

Abstract
Macrophages participate in both the amplification of inflammation at the time of injury, and downregulation of the inflammatory response to avoid excess tissue damage. These divergent functions of macrophages are dictated by their microenvironment, especially cytokines, which promote a spectrum of macrophage phenotypes. The M1 proinflammatory phenotype is induced by LPS, IFN-γ and GM-CSF, and IL-4, IL-13 and M-CSF induce anti-inflammatory M2 macrophages. Suppressors Of Cytokine Signaling (SOCS) proteins function as feedback inhibitors of the JAK/STAT signaling pathway, and can terminate innate and adaptive immune responses. In this study, we have evaluated the influence of SOCS3 on macrophage polarization and function. Macrophages obtained from LysMCre-SOCS3fl/fl mice, which lack SOCS3 in myeloid lineage cells, exhibit enhanced and prolonged activation of JAK/STAT pathway compared to macrophages from SOCS3fl/fl mice. Furthermore, SOCS3-deficient macrophages have higher levels of the M1 genes IL-1β, IL-6, IL-12, IL-23 and iNOS, due to enhanced transcriptional activation and chromatin modifications. SOCS3-deficient M1 macrophages also have a stronger capacity to induce Th1 and Th17 cell differentiation than M1 macrophages from SOCS3fl/fl mice. Lastly, LPS-induced sepsis is exacerbated in LysMCre-SOCS3fl/fl mice, and is associated with enhanced STAT1/3 activation and increased plasma levels of M1 cytokines/chemokines such as IL-1β, TNF-α, IL-6, CCL3, CCL4 and CXCL11. These findings collectively indicate that SOCS3 is involved in repressing the M1 proinflammatory phenotype, thereby deactivating inflammatory responses in macrophages.
Introduction
Macrophage effector function significantly influences the quality, duration and magnitude of innate immune responses. It is now appreciated that there are multiple macrophage phenotypes that carry out differential functions and elicit divergent effects on surrounding cells. This functional diversity reflects a complex interplay between intrinsic differentiation pathways and inputs received from the microenvironment, particularly cytokines (1-3). Furthermore, macrophages are critical for priming and dictating T-cell responses and differentiation, thus extending the function of macrophages to control of the adaptive immune response.
Depending on the microenvironment, macrophages can acquire distinct functional phenotypes, referred to as classically activated, proinflammatory macrophages (M1) and alternatively activated, anti-inflammatory macrophages (M2) (4). Macrophages are polarized to the M1 phenotype by exposure to Th1 cytokines such as IFN-γ and GM-CSF, or in the presence of bacterial products such as LPS. M1 macrophages produce high levels of TNF-α, IL-6, IL-1β, IL-12, IL-23 and CCL2, increased levels of reactive oxygen species, low levels of IL-10, and participate in the induction of Th1 and Th17 responses (4-6). M2 macrophages are polarized by stimulation with Th2 cytokines such as IL-4 and IL-13, as well as M-CSF (7), and have upregulated expression of scavenger and mannose receptors, FIZZ1, the IL-1 receptor antagonist (IL-1RA), arginase-1 and the chitinase family protein Ym1. M2 macrophages are associated with anti-inflammatory and homeostatic functions linked to wound healing and tissue repair, and induce the differentiation of Th2 cells (4, 6, 7). The cytokines that promote polarization of macrophage phenotypes signal predominantly through the JAK/STAT pathway, leading to the activation of transcription factors that dictate M1/M2 polarization (1, 8-11). Macrophage polarization is plastic, suggesting that the M1 to M2 switch during the progression of inflammatory responses enables the dual role of macrophages in orchestrating the onset of inflammation and subsequently promoting healing and repair (4, 7, 8).
Suppressors Of Cytokine Signaling (SOCS) proteins, CIS and SOCS1-7, are feedback inhibitors of the JAK/STAT signaling pathway (12). Through a variety of mechanisms, SOCS proteins negatively regulate both innate and adaptive immune responses. Our previous studies have demonstrated the negative regulatory function of SOCS3 in microglia, astrocytes and CD4+ T cells by inhibiting STAT1 and STAT3 activation (13-15). SOCS3 is essential for the suppression of IL-6/gp130 signaling in macrophages due to the ability of SOCS3 to bind to the Tyr759 region of gp130 (16). In addition, SOCS3 was shown to bind directly to JAK1, JAK2 and TYK2, serving as a non-competitive tyrosine kinase inhibitor (17). We have recently demonstrated that myeloid-specific SOCS3-deficient mice are vulnerable to a neuroinflammation model, which is characterized by enhanced STAT3 signaling, expression of M1-related genes, and an immune response dominated by Th1 and Th17 cells (18). In this study, we have evaluated the polarization of macrophages from wild-type (SOCS3fl/fl) and LysMCre-SOCS3fl/fl mice. We found that SOCS3-deficient bone marrow-derived macrophages (BMDMs) from LysMCre-SOCS3fl/fl mice expressed higher levels of genes related to M1 polarization, such as IL-1β, IL-6, IL-12, IL-23, iNOS, CCL2 and CXCL10, compared to BMDMs from WT mice upon stimulation with M1 inducers. Furthermore, SOCS3 deletion enhanced LPS, IFN-γ and GM-CSF-induced STAT activation, but had no significant effect on LPS-induced NF-κB and MAPK activation. SOCS3-deficient M1 macrophages exhibited a higher phagocytic capacity and a more potent ability to promote Th1 and Th17 cell differentiation than WT M1 macrophages. To investigate the function of myeloid SOCS3 in inflammation, an LPS-induced endotoxic shock model was used. Our findings reveal that in the absence of myeloid SOCS3, LysMCre-SOCS3fl/fl mice are hypersensitive to LPS, which is associated with elevated M1-associated proinflammatory cytokine and chemokine expression, and enhanced STAT activation. These findings identify SOCS3 as a regulator of macrophage activation and M1 polarization via the STAT pathway, and indicate a protective influence of SOCS3 on macrophage inflammatory responses.
