

Abstract
CALM is implicated in the formation of clathrin-coated vesicles, which mediate endocytosis and intracellular trafficking of growth factor receptors and nutrients. We previously found that CALM-deficient mice suffer from severe anemia due to the impaired clathrin-mediated endocytosis of transferrin receptor in immature erythroblast. However, CALM has been supposed to regulate the growth and survival of hematopoietic stem/progenitor cells. So, in this study, we focused on the function of CALM in these cells. We here show that the number of Linage−Sca-1+KIT+ (LSK) cells decreased in the fetal liver of CALM −/− mice. Also, colony forming activity was impaired in CALM−/− LSK cells. In addition, SCF, FLT3, and TPO-dependent growth was severely impaired in CALM−/− LSK cells, while they can normally proliferate in response to IL-3 and IL-6. We also examined the intracellular trafficking of KIT using CALM −/− murine embryonic fibroblasts (MEFs) engineered to express KIT. At first, we confirmed that endocytosis of SCF-bound KIT was not impaired in CALM −/− MEFs by the internalization assay. However, SCF-induced KIT trafficking from early to late endosome was severely impaired in CALM −/− MEFs. As a result, although intracellular KIT disappeared 30 min after SCF stimulation in wild-type (WT) MEFs, it was retained in CALM −/− MEFs. Furthermore, SCF-induced phosphorylation of cytosolic KIT was enhanced and prolonged in CALM −/− MEFs compared with that in WT MEFs, leading to the excessive activation of Akt. Similar hyperactivation of Akt was observed in CALM −/− KIT+ cells. These results indicate that CALM is essential for the intracellular trafficking of KIT and its normal functions. Also, our data demonstrate that KIT located in the early endosome can activate downstream molecules as a signaling endosome. Because KIT activation is involved in the pathogenesis of some malignancies, the manipulation of CALM function would be an attractive therapeutic strategy.
Introduction
Clathrin-mediated endocytosis (CME) is an active cellular process for membrane trafficking, which mediates the entry of nutrients and growth factor receptors into the cells [1]. When the target molecule binds to plasma membrane receptor, clathrin and accessory molecules such as AP2, epsin, clathrin assembly lymphoid myeloid leukemia protein (CALM) are recruited from cytoplasm into the membrane, resulting in the formation of clathrin-coated vesicles. Clathrin-coated vesicles transport target molecules from the membrane to early and late endosomes. Also, it regulates the intercellular trafficking among endosomes, trans-Golgi network (TGN), and lysosomes, thereby regulating the degradation of the target molecules [1]–[3].
Receptor tyrosine kinases (RTKs) such as KIT, epidermal growth factor receptor (EGFR) and MET play crucial roles in the development and maintenance of the cells, on which they are expressed [4]–[7]. Upon the ligand binding, activated RTKs are internalized and transferred to early endosome. Then, clathrin-coated RTKs are partially sorted back to the plasma membrane via recycling endosome (RE). The remaining RTKs are transported to the intraluminal vesicles (ILVs) of late endosomes/multivesicular body (MVB). MVB can be fused to the lysosomes, where RTKs are degraded [8]. Previously, it was believed that several signaling molecules such as MAPKs, JAK/STATs, and PI3K/AKT are activated by RTKs on the plasma membrane. However, several recent reports showed that these signaling molecules can also be activated by clathrin-coated RTKs located in the endosome, leading to the establishment of the concept “signaling endosome” [9]–[11].
CALM encodes a 652 aa protein with multiple domains such as AP180 N-terminal homology (ANTH) domain, DPF motif, NPF motif, and type I and II clathrin-binding sequences (CBS I and II), of which expression is ubiquitously observed in various organs [12]–[18]. Knockdown of CALM by RNA interference leads to the formation of larger and more irregular, clathrin-coated vesicles in HeLa cells, indicating that CALM is required for proper formation of clathrin-coated vesicles [19]. CALM was originally isolated as a part of the fusion gene CALM/AF10, which results from the chromosomal translocation t(10;11) (p13;q14) [20]. This translocation is found in acute lymphoblastic leukemia (ALL), acute myeloid leukemia (AML) and malignant lymphomas [21]. Also, it was shown that overexpression of CALM/AF10 in primary murine bone marrow (BM) cells resulted in the development of an aggressive form of leukemia in a murine BM transplantation model [22], [23]. These results suggest that CALM would play an important role in the growth and differentiation of hematopoietic cells. This hypothesis was subsequently supported by the reports that fit1 mutants, which contain nonsense point mutations in the CALM gene [24], [25]. In these mice, the number of early hematopoietic progenitor cells was severely reduced and numerous morphologic and functional defects were observed in the peripheral blood. However, detailed analysis on the hematopoietic defects in fit1 mutants has not been performed.
To clarify the physiological role of CALM in vivo, we recently generated CALM-deficient mice [26]. Although CALM +/− mice didn’t shown an apparent phenotype, CALM −/− mice exhibited retarded growth in utero and were dwarfed throughout their shortened life-spans. Moreover, CALM-deficient mice suffered from severe anemia due to the impaired CME of transferrin in immature erythroblast. Meanwhile, based on the report of fit1 mutants [24], [25], CALM has been supposed to regulate the growth and survival of hematopoietic stem/progenitor cells. So, in this study, we focused on the molecular mechanism through which CALM regulates their growth and survival. We here show that KIT-mediated growth was impaired in CALM-deficient hematopoietic stem/progenitor cells, probably due to the defect in the KIT trafficking from early to late endosomes.
Materials and Methods
Ethics Statement
This study was approved by the Committee of Animal Experiments, Kinki University Faculty of Medicine (approval ID: 06-13).
Recombinant Growth Factors and Inhibitors
Recombinant murine stem cell factor (SCF), Flt-3 ligand (FL), thrombopoietin (TPO), interleukin-3 (IL-3), and IL-6 were purchased from PeproTech. (Rocky Hill, NJ). Imatinib mesylate (STI571) purchased from Selleck (Houston, TX) was used to inhibit KIT activity and Bafilomycin A1 purchased from Sigma Aldrich (St. Louis, MO) was used to inhibit the protein transport from early to late endosome [27].
Isolation and Immortalization of Murine Embryonic Fibroblasts (MEFs)
Primary MEFs were isolated from wild-type (WT) and CALM-deficient mice on embryonic day 14.5 (E14.5) and cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 15% fetal calf serum (FCS) and 5% CO2 at 37°C. To immortalize MEFs, we transfected the expression vector for SV40 large T antigen into MEFs by Lipofectamine 2000 Reagent (Invitrogen, Life Technologies, Carlsbad, CA) according to the manufacturer’s instructions. Stable immortalized clones were obtained by serial dilution.
Retrovirus Transduction
Murine full-length Kit cDNA kindly provided from Dr. Mizuki M. (Osaka University, Osaka, Japan) was subcloned into pMSCV-IRES-EGFP bicistronic retrovirus vector. The retrovirus vector was transfected into a packaging cell line 293T containing the expression plasmids for gag and pol, which was cultured in DMEM supplemented with 10% FCS. The supernatant was collected 48 h after transfection. MEFs were plated onto the 3.5 cm dish coated with fibronectin fragment (RetroNectin, Takara Bio, Shiga, Japan) and cultured with 1 ml virus supernatant for 72 h. Retrovirus-infected MEFs were isolated as GFP-positive cells by FACS Aria (BD Biosciences, San Jose, CA).
