

Abstract
Respiratory muscle testing is often limited to noninvasive volitional tests such as vital capacity and maximal static pressures. We report the case of a 12-year-old boy with congenital muscular dystrophy (CMD) in whom invasive and non-volitional respiratory muscle tests showed an elective diaphragmatic dysfunction with the preservation of expiratory muscle strength. This finding, coupled with a clinical phenotype associating diffuse muscle atrophy with finger hyperlaxity and proximal contractures, strengthened the suspicion of Ullrich CMD. Skin-cultured fibroblasts showed intracellular retention of collagen 6 (COL6), muscle magnetic resonance imaging was typical of COL6 myopathy, and molecular studies identified a COL6 gene mutation (COL6A2 c.954+2T>C). The diagnosis of a diaphragmatic dysfunction led to a sleep study that evidenced periods of hypoxemia which justified nocturnal noninvasive ventilation. This case report highlights the benefit of assessing respiratory muscles, through invasive procedure, to assist in clinical diagnosis and to guide clinical management.
Keywords: COL6-related myopathy, diaphragm, respiratory function, Ullrich congenital muscular dystrophy, vital capacity
Introduction
Respiratory muscle function is often indirectly assessed by surrogate measures such as vital capacity, maximal static pressures, and the sniff nasal inspiratory pressure. The recording of the esophageal (Pes) and gastric pressures (Pgas) allows the measurement and calculation of numerous volitional and non-volitional parameters that may be helpful to guide the diagnostic and therapeutic approach.
We report the case of a child with a congenital muscular dystrophy (CMD) in whom extensive respiratory muscle testing contributed to diagnosis orientation and clinical management.
Case Report
In January 2008, a 6.5-year-old boy who originated from India with no consanguineous parents was referred for an unclassified CMD. He presented with diffuse muscle atrophy and finger hyperlaxity. He was able to walk at 18 months old, but was never able to run, also with frequent falls, an important muscular fatigue, and the inability to climb the stairs. Frequent lung infections were also noted together with solid-feeding difficulties. Creatine kinase levels were normal. A muscular biopsy performed in 2007 evidenced dystrophic changes. Mutations of the FKRP gene were explored due to irregular alpha-dystroglycan expression, with negative results. Daily sessions of intermittent positive pressure breathing by Alpha-200™ device (Air Liquide Medical Systems, Antony, France) were started from the age of 6.5 years, but regularly done only from the age of 11.7 years. The patient used the Alpha-200™ with a frequency of 30 min/day by means of a facial mask and an abdominal belt. The inspiratory pressure was set at 30 cm H2O, the flow at 25 L/min and the trigger at −1.
At 7.5 years old, important retractions of elbows, hips, knees, and ankles were observed. No scoliosis was reported.
At 8 years old, ambulation was lost.
At 11.7 years old, arterialized capillary blood gases were performed and showed a partial carbon dioxide tension (PCO2) of 5.53 kPa (41.5 mmHg), with a pH of 7.39 and bicarbonates of 26.7. Due to hyperlaxity and proximal contractures, an Ullrich CMD was suspected. Cultured skin fibroblasts were done. Whole-body muscle magnetic resonance imaging showed a typical COL6 pattern (“tigroid” or striated, with alternating bands of normal tissue and fibroadipeous infiltration) [1]. Supine spirometry revealed a significant fall of vital capacity (VC) when compared with sitting spirometry, suggesting a diaphragmatic failure. Consequently at 12 years old, respiratory muscle function was assessed using an esogastric catheter, allowing the recording of the Pes and Pgas. Transdiaphragmatic pressure (Pdi) was obtained by subtracting Pes from Pgas. During spontaneous breathing, tidal volume was decreased with 300 mL for a patient weighing 71 kg, and respiratory rate was moderately increased at 18 breaths/min. A paradoxical breathing pattern was observed, with a ΔPgas/ΔPes of 1.1, indicating a complete diaphragmatic paralysis (Fig. 1A) [2]. The patient's inspiratory effort was assessed by the esophageal (PTPes) and diaphragmatic pressure–time products (PTPdi), showing a nearly normal value of PTPes of 142 cm H2O.s.min−1, and a dramatically reduced PTPdi of 20 cm H2O.s.min−1. Maximal inspiratory pressure during a sniff was reduced at a value of 35 cm H2O, with also a sharply reduced value of 16 cm H2O on the Pdi tracing. A negative deflection was noted on the Pgas tracing during the sniff maneuver, reflecting a diaphragmatic dysfunction (Fig. 1B). Gastric pressure during a maximal cough (Pgas cough) was moderately reduced with a value of 80 cm H2O. Endurance of the diaphragm and the global inspiratory muscles was assessed using the diaphragmatic (TTdi) and esophageal tension–time index (TTes), respectively [3]. TTes was normal with a value of 0.02, while TTdi was above the fatigue threshold (0.15) with a value of 0.22. Daytime PCO2 was 5.63 kPa (42.2 mmHg), with a pH of 7.39 and bicarbonates of 25.1.
Figure 1.
