

Abstract
Orthovanadate (OVA), a protein tyrosine phosphatase (PTPase) inhibitor, exerts contractile effects on smooth muscle in a Rho-kinase-dependent manner, but the precise mechanisms are not elucidated. The aim of this study was to determine the potential roles of Src and epidermal growth factor receptor (EGFR) in the OVA-induced contraction of rat aortas and the phosphorylation of myosin phosphatase target subunit 1 (MYPT1; an index of Rho-kinase activity) in vascular smooth muscle cells (VSMCs). Aortic contraction by OVA was significantly blocked not only by Rho kinase inhibitors Y-27632 [R-[+]-trans-N-[4-pyridyl]-4-[1-aminoethyl]-cyclohexanecarboxamide] and hydroxyfasudil [1-(1-hydroxy-5-isoquinolinesulfonyl)homopiperazine] but also by Src inhibitors PP2 [4-amino-3-(4-chlorophenyl)-1-(t-butyl)-1H-pyrazolo[3,4-d]pyrimidine] and Src inhibitor No. 5 [4-(3′-methoxy-6′-chloro-anilino)-6-methoxy-7(morpholino-3-propoxy)-quinazoline], and the EGFR inhibitors AG1478 [4-(3-chloroanilino)-6,7-dimethoxyquinazoline] and EGFR inhibitor 1 [cyclopropanecarboxylic acid-(3-(6-(3-trifluoromethyl-phenylamino)-pyrimidin-4-ylamino)-phenyl)-amide]. OVA induced rapid increases in the phosphorylation of MYPT1 (Thr-853), Src (Tyr-416), and EGFR (Tyr-1173) in VSMCs, and Src inhibitors abolished these effects. OVA-induced Src phosphorylation was abrogated by Src inhibitors, but not affected by inhibitors of EGFR and Rho-kinase. Inhibitors of Src and EGFR, but not Rho-kinase, also blocked OVA-induced EGFR phosphorylation. Furthermore, a metalloproteinase inhibitor TAPI-0 [N-(R)-[2-(hydroxyaminocarbonyl) methyl]-4-methylpentanoyl-l-naphthylalanyl-l-alanine amide] and an inhibitor of heparin-binding EGF (CRM 197) not only abrogated the OVA-induced aortic contraction, but also OVA-induced EGFR and MYPT1 phosphorylation, suggesting the involvement of EGFR transactivation. OVA also induced EGFR phosphorylation at Tyr-845, one of residues phosphorylated by Src. These results suggest that OVA-induced vasocontraction is mediated by the Rho-kinase-dependent inactivation of myosin light-chain phosphatase via signaling downstream of Src-induced transactivation of EGFR.
Keywords: Vanadate, protein tyrosine phosphatase, Src, EGFR, Rho-kinase, myosin phosphatase target subunit 1, vascular smooth muscle, rat aorta
Introduction
Vanadate anions, which resemble phosphate anions structurally, have various biochemical and pharmacological properties, including the inhibition of ATPases (Cantley et al. 1977) and protein tyrosine phosphatases (PTPases) (Swarup et al. 1982), epidermal growth factor (EGF)-like mitogenic activity (Chen and Chan 1993), insulin-mimetic properties (Mehdi et al. 2006), antiapoptotic activities (Morita et al. 2010), and antitumor or carcinogenic properties (Sabbioni et al. 1991; Wozniak and Blasiak 2004). In addition, a number of studies demonstrated that vanadium compounds, such as sodium orthovanadate (OVA) and pervanadate, exert contractile effects on vascular (Rapp 1981; Fox et al. 1983; Shimada et al. 1986; Sánchez-Ferrer et al. 1988; Laniyonu et al. 1994; Spurrell et al. 2000) and nonvascular smooth muscle, such as the guinea pig taenia coli, trachea, and gall bladder smooth muscle (Nayler and Sparrow 1983; Di Salvo et al. 1993; Alcón et al. 2000), and the rat gastric longitudinal muscle and myometrium (Laniyonu et al. 1994; Boulven et al. 2002). The contractile effects of vanadates have been considered to be due to the inhibition of PTPase (Di Salvo et al. 1993; Laniyonu et al. 1994; Alcón et al. 2000) but not ATPases (Rapp 1981; Sánchez-Ferrer et al. 1988) as genistein, a protein tyrosine kinase inhibitor, attenuated vanadate-induced contraction. Furthermore, Masui and Wakabayashi (2000) showed that OVA augmented Ca2+ -induced contraction in KCl-depolarized guinea pig aortas, implicating a role for the Ca2+ -sensitization in OVA-induced smooth muscle contraction. Subsequently, Mori and Tsushima (2004) reported that OVA activated RhoA/Rho-kinase in ileal longitudinal smooth muscle of guinea pigs, resulting in increased phosphorylation of the 20-kDa myosin light-chain (MLC). Although these studies strongly suggest that the inhibition of PTPases by vanadate compounds increases the Ca2+sensitivity of smooth muscle cells in a Rho-kinase-dependent manner, the precise mechanisms by which vanadate induces smooth muscle contraction are not fully defined.
To determine the mechanisms of vanadate-induced smooth muscle contraction, we first studied the effects of Rho kinase inhibitors on the vasoconstrictor effects of OVA in rat aortic rings, and observed that OVA-induced vasocontraction was blocked completely by these inhibitors. Subsequent studies using a combination of organ bath experiments with aortic rings and western blotting of cultured vascular smooth muscle cells (VSMCs) suggested that OVA induces Rho kinase-dependent phosphorylation of myosin phosphatase target subunit 1 (MYPT1) of myosin light-chain phosphatase (MLCP) through Src-dependent transactivation of the epidermal growth factor receptor (EGFR). We also found that, like OVA, PTPase inhibitor I (PTP-I; α-bromo-4-hydroxyacetophenone), an irreversible inhibitor for SH2 domain-containing SHP-1 and PTPase 1B, also exerted vasoconstrictor effects though the Src/EGFR-dependent phosphorylation of MYPT1. To the best of our knowledge, this study is the first to provide evidence that a downstream signaling from the Src-mediated transactivation of EGFR regulates smooth muscle contractility through the activation of Rho-kinase.
Materials and Methods
Inhibitors
The following inhibitors were purchased from the commercial sources indicated: AG1478 [4-(3-chloroanilino)-6,7-dimethoxyquinazoline], EGFR inhibitor 1 [cyclopropanecarboxylic acid-(3-(6-(3-trifluoromethyl-phenylamino)-pyrimidin-4-ylamino)-phenyl)-amide], hydroxyfasudil [1-(1-hydroxy-5-isoquinolinesulfonyl)homopiperazine], ML-7 [1-(5-iodonaphthalene-1-sulfonyl)homopiperazine], PP2 {[4-amino-3-(4-chlorophenyl)-1-(t-butyl)-1H-pyrazolo[3,4-d]pyrimidine}, PTP-I, TAPI-0 {N-(R)-[2-(hydroxyaminocarbonyl) methyl]-4-methylpentanoyl-l-naphthylalanyl- l-alanine amide}, and Y-27632 {R-[+]-trans-N-[4-pyridyl]-4-[1-aminoethyl]-cyclohexanecarboxamide} from Merck-Millipore (Tokyo, Japan); Src inhibitor No. 5 {5 [4-(3′-methoxy-6′-chloro-anilino)-6-methoxy-7(morpholino-3-propoxy)-quinazoline]} from Biaffin GmbH & Co KG (Kassel, Germany); CRM 197 (an inactive diphtheria toxin) from WAKO (Osaka, Japan); OVA from Nakarai Tesque (Kyoto, Japan). Concentrations of these inhibitors used in this study were as follows: AG1478 (10 μmol/L), EGFR inhibitor 1 (10 μmol/L), hyroxyfasudil (10 μmol/L), ML-7 (10 μmol/L), PP2 (3 μmol/L), PTP-I (100 μmol/L), TAPI-0 (10 μmol/L), Y-27632 (10 μmol/L), Src inhibitor No. 5 (10 μmol/L), CRM 197 (10 μg/mL), and OVA (500 μmol/L).