Purification of Murine Lineage− Sca-1+ KIT+ (LSK) Cells
Murine fetal liver cells were harvested from E14.5 embryos and mononuclear cells (MNCs) were separated by density gradient centrifugation. Then, MNCs were incubated with the antibodies (Abs) as follows: anti-lineage Abs (a cocktail of biotinylated Abs against CD3e (145–2C11), CD45R/B220 (RA3–6B2), Gr-1 (RB6–8C5), and TER-119 (TER-119)), fluorescein isothiocyanate (FITC)-conjugated anti-Sca-1 Ab (D7), allophycocyanin (APC)-conjugated anti-c-KIT Ab (2B8), and streptavidin-phycoerythrin (PE)-cyanin (cy)7 (BD Biosciences). After staining, LSK cells were sorted by FACS Aria. Non-viable cells were eliminated by the staining with 7-amino-actinomycin D (Calbiochem, Merck Millipore, Darmstadt, Germany).
Clonogenic Assay
LSK cells from WT or CALM−/− mice were plated onto Complete Medium with murine Cytokines MethoCult GF M3434 (StemCell Technologies, Vancouver, BC, Canada). The numbers of colony forming unit–mixed (CFU-Mix), CFU-granulocyte macrophage (CFU-GM), and burst-forming unit–erythroid (BFU-E) were counted under the inverted microscope 14 days after plating.
Proliferation Assay
LSK cells were cultured in Roswell Park Memorial Institute-1640 (RPMI-1640) medium (Gibco, Life Technologies, Carlsbad, CA) with 10% FCS containing 100 ng/ml, SCF, 100 ng/ml FL, 100 ng/ml TPO, 100 ng/ml IL-3, 100 ng/ml IL-6 at 37°C. Number of viable LSK cells was measured by the Cell Titer Glo Reagent (Promega, Madison, WI) from the intensity of the luminescence using an Envision plate reader (1420 ARVO MX-2, Wallac, PerkinElmer, Inc., Waltham, MA).
Flow Cytometric Analysis
The expression of surface molecules was examined by FACS Aria using the appropriate Abs and these results were analyzed by BD FACS Diva software (BD Biosciences) or FlowJo software (TreeStar, Ashland, OR). To analyze cytoplasmic AKT phosphorylation in KIT+ hematopoietic cells by flow cytometry, MNCs isolated from BM were fixed with 3.7% (w/v) formaldehyde in PBS for 15 min, and permeabilized with 1% (w/v) bovine serum albumin (BSA) and 0.1% (v/v) Triton X-100 in PBS for 15 min. These cells were incubated with the Alexa647-conjugated anti-phosphorylated Akt Ab (Cell Signaling Technology, Danvers, MA) in combination with the Abs to identify KIT+ cells as described above. After staining, these cells were analyzed by FACS Aria.
Internalization Assay
Internalization assay for KIT was performed as described previously [26]. Briefly, WT and CALM−/− MEFs both engineered to express KIT were cultured with biotinylated SCF (R&D systems, Minneapolis, MN) for 60 min, and further incubated with the APC-conjugated streptavidin (Biolegend, San Diego, CA) for 30 min at 4°C. Then, these cells were incubated at 37°C up to 20 min to allow internalization. After stripping unincorporated SCF with acidic buffer (20 mM MES pH 5, 130 mM NaCl, 2 mM CaCl2 and 0.1% BSA), relative amount of internalized SCF-KIT complex was evaluated from the fluorescence intensity by FACS at the indicated times compared with the initial amount of membrane KIT.
Immunofluorescence Analysis
KIT-transfected MEFs were transferred onto the coverslips and cultured in DMEM supplemented with 15% FCS at 37°C for 48 h. After the stimulation with 100 ng/ml SCF for the indicated times, the cells were washed with ice-cold PBS and then fixed with 3.7% (w/v) formaldehyde in PBS for 15 min. After incubation in blocking buffer with 1% (w/v) bovine serum albumin (BSA) and 0.1% (v/v) Triton X-100 in PBS for 15 min, the fixed cells were incubated with the primary and then with the secondary Abs suspended in the reagents each for 45 min. The utilized Abs and reagent were as follows: biotinylated anti-KIT (2B8, Biolegend), anti-Rab5 (C8B1), anti-Rab7 (D95F2), anti-Rab11 (D4F5) Abs (Cell Signaling Technology), AlexaFluor 488-conjugated anti-rabbit IgG Ab, AlexaFluor 568 streptavidin conjugates (Invitrogen). After washing with PBS, the coverslips were mounted on glass slides using Prolong Gold antifade reagent (Invitrogen) and observed under the confocal microscopy (LSM-410, Nikon, Tokyo, Japan).
Subcellular Protein Fractionation
Subcellular protein fractionation was performed using OptiPrep density gradient medium (Axis-Shield, Oslo, Norway) following their protocol (S23) with some modification. Briefly, cell lysates were centrifuged at 1000 g for 5 min to pellet nuclei and cell debris. Supernatant was loaded on the Opti-Prep discontinuous gradient (30, 25, 20, 15, 10, 5%) and centrifuged at 90,000 g for 16 h. After centrifugation, 50 fractions were collected and subjected to immunoblot analyses.
Subcellular protein fractionation was also performed with a subcellular protein fractionation kit (Thermo scientific, Waltham, MA). Briefly, the cultured cells were incubated in DMEM supplemented with 15% FCS in the presence of 25 µg/ml cycloheximide (Cell Signaling Technology) at 37°C for 1 h, and stimulated with 100 ng/ml SCF for the indicated times. Cultured cells were washed with ice-cold PBS and centrifuged. The pellet was suspended in cytoplasmic extraction buffer at 4°C for 10 min and centrifuged at 500 g for 5 min. The supernatant was transferred and used as a cytoplasmic extract. Then, the remaining pellet was resuspended in membrane extraction buffer at 4°C for 10 min and centrifuged at 500 g for 5 min. This supernatant was used as a membrane extract.
Immunoblot and Immunoprecipitation
Immunoblot analyses were performed as described previously [28]. Briefly, the cultured cells were lysed in lysis buffer containing 1% Nonidet P-40 (NP-40) and protease inhibitors, and insoluble materials were removed by centrifugation. The whole cell lysates (15 µg per each lane) or immunoprecipitated proteins were subjected to SDS–PAGE and electrophoretically transferred onto a polyvinylidene difluoride membrane (Immobilon, Millipore, Bedford, MA). After blocking the residual binding sites on the membrane, immunoblotting was performed with an appropriate Ab. For immunoprecipitation, protein extract was incubated with the appropriate Ab and protein A-sepharose beads at 4°C for 6 h. After SDS-PAGE, protein was transferred to a nitrocellulose membrane. The membranes were incubated in TBST blocking buffer (4% nonfat dry milk in Tris-buffered saline-Tween 20, 0.15 M NaCl, 0.01 M Tris-HCl pH 7.4, 0.05% Tween 20) followed by the incubation with the primary Ab at room temperature for 1 h. The primary Abs utilized in this study were as follows: anti-CALM (G-18), and anti-actin Abs (Santa Cruz Biotech., Santa Cruz, CA), anti-phoshotyrosine (4G10), anti-KIT (D13A2), anti-phosphorylated Akt (Ser473) (587F11), anti-Akt (C67E7), anti-phosphorylated p-44/42MAPK (T202/Y204) (20G11), anti-p44/42MAPK (ERK1/2) (137F5), anti-pan-Cadherin (28E12), anti-HSP90 (E289), anti-EEA1 (C8B1), and ant-LAMP1 (C54H11) Abs (Cell Signaling Technology). Then, the membranes were incubated with the appropriate secondary Abs diluted in blocking buffer, and immunoreactive proteins were visualized by enhanced chemiluminescence (LAS4010, GE Healthcare, Cleveland, OH).