Materials and Methods
Mice
C57BL/6 and OVA-TCRα/β transgenic OT-II mice (19) were bred in the animal facility at the University of Alabama at Birmingham (UAB) (Birmingham, AL). SOCS3 floxed transgenic (SOCS3fl/fl) mice (20) were the generous gift of Dr. Warren Alexander (Cancer and Hematology Division, The Walter and Eliza Hall Institute of Medical Research and the Cooperative Research Centre for Cellular Growth Factors, Victoria, Australia), and were bred at UAB. SOCS3 conditional knockout (LysMCre-SOCS3fl/fl) mice were generated by serial breeding of SOCS3fl/fl mice with mice expressing Cre recombinase under the control of the LysM promoter, in which the conditional SOCS3 allele is excised in myeloid cells (18). All experiments were reviewed and approved by the Institutional Animal Care and Use Committee of UAB.
Recombinant Proteins and Reagents
Escherichia coli LPS was purchased from Sigma-Aldrich (St. Louis, MO) and recombinant mouse IFN-γ, GM-CSF, IL-4, M-CSF, IL-6, IL-23 and human TGF-β1 were from R & D Systems (Minneapolis, MN). Synthetic bacterial Lipoprotein Pam2CSK4 was purchased from InvivoGen (San Diego, CA). Abs against phospho-STAT1 (Tyr701), phospho-STAT3 (Tyr705), phospho-STAT5 (Tyr694), phospho-STAT6 (Tyr641), phospho-p65 (Ser536), phospho-ERK1/ERK2 (Thr202/Tyr204), STAT1, STAT3, STAT5, STAT6, p65 and ERK1/2 were from Cell Signaling Technology (Beverly, MA). Abs against SOCS1 and SOCS3 were from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-CD3 and anti-CD28 Abs were purchased from BioLegend (San Diego, CA), and Ab against GAPDH was from Abcam (Cambridge, MA). Neutralizing Abs to IL-4 and IFN-γ, and conjugated Abs to CD4, IFN-γ and IL-17A were from eBioscience (San Diego, CA).
Bone Marrow-derived Macrophage (BMDM) Preparation
Bone marrow cells were flushed from the femurs and tibiae of 7 to 8-week-old C57BL/6, SOCS3fl/fl or LysMCre-SOCS3fl/fl mice, as previously described (18). These cells were cultured in RPMI 1640 medium containing 10% FBS and 10 ng/ml of murine M-CSF for 5-7 days. Cell purity was determined by FACS analysis for CD11b (>96%).
RNA Isolation, RT-PCR and TaqMan Gene Expression Assays
Total cellular RNA was isolated from untreated or cytokine stimulated BMDMs from C57BL/6, SOCS3fl/fl or LysMCre-SOCS3fl/fl mice, and RT reactions performed as previously described (18). Five hundred ng of RNA was used to reverse transcribe into cDNA and subjected to RT-PCR or quantitative real-time PCR (qRT-PCR) (21). The ABI Prismo 7500 Sequence Detection System, TaqMan Gene Expression Master Mix and TaqMan Gene Expression Assay probes were from Applied Biosystems (Foster City, CA) and were used for qRT-PCR to determine mRNA levels. The data were analyzed using the comparative Ct method to obtain relative quantitation values (21).
Immunoblotting
Thirty μg of cell lysate was separated by electrophoresis on 10% or 12% SDS-polyacrylamide gels, and probed with specific Abs as described previously (18).
ELISA and Multiplex Analysis of Cytokines and Chemokines
Supernatants were collected from untreated or cytokine stimulated BMDMs from C57BL/6, SOCS3fl/fl or LysMCre-SOCS3fl/fl mice, and assayed by ELISA kits (Biolegend, San Diego, CA) or Millipore Mouse Cytokine/Chemokine Panel lI (MPXMCYTO-70K) (Millipore, Billerica, MA) for secretion of murine cytokines (IFN-γ, IL-1α, IL-1β, IL-6, IL-17, TNF-α and G-CSF) and chemokines (CCL2 (MCP-1), CCL3 (MIP-1α), CCL4 (MIP-1β), CCL5 (RANTES), CXCL1 (KC), CXCL2 (MIP-2), CXCL9 (MIG), CXCL10 (IP-10) and CXCL11 (Eotaxin)). Expression levels of cytokines/chemokines were normalized to total protein levels as previously described (18).
Phagocytosis Assay
Macrophage phagocytic activity was determined by cellular uptake of pH-sensitive rhodamine-conjugated E. coli particles (pHrodo™ Phagocytosis Particle Labeling Kit for Flow Cytometry, Invitrogen, Carlsbad, CA). Briefly, 1 × 105 BMDM treated with LPS for 24 h were prepared in 100 μl of Uptake Buffer, Hanks' Balanced Salt Solution (HBSS) with an additional 20 mM HEPES, pH 7.4. Two hundred μl of pHrodo™ dye-labeled E. coli was added to the cells with Uptake Buffer and incubated at 37°C for 30 to 120 min. Phagocytosis was terminated by exposure of tubes to ice, and fluorescence from ingested E. coli particles detected by flow cytometry.