Organ chamber experiments
All animal experiments were carried out in accordance with the Guidelines of the Kobe Gakuin University Experimental Animal Care. Male Wistar rats, 7 weeks old and 150−170 g in body weight, were anesthetized with diethyl ether. The thoracic aorta was excised, placed in Krebs-Henseleit solution (118.4 mmol/L NaC1, 4.7 mmol/L KC1, 2.5 mmol/L CaCl2, 1.2 mmol/L KH2PO4, 1.2 mmol/L MgSO4, 25.0 mmol/L NaHCO3, and 11.1 mmol/L glucose; pH 7.4), and then cleaned of adherent tissue. The aorta was cut into 3-mm rings, and the endothelium was removed by carefully rotating a manipulator inside the ring lumen. Four rings obtained from one animal were fixed vertically under a preload of 1.0 g in 5 mL organ chambers (UC-5A; Medical Kishimoto; Kyoto, Japan) filled with Krebs-Henseleit solution (37°C, pH 7.4), aerated continuously with a gas mixture of 95% O2 and 5% CO2, and equilibrated for 60 min. Isometric tension changes were measured with a force displacement transducer (AP-5; Medical Kishimoto) coupled to a dual-channel chart recorder (SS-250F; SEKONIC; Tokyo, Japan). All rings were exposed to 40 mmol/L KCl for 30 min to obtain the maximal force, and then equilibrated for 60 min before starting experiments. OVA dissolved in Krebs–Henseleit solution (pH 7.4) and PTP-I dissolved in dimethyl sulfoxide were then added to 5 mL organ chambers in a 10-μL volume. Inhibitors used in the present study were dissolved in dimethyl sulfoxide and added to organ chambers in 10-μL volumes 15 min before the addition of OVA or PTP-I. The contractile effects of OVA and PTP-I were expressed as a percentage of the maximal force evoked by 40 mmol/L KCl.
Cell culture
VSMCs were isolated from rat thoracic aortas and cultured in SmGM-2 medium (Lonza; Gaithersburg, MD). Cells at passage 5−10 at ∼90% confluence in 60-mm tissue culture plates were made quiescent by incubating in Dulbecco's modified Eagle medium (DMEM) supplemented with 0.05% fetal bovine serum for 1 day before experiments. Media were replaced with serum-free DMEM 2 h before starting experiments. Cells were then stimulated with or without OVA (500 μmol/L) or PTP-I (100 μmol/L) in serum-free DMEM for 10 min. In some cultures, various inhibitors were added 15 min before the addition of OVA or PTP-I.
Western blotting
After stimulation with OVA or PTP-I, cells were washed with ice-cold saline and lysed in specific buffer (50 mmol/L β-glycerophosphate, 100 mmol/L NaVO3, 2 mmol/L MgCl2, 1 mmol/L ethylene glycol bis(beta-aminoethylether)-N,N,N′,N′-tetraacetic acid, 0.5% Triton X-100, 1 mmol/L dithiothreitol, 20 μmol/L pepstatin, 20 μmol/L leupeptin, 0.1 U/mL aprotinin, and 1 mmol/L phenylmethylsulfonyl fluoride). Samples were centrifuged at 15,000g for 10 min at 4°C, and the concentration of soluble proteins in supernatant was determined using a BCA Protein Assay Kit (Thermo Scientific; Rockford, IL). Proteins were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) using 10% gels, and transferred to polyvinyldifluoride (PVDF) membranes (Immobilon-P; Millipore; Billerica, MA). Separation of proteins for the blots of phosphorylated EGFR at Tyr-845 was carried out by SDS–polyacrylamide gradient (4–15%) gel electrophoresis in a Mini-Protean TGX Gel (Bio-Rad; Hercules, CA). The blots were then blocked in 5% skimmed milk in Tris-buffered saline (0.1% Tween 20 in 10 mmol/L Tris-HCl, pH 7.5, containing 100 mmol/L NaCl) and incubated overnight at 4°C in Tris-buffered saline with rabbit antibodies against MYPT1 (1:200; Santa Cruz Biotechnology; Santa Cruz, CA), phosphorylated MYPT1 at Thr-853 (1:200; Santa Cruz Biotechnology), Src (1:1000; Cell Signaling; Danvers, MA), phosphorylated Src at Tyr-416 (1:1000; Cell Signaling), and EGFR (1:1000; Cell Signaling) and phosphorylated EGFR at Tyr-1173 (1:1000; Cell Signaling) and at Tyr-845 (1:1000; Cell Signaling). The PVDF membranes were then washed with Tris-buffered saline, and incubated with horseradish peroxidase-conjugated goat anti-rabbit (1:2000; Bio-Rad) in Tris-buffered saline for 1 h at room temperature. After washing twice in Tris-buffered saline, the blots were visualized using an enhanced chemiluminescence detection system (GE Healthcare Japan; Tokyo, Japan). Immunoblots were quantified using densitometry with Versa Doc 5000MP (Bio-Rad Laboratories) and Quantity One software (Bio-Rad).
Statistical analysis
All data are expressed as the mean ± SE. Statistical comparisons were performed using one-way analysis of variance with pairwise comparisons conducted by using the Bonferroni-Dunn method. Comparisons of concentration–response curves were carried out by repeated measures analysis of variance followed by the Bonferroni–Dunn method. Differences were considered significant at P < 0.05.
Results
OVA- and PTP-I-induced aortic contraction
OVA (500 μmol/L) evoked rapid contraction of endothelium-denuded aortas, presenting as a hyperbolic rise in tension development within 10 min followed by a slow linear rise (Fig. 1A). After washing the ring and equilibration for 1 h, a repeated exposure to OVA (500 μmol/L) caused reproducible force (Fig. 1A). Therefore, concentration–response curves for OVA (8−1000 μmol/L) were constructed in endothelium-denuded rings using stepwise increases in the concentration of OVA (Fig. 1B), and the maximal force was observed at concentrations of >500 μmol/L. In aortic rings with intact endothelium, OVA-induced contraction was attenuated significantly (P < 0.05; Fig. 1B). Like OVA, PTP-I, an irreversible PTPase inhibitor for SHP-1 and PTPase 1B, also evoked a contractile response in endothelium-denuded rings at a concentration of 100 μmol/L (Fig. 1A). However, after reaching a maximum tension within 20 min, the contractile force decreased rapidly to the initial tension, followed by a prolonged desensitization of the tissue to subsequent contractile actions of PTP-I or 40 mmol/L KCl (Fig. 1A). The mechanism by which a single exposure to PTP-I causes desensitization is unknown, but could be related to irreversible inhibitory effects on PTPases. Therefore, concentration–response curves for PTP-I were constructed by measuring the maximal force development in each ring exposed to a single concentration of PTP-I. As shown in Figure 1B, concentration-dependent contraction was observed in a narrow range of PTP-I concentrations between 50−100 μmol/L (Fig. 1B), and was attenuated significantly in rings with intact endothelium (P < 0.05; Fig. 1B). As OVA is a potent activator of endothelial nitric-oxide synthase (eNOS) (Papapetropoulos et al. 2004), it is likely that endothelium-derived NO attenuated the contractile effects of these PTPase inhibitors on aortic smooth muscle. Therefore, subsequent studies were carried out in endothelium-denuded aortas.
Figure 1.
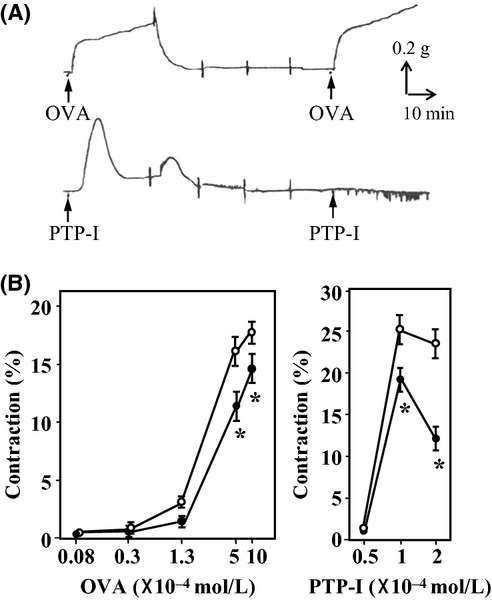
Contractile effects of sodium orthovanadate (OVA) and protein tyrosine phosphatase inhibitor-1 (PTP-I) on rat aortic rings. (A) Representative isometric force tracings of rings without endothelium in response to OVA (500 μmol/L; upper) and PTP-I (100 μmol/L; lower). Changes in force were recorded for 30 min after the addition of OVA or PTP-I. After washing four times every 15 min, the rings were exposed to the same concentration of OVA or PTP-I. (B) Concentration–response curves for OVA and PTP-I in rings with (closed circle) or without (open circle) endothelium. The curves for OVA were constructed by measuring force every 10 min after each stepwise increase in OVA concentration, whereas the curves for PTP-I were constructed by measuring the maximal force developed in each ring within 20 min of exposure to a single concentration of PTP-I. The contractile force was expressed as a percentage of the maximal force evoked by 40 mmol/L KCl. Data for rings taken from 4−5 animals are expressed as mean ± SE; *P < 0.05 vs. rings without endothelium.