Statistical Analysis
Statistical analysis was performed with the Student t test. Error bars indicate the standard deviation (SD) of the mean. P-values less than 0.05 were considered statistically significant.
Results
Number of LSK Cells Decreases in Fetal Liver of CALM −/− Mice
To analyze the role of CALM in hematopoietic stem/progenitor cells, we isolated fetal liver from WT, CALM +/−, and CALM −/− mice on E14.5. Fetal liver from CALM −/− mice was macroscopically small compared with that from WT and CALM +/− mice. In accord with this finding, total number of hematopoietic MNCs significantly decreased in the fetal liver of CALM −/− mice compared with that in WT and CALM +/− mice (Fig. 1A, left). However, the proportions of LSK cells in MNCs were almost same regardless of their genotypes (Fig. 1A, middle). Thus, total number of LSK cells in the fetal liver of CALM −/− mice was about 50% compared with that in WT and CALM +/− mice. (Fig. 1A, right).
Figure 1. Hematopoietic stem/progenitor cells decreases in the fetal liver of CALM −/− mice and their colony forming activities are impaired.

(A) Fetal liver cells were harvested from wild-type (WT) (indicated +/+), CALM +/− (indicated +/−), and CALM −/− (indicated −/−) mice on embryonic day 14.5 (E14.5). Numbers of mononuclear cells (MNCs) in each fetal liver were calculated by cell counter. The proportion of Linage−Sca-1+KIT+ (LSK) cells in MNCs was determined by flow cytometry. Data represent means ± SD (n = 6, *p<0.05). (B) LSK cells isolated from fetal liver of WT, CALM +/−, and CALM −/− mice on E14.5 were subjected to colony assays. All cultures were conducted in triplicate and number of colonies were counted after 14 days. Results are shown as mean ± SD (n = 6, *p<0.05).
Colony Forming Activity and Cytokine-dependent Growth Are Impaired in CALM−/− LSK Cells
We also performed colony assays by plating 1,000 LSK cells into the semisolid medium each containing the cytokine cocktail appropriate for the development of CFU-Mix, CFU-GM, and BFU-E. As shown in Fig. 1B, no apparent difference was observed in clonogenic activity between WT and CALM +/− LSK cells. In contrast, CALM −/− LSK cells yielded significantly less numbers of CFU-GM, BFU-E, and CFF-Mix than WT and CALM +/− LSK cells, indicating that colony forming activities were impaired in CALM −/− LSK cells. In addition, we found that the size of CFU (especially of CFU-Mix) formed from CALM−/− LSK cells was apparently smaller than that from WT and CALM +/− cells (Fig. S1). These findings indicate that CALM −/− LSK cells would have less activity to proliferate in response to cytokines.
To confirm this hypothesis, we isolated LSK cells from fetal liver of WT, CALM +/−, and CALM −/− mice and cultured them with SCF, FL, and TPO. As shown in Fig. 2A, the growth of CALM −/− LSK cells was apparently impaired compared with that of WT and CALM +/− LSKs. However, when IL-3 and IL-6 were added into this culture medium, the growth of CALM −/− LSK cells was not completely but partially recovered compared with that of WT and CALM +/− LSK cells (Fig. 2B). On the other hand, WT, CALM +/− and CALM / LSK cells show similar growth responses to IL-3 and IL-6 (Fig. 2C). These results indicate that CALM plays a crucial role in the transmission of growth signal from SCF, FL, and/or TPO but not from IL-3 or IL-6 whereas its haploinsufficiency doesn’t cause a clear defect.
Figure 2. SCF, FL and TPO-dependent growth is impaired in CALM −/− LSK cells.
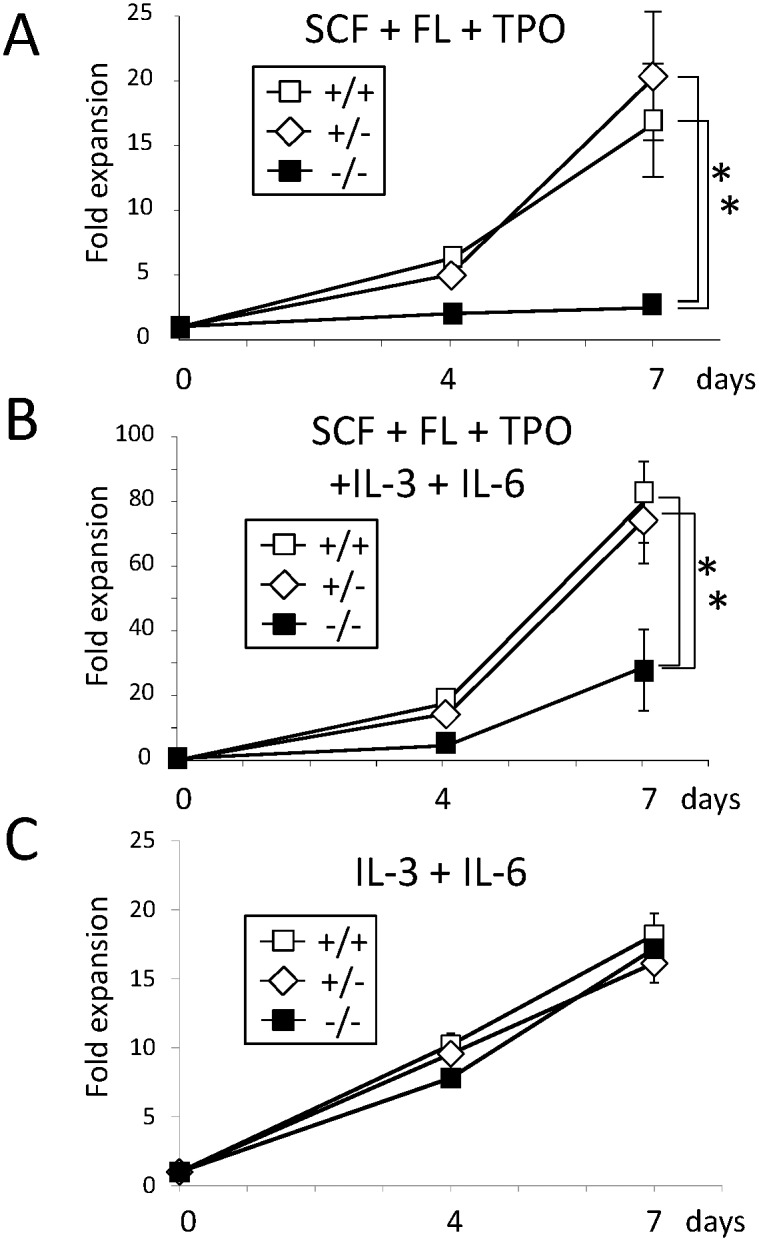
(A) (B) (C) LSK cells isolated from fetal liver of WT, CALM +/−, and CALM −/− mice were cultured in αMEM supplemented with 10% FBS in the presence of the indicated cytokines (100 ng/ml each). Total viable cell number was evaluated by the Cell Titer Glo Reagent (Promega) Data are shown as the mean ± SD of three cultures (n = 3, *p<0.05). SCF, stem cell factor; FL, Flt-3 ligand; TPO, thrombopoetin.