Intracellular Cytokine Staining
BMDMs from SOCS3fl/fl and LysMCre-SOCS3fl/fl mice were polarized to the M1 phenotype with LPS plus IFN-γ for 48 h, and then used as antigen-presenting cells. Naive CD4+ T cells were isolated from the spleen of OVA-TCR transgenic OTII mice (22). M1 macrophages and CD4+ T cells were cultured at a 1:5 ratio for Th1 and Th17 cell differentiation. Th1 cells were differentiated with OVA peptide (5 μg/ml), IL-12 (10 ng/ml) plus anti-IL-4 (10 μg/ml) stimulation for 4 days. Th17 cells were differentiated with OVA peptide (5 μg/ml), TGF-β (5 ng/ml), IL-6 (20 ng/ml), IL-23 (10 ng/ml) plus anti-IFN-γ (10 μg/ml) and anti-IL-4 (10 μg/ml) stimulation for 4 days. At day 4, cells were stimulated with PMA/Ionomycin (25 ng/ml and 1 μg/ml) plus GolgiStop (BD Pharmingen) for 4 h, and were analyzed for intracellular production of cytokines by staining with anti-cytokine Abs and subsequent flow cytometry, as described (18).
Chromatin Immunoprecipitation (ChIP) Assay
ChIP analysis was done following a protocol provided by Upstate Biotechnology with modifications as described previously (13, 23). Primary macrophages were incubated with medium or LPS (10 ng/ml) for 4 h and fixed with 1% formaldehyde for 15 min at room temperature, and then nuclei were isolated. Chromatin was sheared by sonication, and precipitated overnight at 4°C with 5 μg of Abs. Input chromatin and immunoprecipitated chromatin were incubated at 65°C overnight to reverse cross-links. DNA was extracted with the Qiagen Miniprep kit (Qiagen, Valencia, CA). Purified DNA was analyzed by PCR. The following primer pairs were used: 5′-AGTATCTCTGCCTCCTTCCTT-3′ and 5′-GCAACACTGAAAACTAGTGTC-3′ for the murine IL-12p40 promoter; 5′-CCCGACCTAGGCCTCTAGCCCA-3′ and 5′-AAGGTCCCTGCACTGTAAGGCG-3′ for the murine IL-23p19-promoter; 5′-CTAGTGAGTCCCAGTTTTGAAG-3′ and 5′-CCCTGGCAGCAGCCATCAGGTA-3′ for the murine iNOS promoter; and 5′-TCATGCTGGGATCTGAGCTTCT-3′ and 5′-CGGAAGTCACCTTAGCACTCAGT-3′ for the murine IL-10 promoter.
Administration of LPS to Mice
SOCS3fl/fl and LysMCre-SOCS3fl/fl mice were subjected to i.p. injection of repurified LPS at 2.5 mg/kg. In survival studies, mice were monitored over a 4 day time period. In short-term studies, blood was collected from mice by check puncture into EDTA-coated BD Vacutainer® Blood Collection tubes before (0 h time point) and 4 h after injection with LPS, and then plasma was collected to determine expression levels of cytokines and chemokines as described previously (24). At the indicated times, animals were sacrificed, and spleens and livers were collected.
Densitometric and Statistical Analysis
Densitometric quantitation of immunoblotting images in the linear range was performed using an image analysis program (ImageJ 1.41o; National Institutes of Health). All experiments were repeated a minimum of three times. Levels of significance for comparison between samples were determined by the Student's t-test distribution. All results are shown as Mean ± Standard Deviation (S.D.). A value of p ≤ 0.05 was considered to be statistically significant.
Results
M1 and M2 Gene Expression Profiles
LPS, IFN-γ and GM-CSF have been shown to induce the M1 macrophage phenotype to various degrees. We analyzed the influence of LPS, IFN-γ and GM-CSF, alone and in combination, on expression levels of M1 markers in BMDMs. The bacterial product LPS induced expression of IL-1β, IL-6, TNF-α, IL-12p40, IL-23p19, iNOS, CCL2 and CXCL10 compared to untreated cells (Fig. 1A, lanes 1 and 2). IFN-γ induced the expression of TNF-α, iNOS, CCL2 and CXCL10, while not affecting other M1 genes (Fig. 1A, lane 3). However, LPS plus IFN-γ exerted additive and/or synergistic effects on IL-6, IL-12p40 and iNOS gene expression (Fig. 1A, lane 4). GM-CSF promoted expression of IL-1β, TNF-α, CCL2 and CXCL10 (Fig. 1A, lane 5), and had a synergistic effect with LPS on IL-6, IL-12p40, IL-23p19 and iNOS expression (Fig. 1A, lane 6). FIZZ1, PPAR-γ, IL-10, Ym1 and Arginase-1 are genes reflective of the M2 macrophage phenotype, which can be induced by IL-4 and M-CSF (7). IL-4 induced expression of all M2 markers to varying degrees (Fig. 1B, lane 2). M-CSF strongly induced PPAR-γ expression, while not significantly influencing other M2 genes (Fig. 1B, lane 3). The combination of IL-4 and M-CSF promoted a synergistic effect on FIZZ1, PPAR-γ, IL-10 and Arginase-1 expression (Fig. 1B, lane 4). These results indicate that M1 inducers have varying levels of effectiveness on M1 gene expression, with LPS being more potent than IFN-γ or GM-CSF alone, and the combination of LPS and IFN-γ or LPS and GM-CSF having the strongest effect on M1 gene expression. For induction of M2 genes, IL-4 was more potent than M-CSF (except for PPARγ expression), and the combination of IL-4 plus M-CSF had the strongest influence on M2 gene expression.
Figure 1. M1 and M2 Gene Expression in Macrophages.

(A). BMDMs from C57BL/6 mice were cultured with M-CSF (10 ng/ml) for 5 days, and then treated with medium, LPS (10 ng/ml), IFN-γ (10 ng/ml), LPS plus IFN-γ, GM-CSF (50 ng/ml) or LPS plus GM-CSF for 4 h, and mRNA analyzed by RT-PCR for IL-1β, IL-6, TNF-α, IL-12p40, IL-23p19, iNOS, CCL2, CXCL10 and GAPDH expression. Quantification of the data is shown on the right. (B). BMDMs were cultured with M-CSF (10 ng/ml) for 5 days, and then incubated with medium (UN), IL-4 (10 ng/ml), M-CSF (10 ng/ml) or IL-4 plus M-CSF for 4 h, and mRNA analyzed by RT-PCR for FIZZ1, PPAR-γ, IL-10, Ym1, Arginase-1 and GAPDH expression. Quantification of the data is shown on the right. Representative of three independent experiments.