Effects of inhibitors of Src, EGFR, and Rho-Kinase on OVA- and PTP-I-induced aortic contraction
To explore the signaling pathways involved in OVA- or PTP-I-induced vasocontraction, the effects of various inhibitors were investigated by measuring the contractile force 10 min after treatment with OVA (500 μmol/L), or the maximal force developed within 20 min of treatment with PTP-I (100 μmol/L) in aortic rings. A previous study demonstrated that OVA activated RhoA in guinea pig ileal smooth muscle, resulting in increased MLC phosphorylation (Mori and Tsushima 2004). Therefore, we studied effects of Rho-kinase inhibitors on OVA-induced vasocontraction. As shown in Figure 2A, the contractile effects of OVA (500 μmol/L) were abolished completely by the Rho-kinase inhibitors Y-27632 and hydroxyfasudil (P < 0.05). Similarly, the contractile effects of PTP-I (100 μmol/L) were also blocked by Y-27632 (Fig. 2B; P < 0.05). In contrast, the myosin light-chain kinase (MLCK) inhibitor ML-7 did not significantly affect OVA-induced contraction at a concentration of 10 μmol/L (Fig. 2A), which attenuated phenylephrine (1 μmol/L)-induced contraction (96.8 ± 6.0% in phenylephrine alone vs. 68.8 ± 3.3% in phenylephrine plus ML-7, n = 4; P < 0.05).
Figure 2.
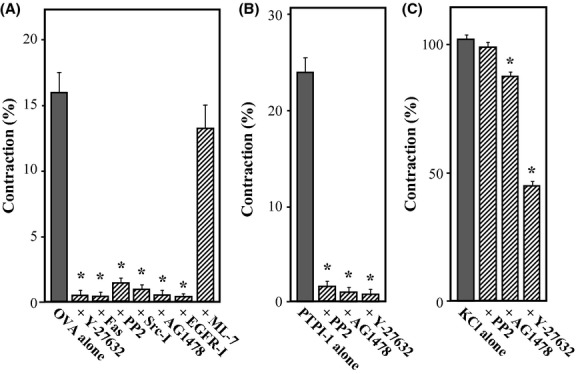
Effects of various inhibitors on aortic contraction induced by sodium orthovanadate (OVA, A), protein tyrosine kinase inhibitor-1 (PTP-I, B) or KCl (C). Contractile forces were measured 10 or 30 min after treatment with OVA (500 μmol/L) or KCl (40 mmol/L), respectively, or the maximal force developed within 20 min of treatment with PTP-I (100 μmol/L) in rings without endothelium. Y-27632 (10 μmol/L), hydroxyfasudil (Fas; 10 μmol/L), PP2 (3 μmol/L), Src inhibitor No. 5 (Src-I; 10 μmol/L), AG1478 (10 μmol/L), epidermal growth factor receptor (EGFR) inhibitor-1 (EGFR-I; 10 μmol/L), or ML-7 (10 μmol/L) was added to the bath 15 min before treatment with OVA or PTP-I. Contractile force was expressed as a percentage of the maximal force evoked by 40 mmol/L KCl. Data for rings were taken from 4−8 animals for experiments using OVA, from 7−8 animals for those using PTP-I, and from four animals for those using KCl. Data are expressed as mean ± SE; *P < 0.05 vs. OVA or PTP-I alone.
To explore the upstream signaling mediators of Rho-kinase activation, we studied the potential role of Src kinase in the contractile effects of these PTPase inhibitors, as it was reported that pervanadate activated Src in rat myometrial cells (Boulven et al. 2002). As expected, specific inhibitors for Src, PP2, and Src inhibitor No. 5, significantly attenuated the contractile effects of OVA (Fig. 2A; P < 0.05). PP2 also blocked the contractile effects of PTP-I (Fig. 2B; P < 0.05).
Previous studies suggested that OVA has EGF-mimetic properties (Chen and Chan 1993; Chien et al. 2006). Therefore, we assessed whether the vasoconstrictor effects of OVA or PTP-I were affected by EGFR kinase inhibitors such as AG1478 and EGFR inhibitor-1. We observed that treating aortic rings with AG1478 or EGFR inhibitor-1 abolished OVA-induced contraction (Fig. 2A; P < 0.05). AG1478 also blocked PTP-I-induced contraction (Fig. 2B; P < 0.05).
In contrast to a remarkable inhibition by these protein kinase inhibitors on OVA- or PTP-I-induced contraction, contraction by KCl was not affected by PP2, but slightly attenuated by AG1478 (P < 0.05) and profoundly inhibited by Y-27632 (P < 0.05). A similar observation was reported in the rabbit aorta in that KCl-induced contraction was blocked ∼50% by Y-27632 (Sakurada et al. 2003). However, it is uncertain whether the modest inhibition of KCl-induced contraction by AG1478 depends on the inhibition of EGFR.
Collectively, these data suggest that protein kinases, including Rho-kinase, Src, and EGFR, are potential signaling molecules that play roles in OVA and PTP-I-induced vasocontraction.
Effect of OVA on phosphorylation of Src, EGFR, and MYPT1 in VSMCs
The data obtained in the organ bath experiments suggested that at least three protein kinases are involved in OVA- and PTP-I-induced vasocontraction. To determine whether OVA induces the activation of Src, EGFR, and Rho-kinase, we measured levels of Src phosphorylated at Tyr-416, EGFR phosphorylated at Tyr-1173, and MYPT1 phosphorylated at Thr-853 in rat aortic VSMCs by western blotting. The levels of phosphorylated MYPT1 are used as an index of Rho-kinase activity (Sakurada et al. 2003). As shown in Figure 3, treating VSMCs with OVA increased the ratio of phosphorylated to total Src, EGFR, and MYPT1 rapidly within 3 min. The increased levels of these phosphorylated proteins were kept for 10 min and then returned to basal levels after 30 min.
Figure 3.
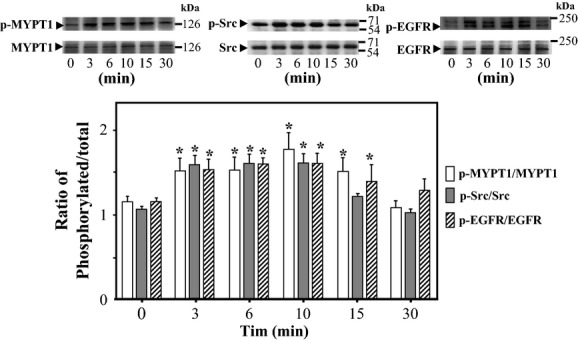
Time courses of sodium orthovanadate (OVA)-induced phosphorylation of Src, epidermal growth factor receptor (EGFR), and myosin phosphatase target subunit 1 (MYPT1) in rat aortic smooth muscle cells (VSMCs). The time course of changes in the phosphorylated forms of MYPT1 (Thr-853), Src (Tyr-416), and EGFR (Tyr-1173) were measured in VSMCs after treatment with OVA (500 μmol/L) by western blotting. (A) Representative blots of phosphorylated and total Src, EGFR, and MYPT1. (B) Bar graphs demonstrating densitometric quantification of the ratios of phosphorylated to total MYPT1, Src, and EGFR. Data are expressed as the mean ± SE of four independent experiments; *P < 0.05 vs. untreated cells (0 min).
Effects of various inhibitors of Src, EGFR, and Rho-kinase on OVA- and PTP-I-induced MYPT1 phosphorylation
To determine whether Rho-kinase is a downstream effector of Src and/or EGFR during PTPase inhibitor-induced vasocontraction, we measured the levels of phosphorylated MYPT1 at Thr-853 in rat VSMCs 10 min after treatment with OVA and PTP-I in the presence or absence of various protein kinase inhibitors. As shown in Figure 4, the ratio of phosphorylated to total MYPTI was increased significantly after treatment with OVA (Fig. 4A; P < 0.05) and PTP-I (Fig. 4B; P < 0.05). These effects of OVA and PTP-I were abolished by inhibitors specific for Rho-kinase (Y-27632 and hydroxyfasudil), Src (PP2 and Src inhibitor No. 5), and EGFR (AG1478 and EGFR inhibitor-1). These results suggest that Rho-kinase is a downstream target of Src and/or EGFR, which are activated by OVA- and PTP-I in VSMCs.
Figure 4.