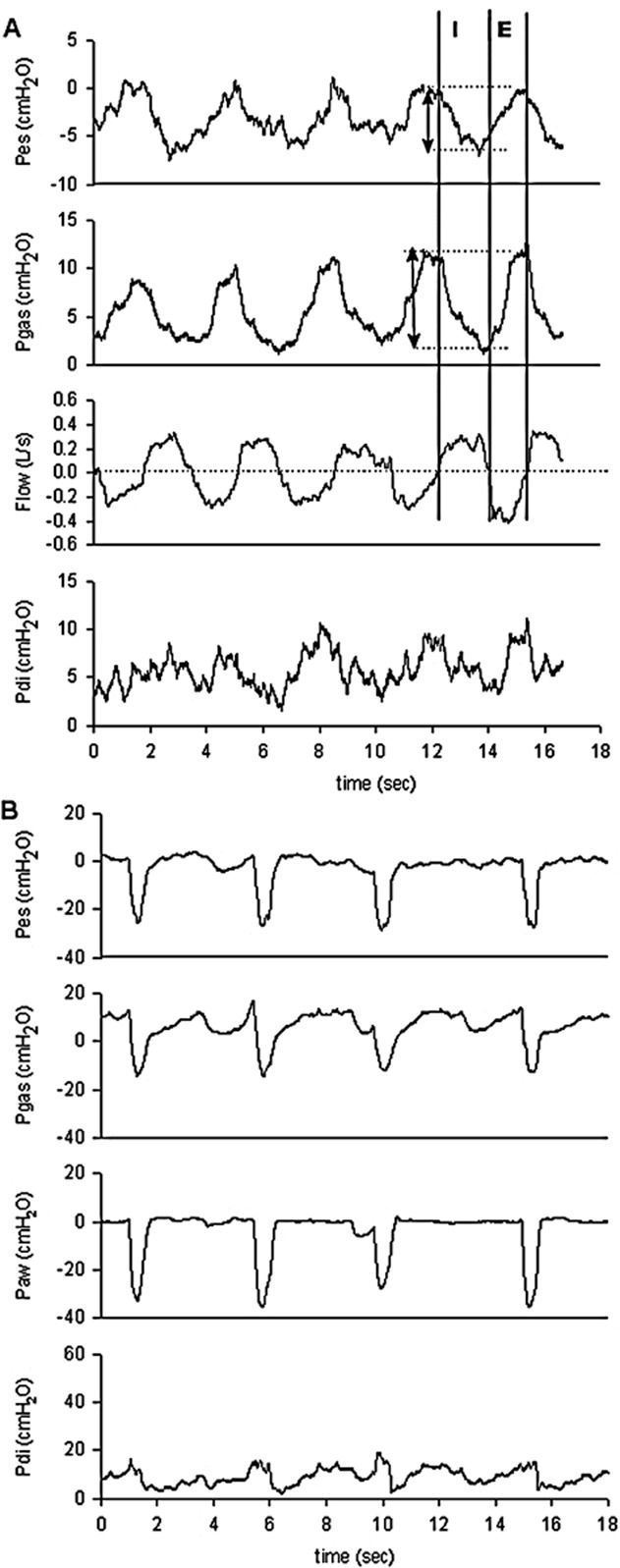
Breathing pattern (A) and sniff test (B) tracings of the subject. Note that Pes and Pgas swings were both negative, contrary to what is normally expected. Esophageal, gastric, nasal, and transdiaphragmatic sniff measures were low. Moreover, SniffPgas was negative, with an initial slight positive deflection, indicating a probable diaphragmatic dysfunction during this volitional maneuver. E, expiration; I, inspiration; Paw: nasal pressure, Pdi, transdiaphragmatic pressure; Pes, esophageal pressure; Pgas, gastric pressure; SniffPgas, gastric pressure during a sniff.
The diagnosis of an elective diaphragmatic dysfunction led to a sleep study that evidenced periods of hypoxemia, justifying the initiation of nocturnal noninvasive ventilation (NIV). A Trilogy 100™ ventilator (Philips-Respironics, Carquefou, France) was used with S/T-AVAPS mode, and optimal settings were an inspiratory airway pressure set between 22 (min) and 30 cm H2O (max), expiratory airway pressure at 4 cm H2O, tidal volume at 400 mL, AVAPS velocity at 1 cm H2O, respiratory rate at 24/min, inspiratory time at 0.8 sec, slope at 2, and an autotrack trigger.
Lung volumes continued to decline with a seated VC of 970 mL, that is 45% predicted, and a supine VC of 860 mL, that is 40% predicted, 1 month after the start of NIV. A polysomnography with NIV was done to assess NIV efficacy and eventually adjust the settings. The patient slept 405 min during which no respiratory events were observed (apnea–hypopnea index of 0 event/h). Patient–ventilator synchronization was excellent. Sleep architecture was normal with an arousal index of 1.5/h. The nocturnal gas exchanges were normal with a mean pulse oximetry of 98% and a maximal transcutaneous PCO2 of 47 mmHg. Figure 2 shows the clinical findings and VC measurements throughout the follow-up period. The very latest molecular studies identified a COL6 gene mutation: COL6A2 c.954+2T>C.