Plasticity of M1 and M2 Macrophage Phenotypes
Macrophages are highly heterogeneous cells that can rapidly change their phenotype and function in response to microenvironmental signals, and studies have documented the flexibility of macrophage activation(25-28). We next investigated whether IL-4 could affect the M1 phenotype induced by LPS, and conversely, whether LPS affects the M2 phenotyp induced by IL-4. As shown in Fig. 2A, IL-4 potently inhibited LPS-induced M1 gene expression (lane 4). Conversely, LPS inhibited IL-4-induced M2 genes (Fig. 2B, lane 4). LPS signaling is complex, and involves activation of numerous signaling cascades, including NF-κB, STATs and ERK1/2 (29). To determine if IL-4 affects LPS-induced signaling pathways, cells were stimulated with LPS, IL-4 or both for 4 h, then pathway activation evaluated. LPS treatment led to the activation of NF-κB p65, ERK1/2, STAT1 and STAT3 (Fig. 2C, lane 2). Inclusion of IL-4 inhibited LPS-induced NF-κB p65, STAT1 and STAT3 activation, with no effect on ERK pathway activation (Fig. 2C, lane 4). IL-4 is a strong activator of STAT6 (Fig. 2C, lane 3), which was inhibited in the presence of LPS (lane 4). These results suggest that antagonism of LPS activation of STAT1/STAT3 and NF-κB by IL-4 correlates with inhibition of LPS-induced M1 gene expression, and LPS suppression of IL-4-induced STAT6 signaling is associated with inhibition of IL-4-induced M2 gene expression.
Figure 2. Plasticity of M1 and M2 Macrophage Phenotype Upon LPS and IL-4 Stimulation.

(A). BMDMs were cultured with M-CSF (10 ng/ml) for 5 days, treated with LPS, IL-4 or LPS plus IL-4 for 4 h, and then mRNA analyzed by RT-PCR for IL-1β, IL-6, IL-12p40, IL-23p19, iNOS, TNF-α and GAPDH expression. (B). BMDMs were cultured with M-CSF (10 ng/ml) for 5 days, treated with LPS, IL-4 or LPS plus IL-4 for 4 h, and then mRNA analyzed by RT-PCR for Arginase-1, FIZZ1, PPAR-γ, Ym1 and GAPDH expression. (C). BMDMs were cultured with M-CSF (10 ng/ml) for 5 days, treated with LPS, IL-4 or LPS plus IL-4 for 4 h, and then protein lysates analyzed by immunoblotting with the specified antibodies. The densitometric quantification of P-NF-κBp65, P-ERK1/ERK2, P-STAT1, P-STAT3 and P-STAT6 was determined using an image analysis program (ImageJ 1.41oh) by comparing to untreated samples. *p<0.05; **p<0.001. Represents three independent experiments.
SOCS3 Deficiency Promotes STAT Activation and M1 Macrophage Polarization
SOCS proteins function as feedback inhibitors for cytokines that utilize the JAK/STAT pathway, and can inhibit inflammatory responses in macrophages (12). To determine whether SOCS proteins are involved in macrophage polarization, we first performed experiments to analyze expression of SOCS1 and SOCS3 in BMDMs. Our results indicate that LPS, IFN-γ and GM-CSF induce SOCS1 protein expression, with IL-4 and M-CSF having a modest effect (Fig. 3A). Interestingly, no synergistic induction of SOCS1 expression was observed with the combination of LPS and IFN-γ (lane 4), and GM-CSF co-incubation with LPS inhibited SOCS1 expression (lane 6). For SOCS3, LPS was the most potent inducer (lane 2), and synergized with IFN-γ (lane 4) and GM-CSF (lane 6) for enhanced SOCS3 expression. IL-4 and M-CSF, alone or in combination, did not influence SOCS3 expression. These results indicate that M1 inducers such as LPS alone and in conjunction with IFN-γ or GM-CSF are potent stimuli for SOCS3 expression.
Figure 3. Absence of SOCS3 Enhances STAT Activation in Macrophages.

(A). BMDMs were treated with LPS, IFN-γ, LPS plus IFN-γ, GM-CSF, LPS plus GM-CSF, IL-4, M-CSF or IL-4 plus M-CSF for 4 h, and then cell lysates were analyzed by immunoblotting with SOCS1, SOCS3 and GAPDH Abs. (B). BMDMs from SOCS3fl/fl and LysMCre-SOCS3fl/fl mice were cultured with M-CSF (10 ng/ml) for 5 days, treated with IFN-γ for up to 4 h, and then mRNA analyzed by RT-PCR for SOCS1, SOCS3 and GAPDH expression. BMDMs were cultured with M-CSF (10 ng/ml) for 5 days, treated with IFN-γ (C)., GM-CSF (D)., LPS (E)., or IL-4 (F). for up to 4 h, and then protein lysates analyzed by immunoblotting with the specified Abs. Represents five independent experiments. **p<0.001.