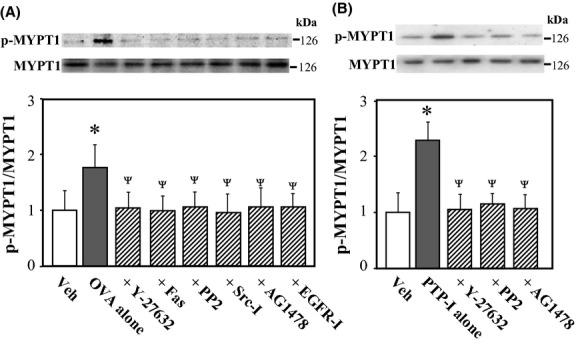
Effects of various inhibitors on sodium orthovanadate (OVA, A)- or protein phosphatase inhibitor-1 (PTP-I, B)-induced phosphorylation of myosin phosphatase target subunit 1 (MYPT1) in rat aortic smooth muscle cells (VSMCs). Phosphorylated (Thr-853) and total MYPT1 were measured in VSMCs 10 min after treatment with OVA (500 μmol/L) or PTP-I (100 μmol/L) by western blotting. Bar graphs show densitometric data as ratios of phosphorylated MYPT1 relative to total MYPT1. Y-27632 (10 μmol/L), hydroxyfasudil (Fas; 10 μmol/L), PP2 (3 μmol/L), Src inhibitor No. 5 (Src-I; 10 μmol/L), AG1478 (10 μmol/L), epidermal growth factor receptor (EGFR) inhibitor-1 (EGFR-I; 10 μmol/L), or vehicle (Veh; 10 μL of dimethyl sulfoxide) was added to cultures 15 min before treatment with OVA or PTP-I. Data represent the mean ± SE from four independent experiments; *P < 0.05 vs. Veh; ΨP < 0.05 vs. OVA or PTP-I alone.
Effects of various inhibitors of Src, EGFR, and Rho-kinase on OVA- and PTP-I-induced Src phosphorylation
To determine whether Src is the upstream effector of EGFR or vice versa during OVA- and PTP-I-induced Rho-kinase activation, we measured levels of Src phosphorylated at Tyr-416 in rat VSMCs 10 min after treatment with OVA and PTP-I in the presence or absence of various inhibitors. The ratio of phosphorylated to total Src was increased significantly after treatment with OVA (Fig. 5A; P < 0.05) and PTP-I (Fig. 5B; P < 0.05). Unlike MYPT1 phosphorylation, OVA- and PTP-I-induced Src phosphorylation was blocked by the Src inhibitors PP2 and Src inhibitor No.5, but not by inhibitors of Rho-kinase or EGFR (Fig. 5), suggesting that Src is a signaling effector acting upstream of Rho-kinase and EGFR.
Figure 5.
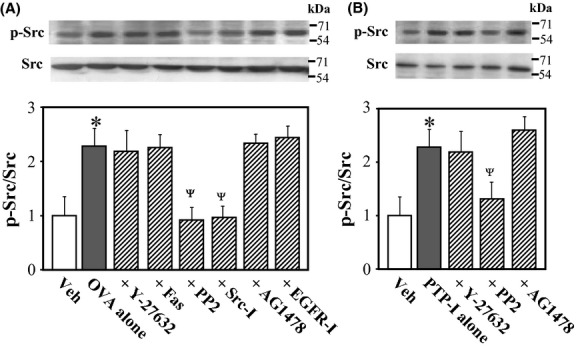
Effects of various inhibitors on sodium orthovanadate (OVA; A)- or protein phosphatase inhibitor-1 (PTP-I; B)-induced Src phosphorylation in rat aortic smooth muscle cells (VSMCs). Phosphorylated (Tyr-416) and total Src were measured in VSMCs 10 min after treatment with OVA (500 μmol/L) or PTP-I (100 μmol/L) by western blotting. Bar graphs represent densitometric quantification of the ratios of phosphorylated to total Src. Y-27632 (10 μmol/L), hydroxyfasudil (Fas; 10 μmol/L), PP2 (3 μmol/L), Src inhibitor No. 5 (Src-I; 10 μmol/L), AG1478 (10 μmol/L), epidermal growth factor receptor (EGFR) inhibitor-1 (EGFR-I; 10 μmol/L), or vehicle (Veh; 10 μL of dimethyl sulfoxide) was added to cultures 15 min before treatment with OVA- or PTP-I. Data represent mean ± SE of four independent experiments. *P < 0.05 vs. Veh; ΨP < 0.05 vs. OVA or PTP-I alone.
Effects of various inhibitors of Src, EGFR, and Rho-kinase on OVA- and PTP-I-induced EGFR phosphorylation
To confirm that EGFR is an intermediate signaling molecule between Src and Rho-kinase in OVA- and PTP-I-induced MYPT1 phosphorylation, we measured levels of EGFR phosphorylated at Tyr-1173 in rat VSMCs 10 min after treatment with OVA- and PTP-I in the presence or absence of various inhibitors. The ratio of phosphorylated to total EGFR was increased significantly after treatment with OVA (Fig. 6A; P < 0.05) and PTP-I (Fig. 6B; P < 0.05). These effects of OVA and PTP-I were abolished by inhibitors of Src (PP2 and Src inhibitor No.5) and EGFR (AG1478 and EGFR inhibitor-1), but not by Rho-kinase inhibitors, confirming that EGFR is a downstream effector of Src, and an upstream effector of Rho-kinase.
Figure 6.
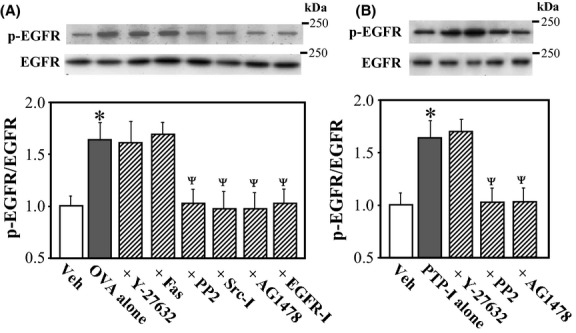
Effects of various inhibitors on sodium orthovanadate (OVA; A)- or protein phosphatase inhibitor-1 (PTP-I; B)-induced phosphorylation of epidermal growth factor receptor (EGFR) in rat aortic smooth muscle cells (VSMCs). Phosphorylated (Tyr-1173) and total EGFR were measured in VSMCs 10 min after treatment with OVA (500 μmol/L) or PTP-I (100 μmol/L) by western blotting. Bar graphs represent densitometric quantification of the ratios of phosphorylated to total EGFR. Y-27632 (10 μmol/L), hydroxyfasudil (Fas; 10 μmol/L), PP2 (3 μmol/L), Src inhibitor No. 5 (Src-I; 10 μmol/L), AG1478 (10 μmol/L), epidermal growth factor receptor (EGFR) inhibitor-1 (EGFR-I; 10 μmol/L), or vehicle (Veh; 10 μL of dimethyl sulfoxide) was added to cultures 15 min before treatment with OVA or PTP-I. Data are presented as the mean ± SE of four independent experiments. *P < 0.05 vs. Veh; ΨP < 0.05 vs. OVA or PTP-I alone.
Transactivation of EGFR in VSMCs by OVA
Transactivation of EGFR can occur through the release of membrane-anchored EGFR ligands, predominantly heparin-binding EGF-like growth factor (HB-EGF) (Prenzel et al. 2001). Interestingly, a previous study showed that pervanadate could induce ectodomain shedding of pro-EGF in Chinese hamster ovary cells (Le Gall et al. 2003). To determine whether pro-EGF shedding contributes to OVA-induced EGFR activation, we tested the effects of two inhibitors on the OVA-induced vascular contraction: TAPI-0, a metalloproteinase inhibitor, and CRM 197, a diphtheria toxin mutant that specifically blocks the action of pro-HB-EGF (Mitamura et al. 1995). These inhibitors significantly attenuated the OVA-induced aortic contraction (Fig. 7A; P < 0.05), while not affected KCl-induced contraction (103.0 ± 0.8% in four rings treated with 40 mmol/L KCl alone vs. 98.7 ± 0.6% or 102.7 ± 1.7% in four rings treated with KCl plus TAPI-0 or CRM 197, respectively; P > 0.05). Furthermore, these inhibitors significantly blocked the OVA-induced phosphorylation of EGFR at Tyr-1173 and MYPT1 at Thr-853 (Fig. 7B; P < 0.05) but not of Src at Tyr-416 in VSMCs (data not shown). These inhibitors did not affect the basal levels of phosphorylated EGFR and MYPT1 in vehicle-treated VSMCs (data not shown).
Figure 7.
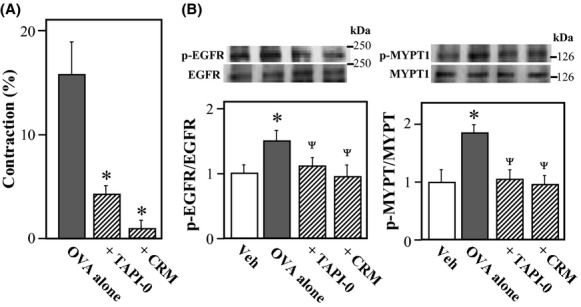
Effects of TAPI-0 and CRM 197 on sodium orthovanadate (OVA)-induced aortic contraction and phosphorylation of epidermal growth factor receptor (EGFR) and myosin phosphatase target subunit 1 (MYPT1) in rat aortic smooth muscle cells (VSMCs). (A) Contractile force was measured 10 min after treatment with OVA (500 μmol/L) in rings without endothelium. TAPI-0 (10 μmol/L) and CRM 197 (10 μg/mL) were added to the bath 15 min before OVA treatment. The contractile force was expressed as a percentage of the maximal force evoked by 40 mmol/L KCl. Data for rings taken from four animals are expressed as mean ± SE *P < 0.05 vs. OVA alone. (B) Phosphorylated EGFR (Tyr-1173), MYPT1 (Thr-853), and the respective total proteins were measured in VSMCs 10 min after treatment with OVA (500 μmol/L) by western blotting. TAPI-0 (10 μmol/L), CRM 197 (10 μg/mL), or vehicle (Veh; 10 μL of dimethyl sulfoxide) was added to cultures 15 min before treatment with OVA. Bar graphs represent densitometric quantification of the ratios of phosphorylated to total EGFR and MYPT1. Data are presented as the mean ± SE of four independent experiments. *P < 0.05 vs. Veh; ΨP < 0.05 vs. OVA alone.