Endocytosis of SCF-bound KIT Is Not Impaired in CALM −/− MEFs
We next focused on the KIT signaling, because CALM has been reported to be involved in the internalization and/or intracellular transport of RTKs [29]. As cytoplasmic area of immature hematopoietic cells was rather small and not suitable for immunofluorescence analysis, we utilized MEFs from WT and CALM −/− mice, both of which were engineered to express KIT by the retrovirus infection.
At first, we confirmed that KIT was expressed at a similar level on the cell surface of WT and CALM −/− MEFs by measuring EGFP, which was expressed together with KIT from a single RNA by the bicistronic promoter of the retrovirus vector (data not shown).
Next, we examined the internalization of KIT in WT and CALM −/− MEFs by incubating them with APC-labeled SCF at 37°C up to 60 min. After stripping unincorporated SCF, we quantified the amount of the internalized SCF from the intensity of fluorescence by FACS. As shown in Figure 3, after treatment with APC-conjugated SCF, the mount of SCF-KIT complex in the cytoplasm peaked at 20 min in both WT and CALM−/− MEFs, indicating that the internalization of KIT wouldn’t be impaired by CALM deficiency. Then, the level of SCF-KIT complex decreased to the near basal level at 30 min and remained low up to 60 min in WT MEFs, suggesting that the internalized KIT was degraded and/or recycled in WT MEFs. On the other hand, it remained high up to 60 min in CALM −/− MEFs, implying that the intracellular trafficking of internalized KIT in CALM −/− MEFs would be rather different from that in WT MEFs.
Figure 3. Endocytosis of SCF-bounded KIT isn’t affected by CALM deficiency.

(A) WT and CALM−/− murine embryonic fibroblasts (MEFs) engineered to express KIT were incubated with biotinylated SCF for 60 min, and then with the APC-conjugated streptavidin for 30 min at 4°C. After stripping unincorporated SCF, the amount of the internalized SCF-KIT complex was quantified from the fluorescence intensity at the indicated times. (B) Uptake of SCF in WT or CALM −/− MEFs. The vertical axis indicates the ratio of mean fluorescence intensity, MFI (internalized SCF-KIT complex/initial surface KIT) (Data represent means ± SD, n = 3, n.s.: not significant (p = 0.079)).
SCF-Induced KIT Trafficking from Early to Late Endosomes Is Impaired in CALM −/− MEFs
Next, we analyzed intracellular KIT transport in WT and CALM −/− MEFs by immunofluorescence analysis. KIT was detected on cell surface prior to SCF stimulation and internalized into cytoplasm 5 min after SCF stimulation in both WT and CALM −/− MEFs (Fig. 4, upper and lower panels, time 0 and 5). Then, the internalized KIT adhered to the intracellular compartments surrounding nucleus, and subsequently formed a numerous punctate pattern in the cytoplasm in WT MEFs. Finally, the majority (about 80%) of KIT disappeared at 30 min in WT MEFs (Fig. 4, upper panel). In contrast, KIT was still easily detectable with about 80% of the basal level in CALM −/− MEFs at 30 min (Fig. 4, lower panel), indicating that the clearance of the internalized KIT from cytoplasm was impaired in CALM −/− MEFs.
Figure 4. Localization of KIT after SCF stimulation is altered in CALM −/− MEFs.

Localization of KIT was analyzed before and after SCF stimulation under confocal microscopy using WT and CALM −/− MEFs engineered to express KIT. KIT was visualized by the biotinylated anti-KIT antibody (Ab) and AlexaFluor 568 streptavidin conjugates.
To analyze cytoplasmic localization of KIT in more detail, we utilized Rab5, Rab7, and Rab11 as a marker of early, late, and recycling endosome, respectively. At first, we confirmed that a significant proportion of CALM was colocalized with Rab5 at 5 min and with Rab7 at 30 min after SCF stimulation (Fig. S2). In accord with the localization of CALM, a substantial proportion of KIT was colocalized with Rab5 in both WT and CALM −/− MEFs 5 min after SCF stimulation (Fig. 5A), indicating that the transport of KIT to the early endosome isn’t disrupted in CALM −/− MEFs. Although only a faint signal of KIT was detectable in WT MEFs after 30-min SCF stimulation (about 20% of the basal level as shown in Fig. 4), the remaining KIT was colocalized with Rab7 in WT MEFs (Fig. 5B, upper panel). In contrast, KIT was scarcely colocalized with Rab7 in CALM −/− MEFs (Fig. 5B, lower panel). As for this reason, we found that KIT still remained in the early endosome in CALM −/− MEFs at this point (Fig. 5C, lower panel). On the other hand, CALM was scarcely colocalized with Rab11 in WT MEFs after SCF-treatment up to 30 min (the lowest panel in Fig. S2). Similarly, KIT wasn’t colocalized with Rab11 neither in WT nor in CALM −/− MEFs (Fig. S3). These results suggest that the internalized KIT isn’t recycled. These findings are in agreement with the previous report by Shimizu Y et al. [30], which indicated that new protein synthesis of KIT is required for its cell surface reappearance after SCF stimulation.
Figure 5. Intracellular trafficking of KIT from early to late endosome is impaired in CALM−/− MEFs.

(A) (B) (C) After SCF stimulation, intracellular localization of KIT was followed by immunofluorescence analyses with the anti-KIT Ab at the indicated times. Rab5 and Rab7 were used as markers of early and late endosome, respectively. Arrows indicate regions of colocalization (Inset shows region of higher magnification).
To quantitatively demonstrate the difference in the intracellular localization of KIT between WT and CALM −/− MEFs after SCF treatment, we performed cell fractionation with OptiPrep density gradient. In this experiment, EEA1 was used as a marker of early endosome and LAMP1 as a marker of late endosome and lysozome, because these markers are more sensitive to discriminate each organella than Rab5 and Rab7 in this method. Treatment with SCF for 15 min led to the formation of a peak of KIT within late endosome to lysosome fractions (fractions 13–23) that are enriched in LAMP1 protein (Fig. 6). In contrast, although a small amount of KIT was detected in LAMP1-positive fractions (fractions 15–21), the majority of KIT was detected in EEA1-positive fractions (fractions 5–15) in CALM −/− MEFs. These results again indicate that SCF-induced KIT trafficking from early to late endosome was impaired in CALM −/− MEFs.
Figure 6. SCF-induced KIT trafficking from early to late endosome is impaired in CALM−/− MEFs.

Whole cell lysates were prepared from WT and CALM−/− MEFs after 15-min SCF stimulation. These lysates were fractionated with OptiPrep density gradient as described in Materials and Methods. Subcellualr localization of KIT was analyzed by immunoblot analysis with the anti-KIT Ab. EEA1 was utilized as an early endosome marker, and LAMP1 as a late endosome to lysosome marker.