To study the involvement of SOCS3 in macrophage polarization, we examined myeloid-specific SOCS3-deficient cells (18). As shown in Fig. 3B, IFN-γ-induced SOCS3 mRNA expression was abolished in BMDM of LysMCre-SOCS3fl/fl mice, with no compensatory expression of SOCS1. The same results were observed using LPS as the stimulus (data not shown). BMDMs from SOCS3fl/fl and LysMCre-SOCS3fl/fl mice were incubated with LPS, IFN-γ or GM-CSF for up to 4 h to examine signaling pathway activation. Deletion of SOCS3 led to enhanced and prolonged IFN-γ-induced STAT1 and STAT3 activation (Fig. 3C) and GM-CSF-induced STAT5 activation (Fig. 3D). We have previously shown that LPS induces rapid activation of NF-κB p65 and ERK1/2 in macrophages, while STAT1 and STAT3 are activated with delayed kinetics, due to IFN-β production and signaling (30). Deletion of SOCS3 had no effect on LPS-induced NF-κB p65 and ERK1/2 activation, but enhanced LPS-induced STAT1 and STAT3 activation (Fig. 3E). There was no difference in the ability of IL-4 to induce STAT6 activation between WT and SOCS3-deficient macrophages (Fig. 3F). These findings indicate that upon SOCS3 deletion, BMDM display an increased sensitivity to M1 classically polarizing stimuli such as IFN-γ, GM-CSF and LPS.
Given that LPS-induced STAT signaling was enhanced in the absence of SOCS3, we next examined M1/M2 gene expression. LPS-induced mRNA expression of the M1 markers IL-1β, IL-6, IL-12p40, IL-23p19 and iNOS was significantly enhanced in SOCS3-deficient BMDMs compared to BMDMs from SOCS3fl/fl mice (Fig. 4A). Interestingly, LPS induction of the M2 marker IL-10 was significantly decreased (Fig. 4A). Two of these genes were examined at the protein level; LPS-induced IL-6 and nitrite expression were significantly enhanced in SOCS3-deficient BMDMs compared to SOCS3fl/fl BMDMs (Figs. 4B and 4C).
Figure 4. SOCS3 Deletion in Macrophages Leads to Enhanced M1 Polarization.

(A). BMDMs from SOCS3fl/fl and LysMCre-SOCS3fl/fl mice were treated with medium or LPS for 4 h. mRNA was analyzed by qRT-PCR for IL-1β, IL-6, IL-12p40, IL-23p19, TNF-α, iNOS, IL-10 and GAPDH expression. (B). BMDMs were treated with medium (UN) or LPS for up to 16 h, and supernatants were analyzed for IL-6 protein by ELISA. (C). BMDMs from SOCS3fl/fl and LysMCre-SOCS3fl/fl mice were treated with medium (UN) or LPS for 24 h. Supernatants were analyzed for production of nitrite, a stable end product of NO production, using the Griess reagent. *p<0.05. (D). BMDMs were treated with medium (UN) or LPS for 4 h, and then cells were cross-linked with formaldehyde. Soluble chromatin was subjected to immunoprecipitation with Abs against histone acetylation (Ac-H3 and Ac-H4) or normal rabbit IgG. PCR analysis of the positive control (input) indicates that soluble chromatin samples obtained from each time point had equal amounts of chromatin fragments containing the IL-12p40, IL-23p19, iNOS and IL-10 promoters. (E). BMDMs from SOCS3fl/fl and LysMCre-SOCS3fl/fl mice were treated with Pam2CSK4 (10 ng/ml) for 1 and 2 h, then protein lysates analyzed by immunoblotting with the specified Abs. (F). BMDMs from SOCS3fl/fl and LysMCre-SOCS3fl/fl mice were treated with Pam2CSK4 (10 ng/ml) for 4 h. mRNA was analyzed by qRT-PCR for IL-1β, IL-6, IL-12p40, IL-23p19, iNOS and GAPDH expression. Represents three independent experiments.
To determine the influence of SOCS3 on M1 and M2 gene transcription, BMDMs were incubated in medium (UN) or LPS for 4 h, and chromatin immunoprecipitation assays were performed. PCR analysis of the positive control (input) indicated that soluble chromatin samples obtained from each treatment had equal amounts of chromatin fragments containing the IL-12p40, IL-23p19, iNOS and IL-10 promoters (Fig. 4D). In BMDM from SOCS3fl/fl mice (WT), acetylation of histones H3 and H4 on all four promoters was enhanced by LPS stimulation (Fig. 4D). H3 and H4 acetylation of the IL-12p40, IL-23p19 and iNOS promoters upon LPS treatment was substantially higher in SOCS3-deficient macrophages compared to WT macrophages (Fig. 4D). LPS-induced H3 and H4 acetylation of the IL-10 promoter was lower in SOCS3-deficient macrophages compared to WT macrophages (Fig. 4D), which correlates with decreased IL-10 mRNA expression (Fig. 4A). These data collectively indicate that deletion of SOCS3 enhances the expression of M1 markers in BMDMs, while inhibiting IL-10 expression, indicating that SOCS3 functions as a modulator of macrophage polarization. Furthermore, the influence of SOCS3 appears to be at the transcriptional level.
As a control, the TLR2 ligand Pam2CSK4 was used to investigate the effect of SOCS3 deletion on JAK/STAT independent signaling pathways. Deletion of SOCS3 had a modest enhancing effect on Pam2CSK4-induced NF-κB p65 and ERK1/2 activation (Fig. 4E), but did not affect Pam2CSK4-induced mRNA expression of IL-1β, IL-6, IL-12p40, IL-23p19 or iNOS (Fig. 4F).
SOCS3 Deficiency Enhances M1-induced Phagocytosis and Th1-Th17 Differentiation
One important function of macrophages is to phagocytose pathogens and foreign materials (3), and M1 macrophages have potent phagocytic capacity (31). LPS-induced M1 macrophages from SOCS3fl/fl and LysMCre-SOCS3fl/fl mice were analyzed for phagocytic function, and the results demonstrate that deletion of SOCS3 enhances the phagocytic capacity of M1 macrophages (Fig. 5A).
Figure 5. Myeloid SOCS3 Influences Phagocytosis and Th1-Th17 Differentiation.