OVA-induced phosphorylation of Tyr-845 in EGFR
Because Src can activate EGFR via the direct phosphorylation at Tyr-845 (Biscardi et al. 1999), we evaluated the effects of OVA on the phosphorylation of this residue in EGFR. Western blotting using a phospho-specific EGFR-Tyr-845 antibody revealed that the phosphorylation of EGFR at Tyr-845 was increased in OVA-treated VSMCs, and that this effect was blocked by Src inhibitor PP2 (Fig. 8; P < 0.05).
Figure 8.
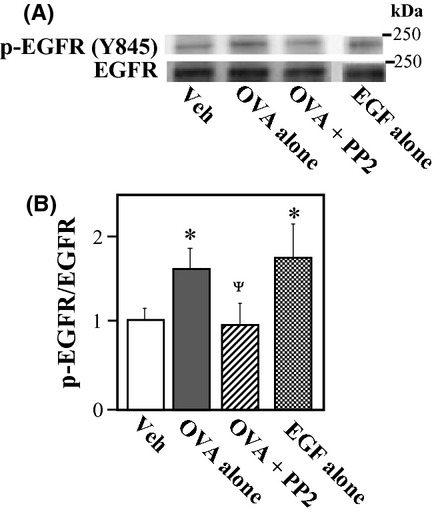
Effect of sodium orthovanadate (OVA) on epidermal growth factor receptor (EGFR) phosphorylation at Tyr-845 in rat aortic smooth muscle cells (VSMCs). VSMCs were treated with OVA (500 μmol/L), EGF (0.1 μmol/L) or vehicle (Veh; 10 μL of dimethyl sulfoxide) for 10 min. PP2 (3 μmol/L) was added 15 min before treatment with OVA. Phosphorylated EGFR at Tyr-845 was measured by western blotting. (A) Representative blots of phosphorylated (Tyr-845) and total EGFR. A blot of phosphorylated EGFR in EGF-treated VSMCs is shown as a positive band. (B) Bar graphs demonstrating densitometric quantification of the ratios of phosphorylated to total EGFR. Data are presented as the mean ± SE of four independent experiments. *P < 0.05 vs. Veh; ΨP < 0.05 vs. OVA alone.
Discussion
The present study demonstrated, for the first time, that the vasoconstrictor actions of OVA are mediated by the Src-dependent transactivation of EGFR, which in turn induces the inactivation of MLCP via the Rho-kinase-dependent phosphorylation of MYPT1. Earlier findings that OVA-induced smooth muscle contraction was inhibited by genistein, a potent protein kinase inhibitor, suggested that OVA-induced smooth muscle contraction is coupled to enhanced protein tyrosine phosphorylation (Di Salvo et al. 1993; Alcón et al. 2000; Mori and Tsushima 2004). This is compatible with the findings of the present study, where the PTPase inhibitor PTP-I also exhibited vasoconstrictor effects via similar mechanisms involving the Src, EGFR, and Rho-kinase signaling pathway.
The Rho kinase pathway is involved in smooth muscle contraction, where activated Rho-kinase inhibits MLCP via MYPT1 phosphorylation, causing Ca2+ -sensitization, and thereby enhanced contraction without changing the levels of cytosolic-free Ca2+ and MLCK activity (Somlyo and Somlyo 2003). A role for Rho-kinase in OVA-induced smooth muscle contraction was demonstrated in a previous study using guinea pig ileal longitudinal smooth muscle, where a Rho-kinase inhibitor, Y-27632, blocked OVA-induced contraction and MLC phosphorylation (Mori and Tsushima 2004). Our present study also identified a primary role for Rho-kinase in OVA-induced aortic contraction, as the Rho-kinase inhibitors Y-27632 and hydoxyfasudil effectively abolished OVA-induced vasocontraction and MYPT1 phosphorylation, whereas the MLCK inhibitor ML-7 did not affect OVA-induced vasocontraction. This is compatible with previous studies showing that the OVA- or pervanadate-induced contractile effects on smooth muscle cells were not accompanied by a rise in cytosolic-free Ca2+, but instead occurred as a result of increased Ca2+ sensitivity of the contractile apparatus (Masui and Wakabayashi 2000; Spurrell et al. 2000; Murphy et al. 2002).
Protein tyrosine phosphorylation is controlled reciprocally by protein tyrosine kinases and PTPases. Because several protein tyrosine kinases, including Src, are themselves activated by phosphorylation at critical tyrosine residues, inhibiting PTPase activity may also directly increase the activity of these endogenous tyrosine kinases (Hubbard and Till 2000). Earlier studies revealed that pervanadate induced the activation of the Src family kinases Ick and Fyn in human T cells (Secrist et al. 1993; Evans et al. 1994) and c-Src in rat myometrial cells (Boulven et al. 2002). In the present study, the structurally distinct Src kinase inhibitors PP2 and Src inhibitor No. 5 abolished OVA- and PTP-I-induced vasocontraction. In addition, both OVA and PTP-I increased the phosphorylation of Src at Tyr-416 in rat VSMCs, which was blocked in the presence of Src inhibitors. This suggests that the inhibition of unidentified PTPase(s) by OVA or PTP-I results in the activation of Src, thereby inducing vasocontraction. This was confirmed by findings that Src inhibitors abrogated OVA- or PTP-I-induced MYPT1 phosphorylation in VSMCs. Src becomes activated as a result of molecular processes that interrupt its negative regulatory interactions, including the binding of the Src SH2 or SH3 domains to a binding protein, such as a tyrosine-phosphorylated growth factor receptor, and dephosphorylation of the carboxyl-terminal negative regulatory site of Src at Tyr-527, resulting in the autophosphorylation of Src at Tyr-416 (Hubbard and Till 2000; Roskoski 2005). Two PTPases, PTPase 1B (Bjorge et al. 2000) and SHP-1 (Somani et al. 1997), can dephosphorylate Src at Tyr-527. However, as shown in the present study, PTP-I which is a specific inhibitor for SHP-1 and PTPase 1B exhibited OVA-like vasoconstrictor effect through the activation of Src. In addition, a previous study showed that OVA was an inhibitor for the dephosphorylation of Src at Tyr-527 by PTPase 1B (Bjorge et al. 2000). Therefore, PTPase(s) which involves the activation of Src by OVA remain unidentified, because these previous studies do not account for the OVA- or PTP-I-induced activation of Src.
We identified EGFR as another signaling molecule involved in OVA-induced vasocontraction, as the EGFR kinase inhibitors AG1478 and EGFR inhibitor-1 blocked OVA-induced aortic contraction and MYPT1 phosphorylation in VSMCs. Western blotting further revealed that treating VSMCs with OVA or PTP-I stimulated the phosphorylation of EGFR at Tyr-1173, one of multiple autophosphorylation sites, which was abolished by EGFR kinase inhibitors. Importantly, EGFR inhibitors did not affect OVA- or PTP-I-induced phosphorylation of Src, whereas Src inhibitors blocked OVA- or PTP-I-induced EGFR phosphorylation at Tyr-1173, suggesting that EGFR is a downstream effector of Src.
EGFR transactivation occurs through two essentially distinct pathways: an intracellular pathway involving protein kinase C, Ca2+, and Src; and the extracellular release of soluble EGFR ligands via ectodomain shedding (Prenzel et al. 2001). Src can activate EGFR either directly through the phosphorylation of EGFR at Tyr-845 in the cytoplasm (Sato et al. 1995; Biscardi et al. 1999), or indirectly via the metalloproteinase-catalyzed release of HB-EGF from pro-HB-EGF (Pai et al. 2002). In the present study, two inhibitors of pro-EGF shedding (TAPI-0 and CRM 197) blocked not only OVA-induced vasocontraction, but also OVA-induced phosphorylation of MYPT1 and EGFR (Tyr-1173). These findings suggest that the OVA-induced vasocontraction depends on the transactivation of EGFR via the shedding of pro-HB-EGF in VSMCs. The observation that OVA-induced EGF phosphorylation at Tyr-1173 was blocked by Src inhibitors suggests that the transactivation of EGF via pro-HB-EGF processing depends on Src kinase activity. Furthermore, we found that treating VSMCs with OVA stimulated the phosphorylation of EGFR at Tyr-845. The Src-catalyzed phosphorylation of EGFR at this residue is thought to be essential for mitogenic signaling by EGFR, as cells expressing Y845F mutant of EGFR showed markedly decreased DNA synthesis in response to EGF (Tice et al. 1999). Therefore, it is likely that the activation of Src by OVA induces the transactivation of EGFR via pro-HB-EGF shedding and the direct phosphorylation of EGFR at Tyr-845, both of which activate EGFR, resulting in the activation of Rho-kinase in VSMCs through unidentified pathway. Nevertheless, it is important to determine whether Src and EGFR act synergistically to regulate the contractile activity of smooth muscles, as occurs during cell growth and tumorigenesis (Haskell et al. 2001).