Figure 2.
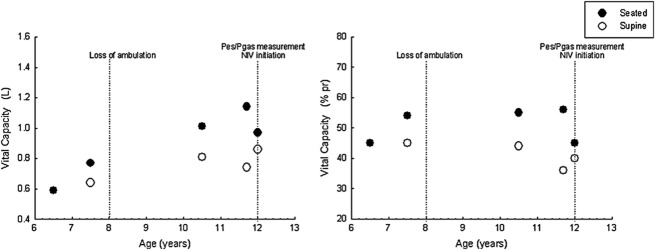
Clinical and lung function evolution. NIV, noninvasive ventilation; Pes, esophageal pressure; Pgas, gastric pressure; VC, vital capacity.
Discussion
This case is an illustration of the clinical and diagnostic interest of extensive respiratory muscle testing [4], [5]. Indeed, the respiratory muscle tests showed the presence of a particular respiratory muscle phenotype with an elective involvement of the diaphragm, observed during spontaneous breathing in the upright position and during a maximal voluntary maneuver (sniff), as noted recently in patients with COL6 myopathies [6].
In the absence of extensive respiratory muscle tests, positional VC remains an informative parameter [7], although this maneuver may be difficult to perform in young patients and reproducibility does not guarantee maximality. The measurement of other parameters, in particular non-volitional and minimally invasive, may then be of great diagnostic and therapeutic interest.
COL6-related myopathies characterized by mutations in the COL6A1-3 genes, going from Bethlem myopathy, the milder form, to Ullrich CMD, the most severe form, appear to be somewhat under-recognized until recently. This is probably due to a difficult diagnostic approach because of an overlapping presentation with other muscular disorders and a large clinical variability within COL6 myopathies. In our subject, the phenotype evoked a “moderate-progressive” form of an Ullrich CMD, which was recently confirmed by genetic studies.
In conclusion, this case report highlights the benefit of assessing extensive respiratory muscles performance, through invasive procedure, to support the diagnosis and to guide clinical management in a child presenting with CMD.
Acknowledgments
Patient and family, AFM, Banque de tissues pour la Recherche – Généthon, Céline Ledeuil and Valérie Jobic (U.F. Myogénétique et Cardiogénétique, GH Pitié-Salpêtrière, Paris, France), Corine Gartioux and Isabelle Nelson (UMRS_974), Dominique Montpoint and Nouha Allani Essid (Pediatric Department, Reference Centre for Neuromuscular Disorders, AP-HP, Hôpital Raymond Poincaré, Garches, France). The research of Brigitte Fauroux is supported by the Association Française contre les Myopathies (AFM); Vaincre la Mucoviscidose (VLM); ADEP ASSISTANCE; ASV Santé; IP Santé Domicile; Assistance Publique-Hôpitaux de Paris, Inserm; and Université Paris Descartes. The research of Susana Quijano-Roy is supported by Université de Versailles Saint-Quentin-en-Yvelines.
Disclosure Statements
No conflict of interest declared.
Appropriate written informed consent was obtained for publication of this case report and accompanying images.
References
- Quijano-Roy S, Avila-Smirnow D, Carlier RY WB-MRI muscle study group. Whole body muscle MRI protocol: pattern recognition in early onset NM disorders. Neuromuscul. Disord. 2012;22(Suppl. 2):S68–S84. doi: 10.1016/j.nmd.2012.08.003. [DOI] [PubMed] [Google Scholar]
- Lisboa C, Paré PD, Pertuzé J, et al. Inspiratory muscle function in unilateral diaphragmatic paralysis. Am. Rev. Respir. Dis. 1986;134:488–492. doi: 10.1164/arrd.1986.134.3.488. [DOI] [PubMed] [Google Scholar]
- Bellemare F, Grassino A. Effect of pressure and timing of contraction on human diaphragm fatigue. J. Appl. Physiol. 1982;53:1190–1195. doi: 10.1152/jappl.1982.53.5.1190. [DOI] [PubMed] [Google Scholar]
- Nicot F, Hart N, Forin V, et al. Respiratory muscle testing: a valuable tool for children with neuromuscular disorders. Am. J. Respir. Crit. Care Med. 2006;174:67–74. doi: 10.1164/rccm.200512-1841OC. [DOI] [PubMed] [Google Scholar]
- Fauroux B, Khirani S. Neuromuscular disease and respiratory physiology in children: putting lung function into perspective. Respirology. doi: 10.1111/resp.12330. doi:10.1111/resp.12330. [DOI] [PubMed] [Google Scholar]
- Quijano-Roy S, Khirani S, Colella M, et al. Diaphragmatic dysfunction in collagen VI myopathies Neuromuscul. Disord 2014. 2014;24:125–133. doi: 10.1016/j.nmd.2013.11.002. [DOI] [PubMed] [Google Scholar]
- Foley AR, Quijano-Roy S, Collins J, et al. Natural history of pulmonary function in collagen VI-related myopathies. Brain. 2013;136:3625–3633. doi: 10.1093/brain/awt284. [DOI] [PMC free article] [PubMed] [Google Scholar]