(A). Macrophages from SOCS3fl/fl and LysMCre-SOCS3fl/fl mice were polarized to the M1 phenotype with LPS for 48 h, and phagocytosis was assessed using the pHrodo™ E. coli BioParticles® Phagocytosis Kit for Flow Cytometry. *p<0.05. (B-E). LPS plus IFN-γ polarized M1 macrophages were used as antigen-presenting cells and cultured with naive CD4+ T cells isolated from the spleen of OVA-TCR transgenic OTII mice at a 1:5 ratio for Th1 and Th17 cell differentiation. Th1 cells were differentiated with IL-12 (10 ng/ml), anti-IL-4 (10 μg/ml) and OVA peptide (5 μg/ml); and Th17 cells were differentiated with TGF-β (5 ng/ml), IL-6 (20 ng/ml), IL-23 (10 ng/ml), anti-IFN-γ (10 μg/ml), anti-IL-4 (10 μg/ml) and OVA peptide (5 μg/ml). At day 4, cells were stimulated with PMA/Ionomycin plus GolgiStop for 4 h, stained for the surface marker CD4 and by intracellular flow for IFN-γ protein (B)., for IFN-γ and T-bet mRNA expression by qRT-PCR (C)., by intracellular flow for IL-17A protein (D)., or for IL-17A and RORγt mRNA expression by qRT-PCR (E). *p<0.05. Represents three independent experiments.
Activated M1 macrophages express MHC class II, CD40, CD80 and CD86 and function as antigen-presenting cells, which leads to efficient Th1 and Th17 responses (9). We next determined whether SOCS3 expression in macrophages influences their ability to promote differentiation of CD4+ T-cells by examining OVA-specific Th1 and Th17 cell differentiation. Macrophages from SOCS3fl/fl and LysMCre-SOCS3fl/fl mice were polarized to the M1 phenotype with LPS plus IFN-γ for 48 h, and then used as antigen-presenting cells. The results demonstrate an enhancement of IFN-γ+ Th1 cells (Fig. 5B) and enhanced expression of IFN-γ and the Th1-specifying transcription factor T-bet mRNA (Fig. 5C) under Th1 cell differentiation conditions with SOCS3-deficient M1 macrophages compared with M1 macrophages from SOCS3fl/fl mice. We also observed an enhancement of IL-17A-producing Th17 cells (Fig. 5D) and higher IL-17A and the Th17-specifying transcription factor RORγt mRNA expression (Fig. 5E) under Th17 cell differentiation conditions with SOCS3-deficient M1 macrophages compared to SOCS3fl/fl M1 macrophages. Hence, the absence of SOCS3 enhances phagocytosis by M1 macrophages, and enhances the ability of M1 macrophages to promote differentiation of CD4+ T-cells into the Th1 and Th17 phenotypes.
Importance of SOCS3 in a Mouse Inflammation Model
Thus far, our data indicate that SOCS3-deficient BMDMs polarized to the M1 phenotype produce significantly higher levels of IL-1β, IL-6, IL-12p40, IL-23p19 and iNOS, and less IL-10, in response to stimulation with LPS than macrophages from SOCS3fl/fl mice (Fig. 4A). To investigate the physiological relevance of these findings, the function of SOCS3 was evaluated in an in vivo model of inflammation, LPS-induced septic shock. SOCS3fl/fl and LysMCre-SOCS3fl/fl mice were challenged with a sub-lethal dose of LPS (2.5 mg/kg) injected intraperitoneally. There was no difference in the levels of cytokines and chemokines in the plasma of SOCS3fl/fl and LysMCre-SOCS3fl/fl mice before LPS injection (Fig. 6A), however, there was a significant enhancement of cytokines/chemokines in the plasma of LysMCre-SOCS3fl/fl mice compared to SOCS3fl/fl mice 4 h after LPS challenge (Fig. 6B). In particular, the M1 cytokine IL-6 was significantly increased, as was the expression of CCL3 (MIP-1α) and CCL4 (MIP1β), two chemokines associated with M1 polarization (32, 33). In vivo, LysMCre-SOCS3fl/fl mice were more susceptible to LPS administration; only 20% of LysMCre-SOCS3fl/fl mice survived at day 4 after LPS administration, compared to 90% of SOCS3fl/fl mice (Fig. 6C). These findings indicate that deletion of SOCS3 in myeloid cells enhances M1-associated in vivo inflammatory responses induced by the TLR agonist LPS.
Figure 6. Function of Myeloid SOCS3 in LPS-induced Septic Shock Model.
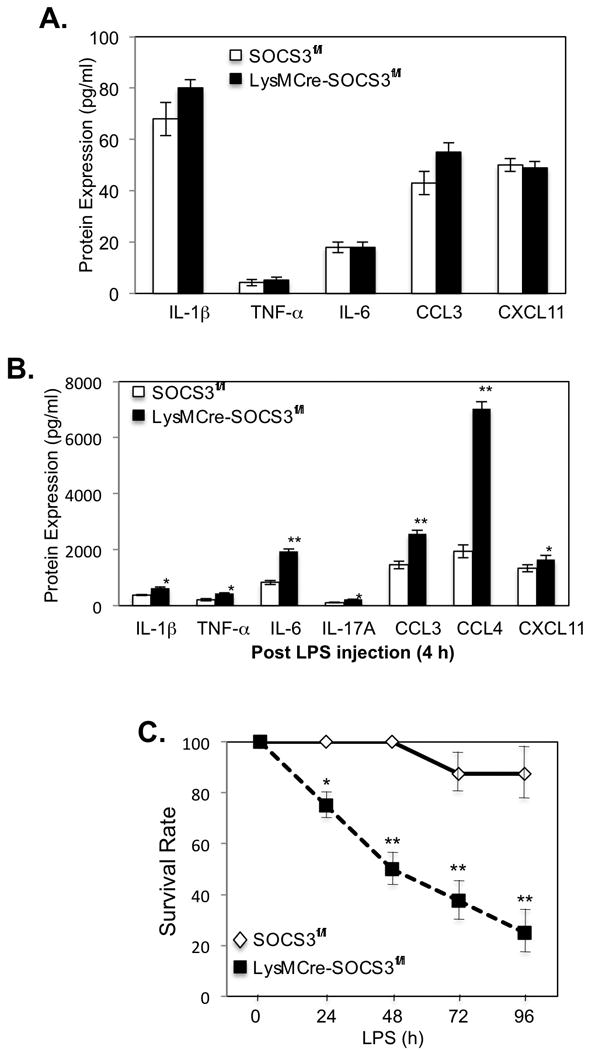
(A). Expression of IL-1β, TNF-α, IL-6, CCL3 and CXCL11 protein in the plasma of SOCS3fl/fl and LysMCre-SOCS3fl/fl mice before LPS administration, as determined by ELISA. (B). Expression of IL-1β, TNF-α, IL-6, IL-17A, CCL3, CCL4 and CXCL11 protein in the plasma of SOCS3fl/fl and LysMCre-SOCS3fl/fl mice 4 h after administration of LPS (2.5 mg/kg body weight), as determined by ELISA. (C). Survival rate of SOCS3fl/fl and LysMCre-SOCS3fl/fl mice (n = 4 per group per experiment) after intraperitoneal (i.p.) injection of LPS (2.5 mg/kg body weight). *p<0.05; **p<0.001. Represents three independent experiments.