SCF-induced Phosphorylation of Cytosolic KIT and Akt Is Enhanced and Prolonged in CALM −/− MEFs
We next examined whether KIT signaling was altered in CALM −/− MEFs because of the impaired trafficking from early to late endosome. For this purpose, we isolated membrane fraction (plasma, mitochondria and ER-Golgi membranes) and cytosolic fraction (containing endosomes) separately, of which separation was confirmed by the blotting with the Abs against pan-cadherin (reactive to only membrane fraction) and HSP90 (reactive to only cytosolic fraction). These lysates were subjected to immunoprecipitation and immunoblot analyses using the Abs indicated in Fig. 7. As shown in Fig. 7, 4th panel, both a mature (fully glycosylated, plasma membrane-bound, and functional) form and an immature (not fully glycosylated, membrane-unbound, and nonfunctional) form of KIT were detected in the membrane fraction with molecular weight 145 kDa and 120 kDa, respectively, of which levels were almost the same between WT and CALM −/− MEFs before SCF treatment. After 30-min SCF stimulation, the amount of mature KIT similarly decreased in both WT MEFs and CALM −/− MEFs (% decrease 17% and 13% by densitometric analysis, p = 0.67 (n = 3) (data not shown)). In addition, membrane KIT revealed similar phosphorylation pattern after SCF stimulation in both WT and CALM −/− MEFs (3rd panel). In contrast, although cytosolic KIT almost disappeared after 5-min SCF stimulation in WT MEFs, it was detected in CALM −/− MEFs (8th panel). This result seems to be inconsistent with our previous result obtained by immunofluoresence analysis indicating that KIT was still detectable in cytoplasm 5 min after SCF stimulation in WT MEFs (shown in Fig. 4, upper panel). As for this reason, we speculate that a substantial proportion of cytosolic KIT might be extracted as membrane protein because this fraction includes ER-Golgi membranes. Nonetheless, SCF-induced phosphorylation of cytosolic KIT (which is mainly located in early endosomes, Fig. 5A, 5B) was enhanced 5 min after SCF stimulation and prolonged up to 30 min in CALM −/− MEFs compared with that in WT MEFs (7th panel). In accord with this finding, SCF-induced phosphorylation of Akt was enhanced and prolonged in CALM −/− MEFs compared with that in WT MEFs (9th panel). In contrast, an apparent difference was not observed in ERK1/2 phosphorylation between these cells (11th panel). To confirm that the enhanced and prolonged phsphorylation of Akt observed in CALM −/− MEFs was dependent on KIT activity, we analyzed the effect of a KIT inhibitor, imatinib, on the activity of Akt after SCF stimulation in CALM −/− MEFs. As a result, we found that imatinib pretreatment inhibited both SCF-induced phosphorylations of KIT and Akt, implying that SCF-induced Akt phosphorylation was dependent on KIT activity (Fig. S4A). Furthermore, we confirmed that Bafilomycin A1, which inhibits protein transport from early to late endosome, enhanced and prolonged phosphorylation of KIT and Akt in SCF-stimulated WT MEFs as seen in CALM −/− MEFs (Fig. S4B). Taken together, these results indicate that KIT located in the early endosome can activate one of downstream molecules, Akt.
Figure 7. Diminution of cytosolic KIT after SCF stimulation is impaired in CALM −/− MEFs, leading to the enhanced and prolonged activation of KIT and Akt.

WT and CALM −/− MEFs were stimulated with SCF and cellular lysates were isolated at the indicated times. Then, cytosolic and membrane fractions were separated, of which separation was confirmed by blotting with the Abs against pan-cadherin (reactive to only membrane fraction) and HSP90 (reactive to only cytosolic fraction). To examine the amounts and phosphorylation status of KIT, iimmunoprecipitated KIT proteins were subjected to immunoblot analyses with the indicated Abs. Phosphorylations of AKT and ERK1/2 were analyzed by immunoblotting using whole cell lysates.
Next, we analyzed whether KIT signaling was also altered in CALM −/− hematopoietic stem/progenitor cells. For this purpose, we isolated fetal liver cells from E14.5 WT and CALM −/− mice. As shown in Fig. 8A, KIT+ fraction increased in the total fetal liver cells from CALM −/− mice compared with that from WT mice (38.1% vs. 23.8%). We stimulated each fetal liver cells with SCF for the indicated times. Because cell components were different between WT and CALM−/− KIT+ fraction, that is, the erythroid progenitor fraction was larger in CALM−/− KIT+ fraction due to anemia than in WT KIT+ fraction (36.1% vs. 17.6%). So, we analyzed Akt phosphorylation in KIT+CD71highTer119low erythroid progenitor (Region I: Ery-P) fraction and KIT+CD71dimTer119low non-erythroid progenitor (Region II: non-Ery-P) fraction separately by gating with these markers. As shown in Fig. 8A and 8B, SCF induced excessive Akt phosphorylation to the similar extent in both erythroid and non- erythroid progenitors. This result indicates that the enhanced and prolonged Akt phosphorylation observed in CALM−/− KIT+ fraction was not due to different cell components. Together, these results indicate that KIT signaling was altered in CALM −/− primary hematopoietic cells as well as in MEFs.
Figure 8. SCF-induced phosphorylation of Akt is enhanced and sustained in primary CALM −/− hematopoietic cells.

(A) KIT+ fractions were compared between WT and CALM −/− fetal liver cells by flow cytometry. After the cultures without growth factors for 16 h, cells were stimulated with 100 ng/ml SCF for the indicated times. After permeabilization, Akt phosphorylation was analyzed with the anti-phospho-Akt (Ser-473) Ab by gating KIT+CD71highTer119low erythroid progenitor (Ery-P) fraction (Region I) and KIT+CD71dimTer119low non-erythroid progenitor (non-Ery-P) fraction (Region II). Unstimulated samples appear as a broken line in the FACS plots. (B) Mean fluorescence intensity (MFI) of phosho-AKT was quantified with an analytical tool, and the results are shown as the mean±SD (n = 3, *p<0.05).
Discussion
Roles of CALM in Endocytosis and Intracellular Transport of KIT
Although cell surface interaction between SCF and KIT has been extensively studied [31]–[33], the precise mechanisms of their endocytosis and intracellular transport have not been clarified. Meanwhile, CME has been reported to be involved in the endocytosis and/or intracellular transport of some receptors such as epidermal growth factors (EGFs) [29], [34], vascular endothelial growth factor (VEGF) [35], [36] and Notch [37], [38]. So, in this study, we analyzed the roles of CALM in the endocytosis and transport of KIT. As a result, we found that CALM plays a critical role for the intracellular transport of KIT from early to late endosome, while neither endocytosis nor transport to the early endosome was affected by CALM deficiency. However, CALM has been shown to regulate proper formation of clathrin-coated vesicles in the process of clathrin-mediated endocytosis (CME), which is the major pathway of the internalization of ligand-bound RTKs, such as KIT and EGFR. So, our result that the internalization of KIT was not impaired by CALM deficiency seems to be strange. However, in agreement with our result, Kon S. et al. recently reported that the internalization of KIT was not impaired in the cells deficient for SMAP1, a member of the small GTPase Arf family, which is implicated in the formation of clathrin-coated vesicles as well as CALM [39]. So, we speculate that even if clathrin-coated vesicles might not be appropriately formed due to the deficiency of CALM or SMAP1, these deformed clathrin-coated vesicles could still work at the step of endocytosis of RTKs.
