleukin 1β (IL-1β) and IL-1β/interleukin 6 (IL-6) combinations on drug metabolizing enzymes in human hepatocyte culture. Curr Drug Metab. 2012;13:930–937. doi: 10.2174/138920012802138642. [DOI] [PubMed] [Google Scholar]
- Eastell R, Christiansen C, Grauer A, Kutilek S, Libanati C, McClung MR, et al. Effects of denosumab on bone turnover markers in postmenopausal osteoporosis. J Bone Miner Res. 2011;26:530–537. doi: 10.1002/jbmr.251. [DOI] [PubMed] [Google Scholar]
- Eklund CM. Proinflammatory cytokines in CRP baseline regulation. Adv Clin Chem. 2009;48:111–136. doi: 10.1016/s0065-2423(09)48005-3. [DOI] [PubMed] [Google Scholar]
- Evers R, Dallas S, Dickmann LJ, Fahmi OA, Kenny JR, Kraynov E, et al. Critical review of preclinical approaches to investigate Cytochrome P450-mediated therapeutic protein drug-drug interactions and recommendations for best practices: a white paper. Drug Metab Dispos. 2013;41:1598–1609. doi: 10.1124/dmd.113.052225. [DOI] [PubMed] [Google Scholar]
- Fleischmann R, Cutolo M, Genovese MC, Lee EB, Kanik KS, Sadis S, et al. Phase IIb dose-ranging study of the oral JAK inhibitor tofacitinib (CP-690,550) or adalimumab monotherapy versus placebo in patients with active rheumatoid arthritis with an inadequate response to disease-modifying antirheumatic drugs. Arthritis Rheum. 2012;64:617–629. doi: 10.1002/art.33383. [DOI] [PubMed] [Google Scholar]
- Food and Drug Administration. Draft Guidance for Industry: Drug Interaction Studies – Study Design, Data Analysis, Implications for Dosing, and Labeling Recommendations. Rockville, MD: US Department of Health and Human Services; 2012. Available at http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM292362.pdf (accessed 20 February 2012) [Google Scholar]
- Girish S, Martin SW, Peterson MC, Zhang LK, Zhao H, Balthasar J, et al. AAPS workshop report: strategies to address therapeutic protein-drug interactions during clinical development. AAPS J. 2011;13:405–416. doi: 10.1208/s12248-011-9285-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorski JC, Vannaprasaht S, Hamman MA, Ambrosius WT, Bruce MA, Haehner-Daniels B, et al. The effect of age, sex, and rifampin administration on intestinal and hepatic cytochrome P450 3A activity. Clin Pharmacol Ther. 2003;74:275–287. doi: 10.1016/S0009-9236(03)00187-5. [DOI] [PubMed] [Google Scholar]
- Greenblatt DJ, Harmatz JS, von Moltke LL, Wright CE, Shader RI. Age and gender effects on the pharmacokinetics and pharmacodynamics of triazolam, a cytochrome P450 3A substrate. Clin Pharmacol Ther. 2004;76:467–479. doi: 10.1016/j.clpt.2004.07.009. [DOI] [PubMed] [Google Scholar]
- Gupta P, Alvey C, Wang R, Dowty ME, Fahmi OA, Walsky RL, et al. Lack of effect of tofacitinib (CP-690,550) on the pharmacokinetics of the CYP3A4 substrate midazolam in healthy volunteers: confirmation of in vitro data. Br J Clin Pharmacol. 2012;74:109–115. doi: 10.1111/j.1365-2125.2012.04168.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RZ, Tsunoda SM, Mroczkowski P, Wong H, Benet LZ. The effects of menopause and hormone replacement therapies on prednisolone and erythromycin pharmacokinetics. Clin Pharmacol Ther. 1996;59:429–435. doi: 10.1016/S0009-9236(96)90112-5. [DOI] [PubMed] [Google Scholar]
- Huang SM, Zhao H, Lee JI, Reynolds K, Zhang L, Temple R, et al. Therapeutic protein-drug interactions and implications for drug development. Clin Pharmacol Ther. 2010;87:497–503. doi: 10.1038/clpt.2009.308. [DOI] [PubMed] [Google Scholar]
- Kenny JR, Liu MM, Chow AT, Earp JC, Evers R, Slatter JG, et al. Therapeutic protein drug-drug interactions: navigating the knowledge gaps-highlights from the 2012 AAPS NBC Roundtable and IQ Consortium/FDA Workshop. AAPS J. 2013;15:933–940. doi: 10.1208/s12248-013-9495-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraynov E, Martin SW, Hurst S, Fahmi OA, Dowty M, Cronenberger C, et al. How current understanding of clearance mechanisms and pharmacodynamics of therapeutic proteins can be applied for evaluation of their drug-drug interaction potential. Drug Metab Dispos. 2011;39:1779–1783. doi: 10.1124/dmd.111.040808. [DOI] [PubMed] [Google Scholar]