In contrast to the vasoconstrictor effects of vanadates in various species and vasculatures (Rapp 1981; Fox et al. 1983; Sánchez-Ferrer et al. 1988; Spurrell et al. 2000; Villalba et al. 2010), the endothelium-dependent vasodilator effect of OVA has also been reported in rat mesenteric artery (Misurski et al. 2000) and porcine coronary artery (Nakaike et al. 1996). As the OVA-induced relaxation of these arteries was reduced by inhibitors of NO synthase and potassium channels, endothelial tyrosine kinases were considered to be involved in the synthesis of NO and endothelial-derived hyperpolarizing factor. Subsequently, Papapetropoulos et al. (2004) demonstrated that OVA caused NO release via a mechanism that involves the phosphoinositide 3-kinase/Akt-mediated eNOS phosphorylation and increased binding of heat shock protein 90 to eNOS. Consistent with these reports, we observed that the OVA- or PTP-I-induced aortic contraction was significantly enhanced by the removal of endothelium (Fig. 1) or treatment with eNOS inhibitor (data not shown), suggesting that the contractile response of VSMCs to OVA is partially neutralized in intact aorta by an indirect action of OVA through endothelium. Upstream events in endothelium involving in the OVA-induced activation of phosphoinositide 3-kinase/Akt pathway are unknown. However, previous findings demonstrating a key role of Src in the activation of eNOS through the phosphoinositide 3-kinase/Akt pathway (Haynes et al. 2003; Jin et al. 2003; Yu et al. 2012) suggest an involvement of Src in the OVA-induced eNOS activation.
The present study has several limitations. First, we carried out the in vitro study using cultured VSMCs to detect signaling events, instead of aortas. A conflicting result was seen in VSMCs 30 min after treatment with OVA in which the levels of phosphorylated MYPT1 returned to basal levels (Fig. 3), despite the sustained contraction of aortic rings (Fig. 1). The reason is unknown, but further studies using aorta will be needed. Second, the evidence obtained in this study may not apply to smooth muscle of resistance arteries, because VSMCs of the large (conduit) arteries are phenotypically and functionally distinct from those of the small (resistance) arteries (Reho et al. 2014). Third, it remains unknown whether the OVA-induced contractile response of nonvascular smooth muscle depends on the same signaling events. Finally, the present study did not identify PTPase(s), a target molecule of OVA involved in Src activation.
In summary, the present study provides novel findings that the downstream signaling from Src-activated EGFR links to Rho-kinase-mediated MLCP inactivation, thereby regulating smooth muscle contractility. Figure 9 illustrates a proposed model delineating the key steps from the inhibition of PTPase by OVA to the increased contractility of VSMCs. The inhibition of unidentified PTPase(s) by vanadate activates Src kinase, which in turn induces the transactivation of EGFR via direct phosphorylation at Tyr-845 and a mechanism involving pro-HB-EGF shedding. Signaling downstream of the activated EGFR stimulates Rho-kinase activity, which inactivates MLCP activity by the phosphorylation of MYPT1, resulting in increased VSMC contractility. Of interest, EGF can stimulate the contraction of vascular and nonvascular smooth muscles, including the rat aorta (Berk et al. 1985), rabbit aorta (Merkel et al. 1993), and guinea pig gastric smooth muscle (Zheng et al. 1997), raising the possibility that the direct stimulation of EGFR by EGF-related growth factors also could induce smooth muscle contraction in a Rho-kinase-dependent manner.
Figure 9.
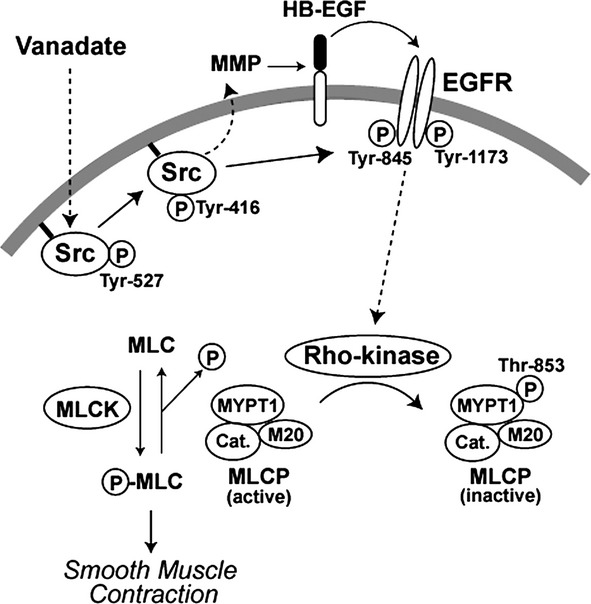
Model depicting the proposed mechanisms underlying vanadate-induced contraction in vascular smooth muscle cells (VSMCs). Inhibition of protein tyrosine phosphatase(s) by vanadate induces the activation of Src (Tyr-416 phosphorylation), which transactivates epidermal growth factor receptor (EGFR) indirectly through the shedding of heparin-binding (HB)-EGF, and directly via the Src-catalyzed phosphorylation of EGFR at Tyr-845. Downstream signaling via unknown pathways from activated EGFR stimulates Rho-kinase activity, which inactivates myosin light chain phosphatase (MLCP) by phosphorylating myosin phosphatase target subunit 1 (MYPT1) at Thr-853. This results in enhanced MLC phosphorylation via the Ca2+-dependent activation of myosin light chain kinase (MLCK).
Perspectives
Several studies have shown that the Src-mediated activation of EGFR plays critical roles not only in cell proliferation (Haskell et al. 2001) but also in the initiation and progression of vascular injury such as atherosclerosis. For instance, vasoconstrictors such as angiotensin II, endothelin, and thrombin induce hyperproliferation or migration of VSMCs via the Src-dependent transactivation of EGFR (Bokemeyer et al. 2000; Hsieh et al. 2009; Li et al. 2010; Mugabe et al. 2010). Furthermore, previous studies have shown that the transactivation of EGFR is involved in the mechanisms of the pressure-induced myogenic tone (Lucchesi et al. 2004) and vasoconstrictor effect of α-adrenoceptor agonist (Hao et al. 2004), suggesting an important role of the signaling pathway from EGFR in the development and progression of hypertensive disorders. Our present study shed light on a link between the Src-dependent EGFR transactivation and contractility in VSMCs, and therefore has important implications in vascular physiology and diseases.
Glossary
- AG1478
4-(3-chloroanilino)-6,7-dimethoxyquinazoline
- DMEM
Dulbecco's modified Eagle medium
- EGF
epidermal growth factor
- EGFR inhibitor 1
cyclopropanecarboxylic acid-(3-(6-(3-trifluoromethyl-phenylamino)-pyrimidin-4-ylamino)-phenyl)-amide
- EGFR
epidermal growth factor receptor
- eNOS
endothelial nitric-oxide synthase
- HB-EGF
heparin-binding EGF-like growth factor
- hydroxyfasudil
1-(1-hydroxy-5-isoquinolinesulfonyl)homopiperazine
- ML-7
1-(5-iodonaphthalene-1-sulfonyl)homopiperazine
- MLCK
myosin light-chain kinase
- MLC
myosin light-chain
- MLCP
myosin light-chain phosphatase
- MYPT1
myosin phosphatase target subunit 1
- OVA
sodium orthovanadate
- PP2
4-amino-3-(4-chlorophenyl)-1-(t-butyl)-1H-pyrazolo[3,4-d]pyrimidine
- PTPase
protein tyrosine phosphatase
- PTP-I
protein tyrosine phosphatase inhibitor-1 α-bromo-4-hydroxyacetophenone
- PVDF
polyvinyldifluoride
- SDS-PAGE
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- Src inhibitor No. 5
4-(3′-methoxy-6′-chloro-anilino)-6-methoxy-7(morpholino-3-propoxy)-quinazoline
- TAPI-0
N-(R)-[2-(hydroxyaminocarbonyl) methyl]-4-methylpentanoyl- l-naphthylalanyl- l-alanine amide
- VSMCs
vascular smooth muscle cells
- Y-27632
R-[+]-trans-N-[4-pyridyl]-4-[1-aminoethyl]-cyclohexanecarboxamide.