In addition, we found that the growth of CALM −/− LSK cells was severely impaired under the culture with SCF, FL, and TPO. In the current study, we focused on the KIT transport and its signaling. However, in our preliminary data, the transport of FLT3, which also belongs to the same class III receptor tyrosine kinase family, is also impaired in CALM −/− MEFs as was the case with KIT (data not shown). So, deregulated FLT3 signal may also be responsible for the impaired growth of CALM −/− LSK cells.
Activating mutations of KIT (KITV816F) and FLT3 (FLT3 internal tandem duplication), which are observed in about 10% and 8% of AML cases, respectively [40]–[42], have been considered to cause AML. Also, AML cases harboring these mutations are known to have poor prognosis [43]–[46]. Based on our data that CALM plays a critical role for the signaling from KIT and FLT3, it was expected that the manipulation of the CALM function would be an attractive new strategy to treat AML cases with such mutations.
Activation of Downstream Molecules by KIT-containing Early Endosomes
Several previous papers demonstrated that PI3K/Akt signaling is confined to the plasma membrane and quickly lost once RTKs are internalized. In contrast, recent studies showed that some cytokine receptors and adaptor proteins located in endosomes can transduce their signals to downstream molecules, proposing the concept of “signaling endosome” [9]–[11]. In addition, it was also reported that oncogenic forms of Met and EGFR are mislocalized in cytoplasm, where they transmit aberrant signals to the downstream molecules. As for KIT, its recruitment to lipid rafts was shown to be necessary to activate PI3K/Akt [47]. However, it was also shown that WT and mutant KIT activate different sets of signaling pathways including Akt and MAPK due to their distinct intracellular localization. In addition, Xiang Z et al. reported that Golgi-localized oncogenic KIT can activate downstream molecules such as Akt, ERK, and STAT3 [48]. In this study, we found that SCF-induced phosphorylation of KIT in early endosomes was enhanced and prolonged in CALM −/− MEFs compared with that in WT MEFs, leading to the excessive activation of Akt. Similar results were also observed in CALM −/− KIT+ hematopoietic cells. These results indicate that KIT-containing early endosomes can indeed transit KIT signals to the downstream molecules as a signaling endosome. A very recent study showed that Bafilomycin A1, which inhibits protein transport from early to late endosome, enhanced FGF (fibroblast growth factor)-induced activation of ERK1/2 in HEK293 cells [27]. However, in contrast to Akt, we didn’t observe the enhanced phosphprylation of ERK1/2 (Fig. 7). So, it was speculated that the signaling pattern from early endosome would be somewhat different among RTKs.
Significance of Enhanced and Prolonged Activation of Akt in CALM −/− LSK Cells
Many of the previous studies provided evidence that PI3K/Akt signaling is essential or important for the growth and survival in various cell types including hematopoietic cells [49]–[51]. In addition, constitutive activation of PI3K/Akt has been reported in AML cells, which is considered to participate in leukemogenesis and to be a good therapeutic target [52]–[54]. In contrast to these reports, in the current study, the impaired cytokine-dependent growth of CALM −/− LSK cells was accompanied by the enhanced and prolonged activation of Akt, suggesting that PI3K/Akt might be a negative regulator of cytokine-dependent growth and survival of hematopoietic cells. However, our result is supported by the fact that most of AML cells with Akt activity in the peripheral blood are in G1 phase of cell cycle [55]–[57], suggesting that constitutive activation of PI3K/Akt isn’t necessarily linked with cell growth. In addition, Akt activation was reported to induce premature senescence and sensitizes cells to ROS (reactive oxygen species)-mediated apoptosis [58]. Furthermore, decreased Akt phosphorylation is observed in a specific type of AML cells, in which the restoration of Akt activity promoted differentiation and disappearance [57]. Together, these results indicate that Akt can act as a both positive and negative regulator of cell growth and survival. To examine the relationship between the enhanced Akt activity and impaired growth of CALM−/− hematopoietic cells, we inhibited PI3K/Akt activity by its inhibitors, LY 294002 and API-2 in CALM−/− hematopoietic cells. In this experiment, we found that complete ablation of Akt activity by its inhibitor didn’t restore the growth but induced apoptosis in CALM−/− hematopoietic cells (data not shown). These results suggest that Akt activity is essential for cell survival in hematopoietic stem/progenitor cells but appropriately regulated PI3K/Akt activity would be required for their full growth and survival.
Supporting Information
Picture of CFU formed from WT, CALM+/− , and CALM−/− fetal liver LSKs. LSK cells isolated from fetal liver of WT, CALM +/−, and CALM −/− mice on E14.5 were subjected to clonogenic assays. The size of colonies was observed under the fluorescence microscopy (BZ-X700, Keyence, Osaka, Japan). The representative results were shown. Mix, CFU-Mix; GM, CFU-GM; E, BFU-E. (+/+), (+/−), and (−/−) represent the origin of LSK cells: WT, CALM +/−, and CALM −/− mice.
(TIFF)
Distribution of CALM after SCF stimulation in WT MEFs. Distribution of CALM was followed at the indicated times after SCF stimulation by immunofluorescence analyses using the anti-CALM Ab. Rab5, Rab7, Rab11 were used as markers of early, late, and recycling endosomes, respectively. Arrows indicate colocalization (Inset shows region of higher magnification).
(TIFF)
Colocalization of KIT and Rab11, a marker of the recycling endosome. KIT and Rab11 were costained with anti-KIT and anti-Rab11 Abs and analyzed by confocal microscopy.
(TIFF)
Activation of downstream molecules by KIT localized at early endosomes. (A) CALM−/− MEFs were preincubated with 5 µM imatinib or vehicle before the treatment with SCF for 6 h. After SCF-stimulation, the amounts and phosphorylation status of KIT were analyzed at the indicated time points using immunoprecipitated cell lysates. Also, whole cell lysates were subjected to immunoblot analyses using anti-Akt and anti-phosho-Akt Abs. (B) WT MEFs were incubated with 1 µM Bafilomycin A1 or vehicle during the treatment with SCF and whole cell lysates were subjected to the same experiment as Fig. 7A.
(TIFF)
Acknowledgments
We would like to thank to Drs M Mizuki, J Fujita (Osaka University) for providing DNA constructs. The technical help of Mrs. K Furukawa, K Niki (Kinki University Faculty of Medicine) is acknowledged.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by Grants-in-aid for Scientific Research (C) (25461439) and Grant-in-Aid for Scientific Research on Innovative Areas (25130712) from the Japan Society for the Promotion of Science. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. (http://kaken.nii.ac.jp/).