- Kremer JM, Cohen S, Wilkinson BE, Connell CA, French JL, Gomez-Reino J, et al. A phase IIb dose-ranging study of the oral JAK inhibitor tofacitinib (CP-690,550) versus placebo in combination with background methotrexate in patients with active rheumatoid arthritis and an inadequate response to methotrexate alone. Arthritis Rheum. 2012;64:970–981. doi: 10.1002/art.33419. [DOI] [PubMed] [Google Scholar]
- Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T, et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998;93:165–176. doi: 10.1016/s0092-8674(00)81569-x. [DOI] [PubMed] [Google Scholar]
- Lee JI, Zhang L, Men AY, Kenna LA, Huang SM. CYP-mediated therapeutic protein-drug interactions: clinical findings, proposed mechanisms and regulatory implications. Clin Pharmacokinet. 2010;49:295–310. doi: 10.2165/11319980-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Lewiecki EM, Miller PD, McClung MR, Cohen SB, Bolognese MA, Liu Y, et al. Two-year treatment with denosumab (AMG 162) in a randomized phase 2 study of postmenopausal women with low BMD. J Bone Miner Res. 2007;22:1832–1841. doi: 10.1359/jbmr.070809. [DOI] [PubMed] [Google Scholar]
- Lilja JJ, Neuvonen M, Neuvonen PJ. Effects of regular consumption of grapefruit juice on the pharmacokinetics of simvastatin. Br J Clin Pharmacol. 2004;58:56–60. doi: 10.1111/j.1365-2125.2004.02095.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClung MR, Lewiecki EM, Cohen SB, Bolognese MA, Woodson GC, Moffett AH, et al. Denosumab in postmenopausal women with low bone mineral density. N Engl J Med. 2006;354:821–831. doi: 10.1056/NEJMoa044459. [DOI] [PubMed] [Google Scholar]
- Miller PD, Bolognese MA, Lewiecki EM, McClung MR, Ding B, Austin M, et al. Effect of denosumab on bone density and turnover in postmenopausal women with low bone mass after long-term continued, discontinued, and restarting of therapy: a randomized blinded phase 2 clinical trial. Bone. 2008;43:222–229. doi: 10.1016/j.bone.2008.04.007. [DOI] [PubMed] [Google Scholar]
- Miller PD, Wagman RB, Peacock M, Lewiecki EM, Bolognese MA, Weinstein RL, et al. Effect of denosumab on bone mineral density and biochemical markers of bone turnover: six-year results of a phase 2 clinical trial. J Clin Endocrinol Metab. 2011;96:394–402. doi: 10.1210/jc.2010-1805. [DOI] [PubMed] [Google Scholar]
- Morgan ET. Regulation of cytochrome p450 by inflammatory mediators: why and how? Drug Metab Dispos. 2001;29:207–212. [PubMed] [Google Scholar]
- Morgan ET. Impact of infectious and inflammatory disease on cytochrome P450-mediated drug metabolism and pharmacokinetics. Clin Pharmacol Ther. 2009;85:434–438. doi: 10.1038/clpt.2008.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan ET, Li-Masters T, Cheng PY. Mechanisms of cytochrome P450 regulation by inflammatory mediators. Toxicology. 2002;181–182:207–210. doi: 10.1016/s0300-483x(02)00283-4. [DOI] [PubMed] [Google Scholar]
- Mousa O, Brater DC, Sunblad KJ, Hall SD. The interaction of diltiazem with simvastatin. Clin Pharmacol Ther. 2000;67:267–274. doi: 10.1067/mcp.2000.104609. [DOI] [PubMed] [Google Scholar]
- Özmen B, Kirmaz C, Aydin K, Kafesciler SO, Guclu F, Hekimsoy Z. Influence of the selective oestrogen receptor modulator (raloxifene hydrochloride) on IL-6, TNF-alpha, TGF-beta1 and bone turnover markers in the treatment of postmenopausal osteoporosis. Eur Cytokine Netw. 2007;18:148–153. doi: 10.1684/ecn.2007.0097. [DOI] [PubMed] [Google Scholar]
- Padhi D, Salfi M, Emery M. Cinacalcet does not affect the activity of cytochrome P450 3A enzymes, a metabolic pathway for common immunosuppressive agents: a randomized, open-label, crossover, single-centre study in healthy volunteers. Drugs R D. 2008;9:335–343. doi: 10.2165/00126839-200809050-00004. [DOI] [PubMed] [Google Scholar]
- Parmentier JH, Schohn H, Bronner M, Ferrari L, Batt AM, Dauca M, et al. Regulation of CYP4A1 and peroxisome proliferator-activated receptor alpha expression by interleukin-1beta, interleukin-6, and dexamethasone in cultured fetal rat hepatocytes. Biochem Pharmacol. 1997;54:889–898. doi: 10.1016/s0006-2952(97)00256-6. [DOI] [PubMed] [Google Scholar]