Disclosure
None declared.
References
- Alcón S, Camello PJ, Garcia LJ, Pozo MJ. Activation of tyrosine kinase pathway by vanadate in gallbladder smooth muscle. Biochem Pharmacol. 2000;59:1077–1089. doi: 10.1016/s0006-2952(00)00237-9. [DOI] [PubMed] [Google Scholar]
- Berk BC, Brock TA, Webb RC, Taubman MB, Atkinson WJ, Gimbrone MA, et al. Epidermal growth factor, a vascular smooth muscle mitogen, induces rat aortic contraction. J Clin Invest. 1985;75:1083–1086. doi: 10.1172/JCI111772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biscardi JS, Maa M-C, Tice DA, Cox ME, Leu T-H, Parsons SJ. C-Src-mediated phosphorylation of the epidermal growth factor receptor on Tyr845 and Tyr1101 is associated with modulation of receptor function. J Biol Chem. 1999;274:8335–8343. doi: 10.1074/jbc.274.12.8335. [DOI] [PubMed] [Google Scholar]
- Bjorge JD, Pang A, Fujita DJ. Identification of protein-tyrosine phosphatase B as the major tyrosine phosphatase activity capable of dephosphorylating and activating c-Src in several human breast cancer cell lines. J Biol Chem. 2000;275:41439–41446. doi: 10.1074/jbc.M004852200. [DOI] [PubMed] [Google Scholar]
- Bokemeyer D, Schmitz U, Kramer HJ. Angiotensin II-induced growth of vascular smooth muscle cells requires an Src-dependent activation of the epidermal growth factor receptor. Kidney Int. 2000;58:549–558. doi: 10.1046/j.1523-1755.2000.t01-1-00201.x. [DOI] [PubMed] [Google Scholar]
- Boulven I, Robin P, Desmyter C, Harbon S, Leiber D. Differential involvement of Src family kinases in pervamadate-mediated responses in rat myometrial cells. Cell Signal. 2002;14:341–349. doi: 10.1016/s0898-6568(01)00269-8. [DOI] [PubMed] [Google Scholar]
- Cantley LC, Jr, Josephson L, Warner R, Yanagisawa M, Lechene C, Guidotti G. Vanadate is a potent (Na, K)-ATPase inhibitor found in ATP derived from muscle. J Biol Chem. 1977;252:7421–7423. [PubMed] [Google Scholar]
- Chen Y, Chan TM. Orthovanadate and 2, 3-dimethoxy-1, 4-naphthoquinone augment growth factor-induced cell proliferation and c-fos gene expression in 3T3-L1 cells. Arch Biochem Biophys. 1993;305:9–16. doi: 10.1006/abbi.1993.1387. [DOI] [PubMed] [Google Scholar]
- Chien PS, Mak OT, Huang HJ. Induction of COX-2 protein expression by vanadate in A549 human lung carcinoma cell line through EGF receptor and p38 MAPK-mediated pathway. Biochem Biophys Res Commun. 2006;339:562–568. doi: 10.1016/j.bbrc.2005.11.045. [DOI] [PubMed] [Google Scholar]
- Di Salvo J, Semenchuk LA, Lauer J. Vanadate-induced contraction of smooth muscle and enhanced protein tyrosine phosphorylation. Arch Biochem Biophys. 1993;304:386–391. doi: 10.1006/abbi.1993.1366. [DOI] [PubMed] [Google Scholar]
- Evans GA, Garcia GG, Erwin R, Howard OM, Farrar WL. Pervanadate stimulates the effects of interleukin-2 (IL-2) in human T cells and provides evidence for the activation of two distinct tyrosine kinase pathways by IL-2. J Biol Chem. 1994;269:23407–23412. [PubMed] [Google Scholar]
- Fox AA, Borchard U, Neumann M. Effects of vanadate on isolated vascular tissue: biochemical and functional investigations. J Cardiovasc Pharmacol. 1983;5:309–316. doi: 10.1097/00005344-198303000-00024. [DOI] [PubMed] [Google Scholar]
- Hao L, Du M, Lopez-Campistrous A, Fernandez-Patron C. Agonist-induced activation of matrix metalloproteinase-7 promotes vasoconstriction through the epidermal growth factor-receptor pathway. Circ Res. 2004;94:68–76. doi: 10.1161/01.RES.0000109413.57726.91. [DOI] [PubMed] [Google Scholar]
- Haskell MD, Slack JK, Parsons JT, Parsons SJ. c-Src tyrosine phosphorylation of epidermal growth factor receptor, p190 RhoGAP, and focal adhesion kinase regulates diverse cellular processes. Chem Rev. 2001;101:2425–2440. doi: 10.1021/cr0002341. [DOI] [PubMed] [Google Scholar]
- Haynes MP, Li L, Sinha D, Russell KS, Hisamoto K, Baron R, et al. Src kinase mediates phosphatidylinositol 3-kinase/Akt-dependent rapid endothelial nitric-oxide synthase activation by estrogen. J Biol Chem. 2003;278:2118–2123. doi: 10.1074/jbc.M210828200. [DOI] [PubMed] [Google Scholar]
- Hsieh H-L, Tung W-H, Wu C-Y, Wang H-H, Lin C-C, Wang T-S, et al. Thrombin induces EGF receptor expression and cell proliferation via a PKC(δ)/c-Src-dependent pathway in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2009;29:1594–1601. doi: 10.1161/ATVBAHA.109.185801. [DOI] [PubMed] [Google Scholar]
- Hubbard SR, Till JH. Protein tyrosine kinase structure and function. Ann Rev Biochem. 2000;69:373–398. doi: 10.1146/annurev.biochem.69.1.373. [DOI] [PubMed] [Google Scholar]
- Jin ZG, Ueba H, Tanimoto T, Lungu AO, Frame MD, Berk BC. Ligand-independent activation of vascular endothelial growth factor receptor 2 by fluid shear stress regulates activation of endothelial nitric oxide synthase. Cric Res. 2003;93:354–363. doi: 10.1161/01.RES.0000089257.94002.96. [DOI] [PubMed] [Google Scholar]
- Laniyonu A, Saifeddine M, Ahmad S, Hollenberg MD. Regulation of vascular and gastric smooth muscle contractility by pervanadate. Br J Pharmacol. 1994;113:403–410. doi: 10.1111/j.1476-5381.1994.tb17003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Gall SM, Auger R, Dreux C, Mauduit P. Regulated cell surface pro-EGF ectodomain shedding is a zinc metalloprotease-dependent process. J Biol Chem. 2003;278:45255–45268. doi: 10.1074/jbc.M307745200. [DOI] [PubMed] [Google Scholar]
- Li Y, Lévesque L-O, Anand-Srivastava MB. Epidermal growth factor receptor transactivation by endogenous vasoactive peptides contributes to hyperproliferation of vascular smooth muscle cells of SHR. Am J Physiol Heart Circ Physiol. 2010;299:H1959–H1967. doi: 10.1152/ajpheart.00526.2010. [DOI] [PubMed] [Google Scholar]
- Lucchesi PA, Sabri A, Belmadani S, Matrougui K. Involvement of metalloproteinases 2/9 in epidermal growth factor receptor transactivation in pressure-induced myogenic tone in mouse mesenteric resistance arteries. Circulation. 2004;110:3587–3593. doi: 10.1161/01.CIR.0000148780.36121.47. [DOI] [PubMed] [Google Scholar]
- Masui H, Wakabayashi I. Tyrosine phosphorylation increases Ca2+ sensitivity of vascular smooth muscle contraction. Life Sci. 2000;68:363–372. doi: 10.1016/s0024-3205(00)00942-5. [DOI] [PubMed] [Google Scholar]
- Mehdi MZ, Pandey SK, Theberge JF, Srivastava AK. Insulin signal mimicry as a mechanism for the insulin-like effects of vanadium. Cell Biochem Biophys. 2006;44:73–81. doi: 10.1385/CBB:44:1:073. [DOI] [PubMed] [Google Scholar]
- Merkel LA, Rivera LM, Colussi DJ, Perrone MH. Inhibition of EGF-induced vasoconstriction in isolated rabbit aortic rings with the tyrosine kinase inhibitor RG50864. Biochem Biophys Res Commun. 1993;192:1319–1326. doi: 10.1006/bbrc.1993.1560. [DOI] [PubMed] [Google Scholar]
- Misurski D, Tatchum-Talom R, McNeill JR, Gopalakrishnan V. Vanadate-evoked relaxation of the perfused rat mesenteric vascular bed. Life Sci. 2000;67:1369–1379. doi: 10.1016/s0024-3205(00)00725-6. [DOI] [PubMed] [Google Scholar]