Splenomegaly and Enhanced STAT1/3 Activation in LysMCre-SOCS3fl/fl Mice
The spleen and liver are important organs in monitoring infections, especially polysaccharide encapsulated bacterial infections (34, 35). Enlarged spleens were observed in LysMCre-SOCS3fl/fl mice after LPS administration, as evidenced by comparing spleen weight (∼2.0 fold increase) (Figure 7A). STAT1, STAT3 and ERK1/2 activation, and inflammatory gene expression in spleen and liver was examined in SOCS3fl/fl and LysMCre-SOCS3fl/fl mice with systemic LPS challenge. Immunoblotting demonstrated that STAT1 and STAT3 tyrosine phosphorylation was exaggerated and prolonged in the liver and spleen of LysMCre-SOCS3fl/fl compared to SOCS3fl/fl mice (Figs. 7B and 7C). The activation of ERK1/2 was enhanced in the spleen, but not in liver, of LysMCre-SOCS3fl/fl compared to SOCS3fl/fl mice (Figs. 7B and 7C). Furthermore, significant enhancement of IL-1β, TNF-α and IL-6 mRNA expression was observed in the spleen and liver from LysMCre-SOCS3fl/fl mice in response to LPS (Fig. 7D), with IL-6 expression being the most elevated. This correlates with LPS-induced hypersensitivity and exaggerated activation of STAT1 and STAT3 in LysMCre-SOCS3fl/fl mice. Collectively, these data demonstrate that SOCS3 is an important negative regulator of LPS-driven inflammatory responses in vivo, affecting activation of the STAT pathway.
Figure 7. Enhanced Activation of STATs and Increased Cytokine/Chemokine Expression in LysMCre-SOCS3fl/fl Mice.

(A). Relative size of LysMCre-SOCS3fl/fl (right) spleen compared to SOCS3fl/fl mice (left) after i.p. administration of 2.5 mg/kg of LPS for 16 h. Average spleen weight is represented as mean ± SD (n=3). *p <0.05. (B). SOCS3fl/fl and LysMCre-SOCS3fl/fl mice were administered LPS (2.5 mg/kg), and STAT1, STAT3 and ERK1/2 phosphorylation was measured by immunoblotting in liver lysates at the indicated time points. Quantification of the data is shown on the right. *p<0.05; **p<0.001. (C). Spleen lysates from SOCS3fl/fl and LysMCre-SOCS3fl/fl mice were immunoblotted as in (B) at the indicated time points. Quantification of the data is shown on the right. *p<0.05; **p<0.001. (D). QRT-PCR analysis of IL-1β, TNF-α and IL-6 mRNA expression in liver and spleen tissue from mice after i.p. administration of LPS (2.5 mg/kg) (0-6 h). *p <0.05; **p <0.001 versus data from SOCS3fl/fl mice at the corresponding time points. Represents three independent experiments.
Discussion
We identify a novel role for the SOCS3 protein in regulation of macrophage polarization in vitro and in vivo. A deficiency of SOCS3 in macrophages promotes a heightened responsiveness to IFN-γ, LPS and GM-CSF with respect to STAT activation and expression of genes that characterize M1 classically activated macrophages. Furthermore, in vivo studies utilizing an LPS-induced septic shock model reveal that SOCS3 deficiency in myeloid cells promotes exacerbation of disease, which is associated with increased expression of M1 cytokines and chemokines, and aberrant STAT activation. Collectively, these findings identify SOCS3 as a modulator of macrophage activation and M1 polarization via the STAT signaling pathway.