References
- 1. Schmid EM, McMaho HT (2007) Integrating molecular and network biology to decode endocytosis. Nature 448: 883–888. [DOI] [PubMed] [Google Scholar]
- 2. McMahon HT, Boucrot E (2011) Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat Rev Mol Cell Biol 12: 517–533. [DOI] [PubMed] [Google Scholar]
- 3. Godlee C, Kaksonen M (2013) From uncertain beginnings: initiation mechanisms of clathrin-mediated endocytosis. J Cell Biol 203: 717–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Casaletto JB, McClatchey AI (2012) Spatial regulation of receptor tyrosine kinases in development and cancer. Nat Rev Cancer 12: 387–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Matsumura I, Mizuki M, Kanakura Y (2008) Roles for deregulated receptor tyrosine kinases and their downstream signaling molecules in hematologic malignancies. Cancer Sci 99: 479–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Higashiyama S, Iwabuki H, Morimoto C, Hieda M, Inoue H, et al. (2008) Membrane-anchored growth factors, the epidermal growth factor family: beyond receptor ligands. Cancer Sci 99: 214–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Graveel CR, Tolbert D, Vande Woude GF (2013) MET: a critical player in tumorigenesis and therapeutic target. Cold Spring Harb Perspect Biol 5: a009209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Goh LK, Sorkin A (2013) Endocytosis of receptor tyrosine kinases. Cold Spring Harb Perspect Biol 5: a017459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dobrowolski R, De Robertis EM (2011) Endocytic control of growth factor signaling: multivesicular bodies as signalling organelles. Nat Rev Mol Cell Biol 13: 53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Platta HW, Stenmark H (2011) Endocytosis and signaling. Current Opinion in Cell Biology 23: 393–03. [DOI] [PubMed] [Google Scholar]
- 11. Miaczynska M (2013) Effects of membrane trafficking on signalling by receptor tyrosine kinases. Cold Spring Harb Perspect Biol 5: a009035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tebar F, Bohlander SK, Sorkin A (1999) Clathrin assembly lymphoid myeloid leukemia (CALM) protein: localization in endocytic-coated pits, interactions with clathrin, and the impact of overexpression on clathrin-mediated traffic. Mol Biol Cell 10: 2687–2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ford MG, Pearse BM, Higgins MK, Vallis Y, Owen DJ, et al. (2001) Simultaneous binding of PtdIns(4,5)P2 and clathrin by AP180 in the nucleation of clathrin lattices on membranes. Science 291: 1051–1055. [DOI] [PubMed] [Google Scholar]
- 14. Gillooly DJ, Stenmark H (2001) A lipid oils the endocytosis machine. Science 291: 993–994. [DOI] [PubMed] [Google Scholar]
- 15. Ford MG, Mills IG, B. Peter BJ, Vallis Y, Praefcke GJ, et al. (2002) Curvature of clathrin-coated pits driven by epsin. Nature 419: 361–366. [DOI] [PubMed] [Google Scholar]
- 16. Hom RA, Vora M, Regner M, Subach OM, Verkhusha VV, et al. (2007) pH-dependent binding of the Epsin ENTH domain and the AP180 ANTH domain to PI(4,5)P2-containing bilayers. J Mol Biol 373: 412–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Legendre-Guillemin V, Wasiak S, Hussain NK, Angers A, McPherson PS (2004) ENTH/ANTH proteins and clathrin-mediated membrane budding. J Cell Sci 117: 9–18. [DOI] [PubMed] [Google Scholar]
- 18. Miller SE, et al. (2011) The molecular basis for the endocytosis of small R-SNAREs by the clathrin adaptor CALM. Cell 147: 1118–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Meyerholz A, Hinrichsen L, Groos S, Esk PC, Brandes G, et al. (2005) Effect of clathrin assembly lymphoid myeloid leukemia protein depletion on clathrin coat formation. Traffic 6: 1225–1234. [DOI] [PubMed] [Google Scholar]
- 20. Dreyling MH, Martinez-Climent JA, Zheng M, Mao J, Rowley JD, et al. (1996) The t(10;11) (p13;q14) in the U937 cell line results in the fusion of the AF10 gene and CALM, encoding a new member of the AP-3 clathrin assembly protein family. Proc Natl Acad Sci USA 93: 4804–4809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Caudell D, Aplan PD (2008) The role of CALM-AF10 gene fusion in acute leukemia. Leukemia 22: 678–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Deshpande AJ, Cusan M, Rawat VP, Reuter H, Krause A, et al. (2006) Acute myeloid leukemia is propagated by a leukemic stem cell with lymphoid characteristics in a mouse model of CALM/AF10-positive leukemia. Cancer Cell 10: 363–374. [DOI] [PubMed] [Google Scholar]
- 23. Okada Y, Jiang Q, Lemieux M, Jeannotte L, Su L, et al. (2006) Leukaemic transformation by CALM-AF10 involves upregulation of Hoxa5 by hDOT1L. Nat Cell Biol 8: 1017–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Potter MD, Shinpock SG, Popp RA, Godfrey V, Carpenter DA, et al. (1997) Mutations in the murine fitness 1 gene result in defective hematopoiesis. Blood 90: 1850–1857. [PubMed] [Google Scholar]
- 25. Klebig ML, Wall MD, Potter MD, Rowe EL, Carpenter DA, et al. (2003) Mutations in the clathrin-assembly gene Picalm are responsible for the hematopoietic and iron metabolism abnormalities in fit1 mice. Proc Natl Acad Sci U S A 100: 8360–8365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Suzuki M, Tanaka H, Tanimura A, Tanabe K, Oe N, et al. (2012) The clathrin assembly protein PICALM is required for erythroid maturation and transferrin internalization in mice. PLoS One 7: e31854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rengarajan C, Matzke A, Reiner L, Orian-Rousseau V, Scholpp S (2014) Endocytosis of Fgf8 is a double-stage process and regulates spreading and signaling. PLoS One 9: e86373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tanaka H, Matsumura I, Ezoe S, Satoh Y, Sakamaki T, et al. (2002) E2F1 and c-Myc potentiate apoptosis through inhibition of NF-kappaB activity that facilitates MnSOD-mediated ROS elimination. Mol Cell 9: 1017–1029. [DOI] [PubMed] [Google Scholar]
- 29. Huang F, Khvorova A, Marshall W, Sorkin A (2004) Analysis of clathrin-mediated endocytosis of epidermal growth factor receptor by RNA interference. J Biol Chem 279: 16657–16661. [DOI] [PubMed] [Google Scholar]
- 30. Shimizu Y, Ashman LK, Du Z, Scwartz LB (1996) Internalization of Kit Together with Stem Cell Factor on Human Fetal liver-Derived Mast Cells. J. Immunol 156: 3443–3449. [PubMed] [Google Scholar]
- 31. Gommerman JL, Rottapel R, Berger SA (1997) Phosphatidylinositol 3-kinase and Ca2+ influx dependence for ligand-stimulated internalization of the c-Kit receptor. J Biol Chem 272: 30519–30525. [DOI] [PubMed] [Google Scholar]
- 32. Jahn T, Seipel P, Coutinho S, Urschel S, Schwarz K, et al. (2002) Analysing c-kit internalization using a functional c-kit-EGFP chimera containing the fluorochrome within the extracellular domain Oncogene. 21: 4508–4520. [DOI] [PubMed] [Google Scholar]