- Mitamura T, Higashiyama S, Taniguchi N, Klagsbrun M, Mekada E. Diphtheria toxin binds to the epidermal growth factor (EGF)-like domain of human heparin-binding EGF-like growth factor/diphtheria toxin receptor and inhibits specifically its mitogenic activity. J Biol Chem. 1995;270:1015–1019. doi: 10.1074/jbc.270.3.1015. [DOI] [PubMed] [Google Scholar]
- Mori M, Tsushima H. Vanadate activates Rho A translocation in association with contracting effects in ileal longitudinal smooth musucle of guinea pig. J Pharmacol Sci. 2004;95:443–451. doi: 10.1254/jphs.fp0030576. [DOI] [PubMed] [Google Scholar]
- Morita A, Yamamoto S, Wang B, Tanaka K, Suzuki N, Aoki S, et al. Sodium orthovanadate inhibits p53-mediated apoptosis. Cancer Res. 2010;70:257–265. doi: 10.1158/0008-5472.CAN-08-3771. [DOI] [PubMed] [Google Scholar]
- Mugabe BE, Yaghini FA, Song CY, Buharalioglu CK, Water CM, Malik KU. Angiotensin II-induced migration of vascular smooth muscle cells is mediated by p38 mitogen-activated protein kinase-activated c-Src through spleen tyrosine kinase and epidermal growth factor receptor transactivation. J Phamacol Exp Ther. 2010;332:16–124. doi: 10.1124/jpet.109.157552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy TV, Spurrell BE, Hill MA. Mechanisms underlying pervanadate-induced contraction of rat cremaster muscle arterioles. Eur J Pharmacol. 2002;442:107–114. doi: 10.1016/s0014-2999(02)01498-x. [DOI] [PubMed] [Google Scholar]
- Nakaike R, Shimokawa H, Owada MK, Tokunaga O, Yasutake H, Kishimoto T, et al. Vanadate causes synthesis of endothelium-derived NO via pertussis toxin-sensitive G protein in pigs. Am J Physiol. 1996;271:H296–H302. doi: 10.1152/ajpheart.1996.271.1.H296. [DOI] [PubMed] [Google Scholar]
- Nayler RA, Sparrow MP. Mechanism of vanadate-induced contraction of airway smooth muscle of the guinea-pig. Br J Pharmacol. 1983;80:163–172. doi: 10.1111/j.1476-5381.1983.tb11062.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pai R, Soreghan B, Szabo IL, Pavelka M, Baatar D, Tarnawski AS. Prostaglandin E2 transactivates EGF receptor: a novel mechanism for promoting colon cancer growth and gastrointestinal hypertrophy. Nat Med. 2002;8:289–293. doi: 10.1038/nm0302-289. [DOI] [PubMed] [Google Scholar]
- Papapetropoulos A, Fulton D, Lin MI, Fontana J, McCabe TJ, Zoellner S, et al. Vanadate is a potent activator of endothelial nitric-oxide synthase: evidence for the role of the serine/threonine kinase Akt and the 90-kDa heat shock protein. Mol Pharmacol. 2004;65:407–415. doi: 10.1124/mol.65.2.407. [DOI] [PubMed] [Google Scholar]
- Prenzel N, Fisher OM, Streit S, Hart S, Ullrich A. The epidermal growth factor receptor family as a central element for cellular signal transduction and diversification. Endocr Relat Cancer. 2001;8:11031. doi: 10.1677/erc.0.0080011. [DOI] [PubMed] [Google Scholar]
- Rapp J. Aortic responses to vanadate: independence from (Na, K)-ATPase and comparison of Dahl salt-sensitive and salt-resistant rats. Hypertension. 1981;3:I168–I172. doi: 10.1161/01.hyp.3.3_pt_2.i168. [DOI] [PubMed] [Google Scholar]
- Reho JJ, Zheng X, Fisher SA. Smooth muscle contractile diversity in the control of regional circulations. Am J Physiol Heart Circ Physiol. 2014;306:H163–H172. doi: 10.1152/ajpheart.00493.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roskoski R. Src kinase regulation by phosphorylation and dephosphorylation. Biochem Biophys Res Commun. 2005;331:1–14. doi: 10.1016/j.bbrc.2005.03.012. [DOI] [PubMed] [Google Scholar]
- Sabbioni E, Pozzi G, Pintar A, Casella L, Garattini S. Cellular retention, cytotoxicity and morphological transformation by vanadium(IV) and vanadium(V) in BALB/3T3 cell lines. Carcinogenesis. 1991;12:47–52. doi: 10.1093/carcin/12.1.47. [DOI] [PubMed] [Google Scholar]
- Sakurada S, Takuwa N, Sugimoto N, Wang Y, Seto M, Sasaki Y, et al. Ca2+ -dependent activation of Rho and Rho kinase in membrane depolarization-induced and receptor stimulation-induced vascular smooth muscle contraction. Circ Res. 2003;93:548–556. doi: 10.1161/01.RES.0000090998.08629.60. [DOI] [PubMed] [Google Scholar]
- Sánchez-Ferrer CF, Marín J, Lluch M, Valverde A, Salaices M. Actions of vanadate on vascular tension and sodium pump activity in cat isolated cerebral and femoral arteries. Br J Pharmacol. 1988;93:53–60. doi: 10.1111/j.1476-5381.1988.tb11404.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K, Sato A, Aoto M, Fukami Y. c-Src phosphorylates epidermal growth factor receptor on tyrosine 845. Biochem Biophys Res Commun. 1995;215:1078–1087. doi: 10.1006/bbrc.1995.2574. [DOI] [PubMed] [Google Scholar]
- Secrist JP, Burns LA, Karnitz L, Koretzky GA, Abraham RT. Stimulatory effects of the protein tyrosine phosphatase inhibitor, pervanadate, on T-cell activation events. J Biol Chem. 1993;268:5886–5893. [PubMed] [Google Scholar]
- Shimada T, Shimamura K, Sunano S. Effects of sodium vanadate on various types of vascular smooth muscles. Blood Vessels. 1986;23:113–124. doi: 10.1159/000158628. [DOI] [PubMed] [Google Scholar]
- Somani A-K, Bignon JS, Mills GB, Siminovitch KA, Branch DR. Src kinase activity is regulated by the SHP-1 protein-tyrosine phosphatase. J Biol Chem. 1997;272:21113–21119. doi: 10.1074/jbc.272.34.21113. [DOI] [PubMed] [Google Scholar]
- Somlyo AP, Somlyo AV. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatases. Physiol Rev. 2003;83:1325–1358. doi: 10.1152/physrev.00023.2003. [DOI] [PubMed] [Google Scholar]
- Spurrell BE, Murphy TV, Hill MA. Tyrosine phosphorylation modulates arteriolar tone but is not fundamental to myogenic response. Am J Physiol Heart Circ Physiol. 2000;278:H373–H382. doi: 10.1152/ajpheart.2000.278.2.H373. [DOI] [PubMed] [Google Scholar]
- Swarup G, Cohen S, Garbers DL. Inhibition of membrane phosphotyrosyl-protein phosphatase activity by vanadate. Biochem Biophys Res Commun. 1982;107:1104–1109. doi: 10.1016/0006-291x(82)90635-0. [DOI] [PubMed] [Google Scholar]
- Tice DA, Giscardi JS, Nickles AL, Parsons SJ. Mechanism of biological synergy between cellular Src and epidermal growth factor receptor. Proc Natl. Acad Sci USA. 1999;96:1415–1420. doi: 10.1073/pnas.96.4.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalba N, Kun A, Stankevicius E, Simonsen U. Role of tyrosine kinase in contraction of rat penile small arteries. J Sex Med. 2010;7:2086–2095. doi: 10.1111/j.1743-6109.2010.01788.x. [DOI] [PubMed] [Google Scholar]
- Wozniak K, Blasiak J. Vanadyl sulfate can differentially damage DNA in human lymphocytes and HeLa cells. Arch Toxicol. 2004;78:7–15. doi: 10.1007/s00204-003-0506-3. [DOI] [PubMed] [Google Scholar]
- Yu J, Akishita M, Eto M, Koizumi H, Hashimoto R, Ogawa S, et al. Src kinase-mediates androgen receptor-dependent non-genomic activation of signaling cascade leading to endothelial nitric oxide synthase. Biochem Biophys Res Commun. 2012;424:538–543. doi: 10.1016/j.bbrc.2012.06.151. [DOI] [PubMed] [Google Scholar]
- Zheng X-L, Mokashi S, Hollenberg MD. Contractile action of ethanol in guinea pig smooth muscle: inhibition by tyrosine kinase inhibitors and comparison with the contractile action of epidermal growth factor-urogastron. J Pharmacol Exp Ther. 1997;282:484–495. [PubMed] [Google Scholar]