- Prolia. Denosumab injection for subcutaneous use. Thousand Oaks, CA: Amgen Inc; 2012. Full prescribing information. [Google Scholar]
- Schmitt C, Kuhn B, Zhang X, Kivitz AJ, Grange S. Disease-drug-drug interaction involving tocilizumab and simvastatin in patients with rheumatoid arthritis. Clin Pharmacol Ther. 2011;89:735–740. doi: 10.1038/clpt.2011.35. [DOI] [PubMed] [Google Scholar]
- Slatter JG, Wienkers LC, Dickmann LJ. Drug interactions of cytokines and anti-cytokine therapeutic proteins. In: Zhou H, Meibohm B, editors. Drug-drug interactions for therapeutic biologics. Hoboken, NJ: John Wiley & Sons Inc; 2013. pp. 215–237. [Google Scholar]
- Stolina M, Dwyer D, Ominsky MS, Corbin T, Van G, Bolon B, et al. Continuous RANKL inhibition in osteoprotegerin transgenic mice and rats suppresses bone resorption without impairing lymphorganogenesis or functional immune responses. J Immunol. 2007;179:7497–7505. doi: 10.4049/jimmunol.179.11.7497. [DOI] [PubMed] [Google Scholar]
- Stolina M, Schett G, Dwyer D, Vonderfecht S, Middleton S, Duryea D, et al. RANKL inhibition by osteoprotegerin prevents bone loss without affecting local or systemic inflammation parameters in two rat arthritis models: comparison with anti-TNFalpha or anti-IL-1 therapies. Arthritis Res Ther. 2009;11:R187. doi: 10.1186/ar2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su AI, Wiltshire T, Batalov S, Lapp H, Ching KA, Block D, et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci USA. 2004;101:6062–6067. doi: 10.1073/pnas.0400782101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutjandra L, Rodriguez RD, Doshi S, Ma M, Peterson MC, Jang GR, et al. Population pharmacokinetic meta-analysis of denosumab in healthy subjects and postmenopausal women with osteopenia or osteoporosis. Clin Pharmacokinet. 2011;50:793–807. doi: 10.2165/11594240-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Suzuki M, Nakamura H, Toyoizumi S, Zwillich SH. Phase II study of tofacitinib (CP-690,550) combined with methotrexate in patients with rheumatoid arthritis and an inadequate response to methotrexate. Arthritis Care Res (Hoboken) 2011;63:1150–1158. doi: 10.1002/acr.20494. [DOI] [PubMed] [Google Scholar]
- Tato CM, Cua DJ. SnapShot: Cytokines I. Cell. 2008a;132:324. doi: 10.1016/j.cell.2008.01.001. [DOI] [PubMed] [Google Scholar]
- Tato CM, Cua DJ. SnapShot: Cytokines II. Cell. 2008b;132:500. doi: 10.1016/j.cell.2008.01.002. [DOI] [PubMed] [Google Scholar]
- Tato CM, Cua DJ. SnapShot: Cytokines III. Cell. 2008c;132:900. doi: 10.1016/j.cell.2008.02.023. [DOI] [PubMed] [Google Scholar]
- Tato CM, Cua DJ. SnapShot: Cytokines IV. Cell. 2008d;132:1062. doi: 10.1016/j.cell.2008.02.024. [DOI] [PubMed] [Google Scholar]
- Winter H, Egizi E, Erondu N, Ginsberg A, Rouse DJ, Severynse-Stevens D, et al. Evaluation of pharmacokinetic interaction between PA-824 and midazolam in healthy adult subjects. Antimicrob Agents Chemother. 2013;57:3699–3703. doi: 10.1128/AAC.02632-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong BR, Rho J, Arron J, Robinson E, Orlinick J, Chao M, et al. TRANCE is a novel ligand of the tumor necrosis factor receptor family that activates c-Jun N-terminal kinase in T cells. J Biol Chem. 1997;272:25190–25194. doi: 10.1074/jbc.272.40.25190. [DOI] [PubMed] [Google Scholar]
- XGEVA. Denosumab injection for subcutaneous use. Thousand Oaks, CA: Amgen Inc; 2013. Full prescribing information. [Google Scholar]
- Zhang X, Quinney SK, Gorski JC, Jones DR, Hall SD. Semiphysiologically based pharmacokinetic models for the inhibition of midazolam clearance by diltiazem and its major metabolite. Drug Metab Dispos. 2009;37:1587–1597. doi: 10.1124/dmd.109.026658. [DOI] [PubMed] [Google Scholar]
- Zhou H, Davis HM. Risk-based strategy for the assessment of pharmacokinetic drug-drug interactions for therapeutic monoclonal antibodies. Drug Discovery Today. 2009;14:891–898. doi: 10.1016/j.drudis.2009.05.014. [DOI] [PubMed] [Google Scholar]
- ZOCOR. Simvastatin tablets. Whitehouse Statin, NJ: Merck & Co., Inc; 2012. Full prescribing information. [Google Scholar]