- 33. Sun J, Pedersen M, Bengtsson S, Rönnstrand L (2007) Grb2 mediates negative regulation of stem cell factor receptor/c-Kit signaling by recruitment of Cbl. Exp Cell Res 313: 3935–3942. [DOI] [PubMed] [Google Scholar]
- 34. Kazazic M, Bertelsen W, Pedersen KW, Vuong TT, Grandal MV, et al. (2009) Epsin 1 is involved in recruitment of ubiquitinated EGF receptors into clathrincoated pits. Traffic 10: 235–245. [DOI] [PubMed] [Google Scholar]
- 35. Lampugnani MG, Orsenigo F, Gagliani MC, Tacchetti C, Dejana E (2006) Vascular endothelial cadherin controls VEGFR-2 internalization and signaling from intracellular compartments. J Cell Biol 174: 593–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nakayama M, Nakayama A, van Lessen M, Yamamoto H, Hoffmann S, et al. (2013) Spatial regulation of VEGF receptor endocytosis in angiogenesis. Nat Cell Biol 15: 249–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Windler SL, Bilder D (2010) Endocytic internalization routes required for delta/notch signalling Curr Biol. 20: 538–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kandachar V, Roegiers F (2012) Endocytosis and control of Notch signaling. Curr Opin Cell Biol 24: 534–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kon S, Minegishi N, Tanabe K, Watanabe T, Funaki T, et al. (2013) Smap1 deficiency perturbs receptor trafficking and predisposes mice to myelodysplasia. J Clin Invest. 123: 1123–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang YY, Zhao LJ, Wu CF, Liu P, Shi L, et al. (2011) C-KIT mutation cooperates with full-length AML1-ETO to induce acute myeloid leukemia in mice. Proc Natl Acad Sci U S A 108: 2450–2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhao L, Melenhorst JJ, Alemu L, Kirby M, Anderson S, et al. (2012) KIT with D816 mutations cooperates with CBFB-MYH11 for leukemogenesis in mice. Blood 119: 1511–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kiyoi H, Naoe T (2006) Biology, clinical relevance, and molecularly targeted therapy in acute leukemia with FLT3 mutation. Int J Hematol. 83: 301–308. [DOI] [PubMed] [Google Scholar]
- 43. Schnittger S, Kohl TM, Haferlach T, Kern W, Hiddemann W, et al. (2006) KIT-D816 mutations in AML1-ETO-positive AML are associated with impaired event-free and overall survival. Blood 107: 1791–1799. [DOI] [PubMed] [Google Scholar]
- 44. Paschka P (2008) Core binding factor acute myeloid leukemia. Semin Oncol 35: 410–417. [DOI] [PubMed] [Google Scholar]
- 45. Al-Mawali A, Gillis D, Lewis I (2013) Characteristics and Prognosis of Adult Acute Myeloid Leukemia with Internal Tandem Duplication in the FLT3 Gene. Oman Med J 28: 432–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pratcorona M, Brunet S, Nomdedéu J, Ribera JM, Tormo M, et al. (2013) Favorable outcome of patients with acute myeloid leukemia harboring a low-allelic burden FLT3-ITD mutation and concomitant NPM1 mutation: relevance to post-remission therapy. Blood 121: 2734–2738. [DOI] [PubMed] [Google Scholar]
- 47. Jahn T, Leifheit E, Gooch S, Sindhu S, Weinberg K (2007) Lipid rafts are required for Kit survival and proliferation signals. Blood 110: 1739–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Xiang Z, Kreisel F, Cain J, Colson A, Tomasson MH (2007) Neoplasia driven by mutant c-KIT is mediated by intracellular, not plasma membrane, receptor signaling. Mol Cell Biol. 27: 267–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chow LM, Baker SJ (2006) PTEN function in normal and neoplastic growth. Cancer Lett 241: 184–196. [DOI] [PubMed] [Google Scholar]
- 50. Buitenhuis M, Coffer PJ (2009) The role of the PI3K–PKB signaling module in regulation of hematopoiesis. Cell Cycle 8: 560–566. [DOI] [PubMed] [Google Scholar]
- 51. Polak R, Buitenhuis M (2012) The PI3K/PKB signaling module as key regulator of hematopoiesis: implications for therapeutic strategies in leukemia. Blood 119: 911–923. [DOI] [PubMed] [Google Scholar]
- 52. Kharas MG, Okabe R, Ganis JJ, Gozo M, Khandan T, et al. (2010) Constitutively active AKT depletes hematopoietic stem cells and induces leukemia in mice. Blood 115: 1406–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Martelli AM, Evangelisti C, Chappell, Abrams SL, Bäsecke J, et al. (2011) Targeting the translational apparatus to improve leukemia therapy: roles of the PI3K/PTEN/Akt/mTOR pathway Leukemia. 25: 1064–79. [DOI] [PubMed] [Google Scholar]
- 54. Rodon J, Dienstmann R, Serra V, Tabernero J (2013) Development of PI3K inhibitors: lessons learned from early clinical trials. Nat Rev Clin Oncol 10: 143–53. [DOI] [PubMed] [Google Scholar]
- 55. Jordan CT (2010) Targeting myeloid leukemia stem cells. Sci Transl Med May 2: 31ps21. [DOI] [PubMed] [Google Scholar]
- 56. Essers MA, Trumpp A (2010) Targeting leukemic stem cells by breaking their dormancy. Mol Oncol 4: 443–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sykes SM, Lane SW, Bullinger L, Kalaitzidis D, Yusuf R, et al. (2011) AKT/FOXO signaling enforces reversible differentiation blockade in myeloid leukemias. Cell 146: 697–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Nogueira V, Park Y, Chen CC, Xu PZ, Chen ML, Tonic I, et al. (2008) Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis Cancer Cell. 14: 458–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Picture of CFU formed from WT, CALM+/− , and CALM−/− fetal liver LSKs. LSK cells isolated from fetal liver of WT, CALM +/−, and CALM −/− mice on E14.5 were subjected to clonogenic assays. The size of colonies was observed under the fluorescence microscopy (BZ-X700, Keyence, Osaka, Japan). The representative results were shown. Mix, CFU-Mix; GM, CFU-GM; E, BFU-E. (+/+), (+/−), and (−/−) represent the origin of LSK cells: WT, CALM +/−, and CALM −/− mice.
(TIFF)
Distribution of CALM after SCF stimulation in WT MEFs. Distribution of CALM was followed at the indicated times after SCF stimulation by immunofluorescence analyses using the anti-CALM Ab. Rab5, Rab7, Rab11 were used as markers of early, late, and recycling endosomes, respectively. Arrows indicate colocalization (Inset shows region of higher magnification).
(TIFF)
Colocalization of KIT and Rab11, a marker of the recycling endosome. KIT and Rab11 were costained with anti-KIT and anti-Rab11 Abs and analyzed by confocal microscopy.
(TIFF)
Activation of downstream molecules by KIT localized at early endosomes. (A) CALM−/− MEFs were preincubated with 5 µM imatinib or vehicle before the treatment with SCF for 6 h. After SCF-stimulation, the amounts and phosphorylation status of KIT were analyzed at the indicated time points using immunoprecipitated cell lysates. Also, whole cell lysates were subjected to immunoblot analyses using anti-Akt and anti-phosho-Akt Abs. (B) WT MEFs were incubated with 1 µM Bafilomycin A1 or vehicle during the treatment with SCF and whole cell lysates were subjected to the same experiment as Fig. 7A.
(TIFF)
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.