Classical M1 macrophages are activated by proinflammatory cytokines and pathogen-associated molecular patterns, and produce proinflammatory cytokines/chemokines and an array of cytotoxic molecules that aid in the clearance of invading pathogens (1). SOCS3 is implicated in regulating innate immune responses and in attenuating pro-inflammatory cytokine signals, leading to inhibition of STAT activation (12, 36, 37). We observed several striking features of SOCS3-deficient macrophages. In absence of SOCS3, IFN-γ, LPS and GM-CSF-induced activation of the STAT pathway was enhanced and prolonged, indicating increased sensitivity to M1-inducing stimuli. Activation of the NF-κB and ERK1/2 pathways in response to LPS was not affected by the absence of SOCS3. Although SOCS3 was initially identified as an inhibitor of IL-6 cytokine family signaling via interaction with the gp130 receptor subunit (36, 37), it is appreciated that SOCS3 can negatively impact signaling by other cytokines, including IFN-γ and GM-CSF (38). SOCS3 was recently shown to directly inhibit JAK1, JAK2 and TYK2 via a conserved three-residue motif (17). IFN-γ signaling utilizes JAK1 and JAK2, and GM-CSF signals through a JAK2 homodimer. We have previously shown that LPS stimulation of macrophages results in rapid activation of the NF-κB pathway, subsequent IFN-β production, and delayed activation of STAT1 and STAT3 (30). IFN-β signals through JAK1 and TYK2, thus explaining how SOCS3 deficiency allows heightened LPS-activation of the STAT pathway. Interestingly, no differences were observed in the ability of IL-4 to activate STAT6 in the absence/presence of SOCS3. IL-4 signals through JAK1 and JAK3, thus the inability of SOCS3 to repress JAK3 activation may explain the comparable levels of STAT6 activation in SOCS3fl/fl and SOCS3-deficient macrophages. We have previously shown that IL-4 can promote M2 polarization in SOCS3-deficient macrophages to the same extent as SOCS3fl/fl macrophages (18), which is likely the result of comparable activation of the STAT6 transcription factor. These results indicate that in absence of SOCS3, macrophages have a heightened sensitivity to inducers of the M1 phenotype such as LPS, IFN-γ and GM-CSF. Conversely, the ability to respond to the M2-phenotype inducer IL-4 is not altered.
We further demonstrate that in response to LPS plus IFN-γ, SOCS3-deficient macrophages display a gene signature of the M1 phenotype, which is more pronounced than that of WT M1 macrophages. This is attributed to the enhanced STAT activation observed in the absence of SOCS3, resulting in downstream gene expression. The influence of SOCS3 appears to be at the level of gene transcription, as the IL-12p40, IL-23p19 and iNOS promoters, reflective of the M1 phenotype, all displayed enhanced acetylation of histones H3 and H4, characteristics of actively transcribing promoters (39). IRF5 was recently demonstrated as a transcription factor that promotes M1 macrophage polarization by directly binding to IL-12 and IL-23 promoters (9). We previously demonstrated that IRF5 expression was enhanced in the absence of SOCS3 (18), thus, this may be one mechanism contributing to the heightened M1 phenotype observed in our studies. Notably, IL-10 mRNA expression was reduced in SOCS3-deficient macrophages, and histone H3 and H4 acetylation of the IL-10 promoter was also diminished in the absence of SOCS3. Again, this may reflect the role of IRF5, which functions as a direct repressor of the IL-10 promoter (9).
With respect to macrophage function, we examined two parameters, phagocytosis and Th cell differentiation. The phagocytic capacity of SOCS3-deficient M1 macrophages was enhanced compared to WT M1 macrophages, and their ability to promote Th1 and Th17 cell differentiation was also enhanced. This is attributed to heightened IL-12 production by SOCS3-deficient macrophages, which will promote Th1 cell differentiation (40), and enhanced secretion of IL-1β, IL-6 and/or IL-23, which will promote the differentiation of Th17 cells (40). Th1 cells express IFN-γ and the transcription factor T-bet (40), while Th17 cells express IL-17A and the transcription factor RORγt (40). All these Th1 and Th17 markers were upregulated in M1 SOCS3-deficient macrophages compared to WT M1 cells. Thus, SOCS3-deficient macrophages provide the microenvironment for differentiation of Th1 and Th17 cells, which function as immune effector cells in many autoimmune diseases including arthritis, inflammatory bowel disease and multiple sclerosis (38). In this regard, we have recently shown that mice with conditional SOCS3 deletion in myeloid cells are vulnerable to neuroinflammation characterized by M1 polarization, enhanced STAT activation and Th1/Th17 cell differentiation (18). Collectively, these findings document an important role for SOCS3 in limiting polarization to the M1 phenotype, and ultimately attenuating the inflammatory potential of these cells.
SOCS3 expression has been shown to have a beneficial role in attenuating inflammatory and autoimmune diseases (18, 41-45). SOCS3 expression is up-regulated in immune cells (macrophages and neutrophils) in response to a septic challenge induced by cecal ligation and puncture (CLP) (42), which may protect from a fulminant immune response. Gene delivery of SOCS3 protected mice from endotoxic septic shock and was associated with decreased levels of TNF-α in the serum (43). Adenovirus-mediated gene delivery of SOCS3 significantly reduced parameters of arthritic inflammation (44), while a recombinant cell-penetrating form of SOCS3 inhibited the lethal effects of LPS by reducing production of inflammatory cytokines and attenuating liver apoptosis and hemorrhagic necrosis (45). In an acute lung injury model induced by LPS, SOCS3 expression in alveolar macrophages was protective by regulating oxidative stress (46). Our findings demonstrate that in the absence of myeloid SOCS3, LysMCre-SOCS3fl/fl mice exhibit exacerbated inflammation in response to LPS-mediated septic shock, which is associated with increased expression of M1 macrophage products such as IL-6, TNF-α, IL-1β, CCL3, CCL4 and CXCL11, and STAT1 and STAT3 activation in the liver and spleen. Our results in agreement with those of Greenhill et al., (24), who demonstrated that LPS-induced septic shock is associated with IL-6 trans-signaling and inappropriate STAT3 activation, which sensitizes mice to LPS/TLR4-driven inflammatory responses. In contrast, our findings differ from Yasukawa et al., (36), in which LysMCre-SOCS3fl/fl mice were resistant to LPS-induced septic shcok. A possible explanation for the discrepancy is that Yasukawa et al., used a low-dose LPS model (250-500 ng) in conjunction with galactosamine, in contrast to our high-dose LPS model. LPS toxicity in galactosamine-treated mice involves TNF-α induced caspase 3 dependent acute liver injury, not systemic inflammatory response (47).
Previous studies demonstrated that myeloid STAT3 functions in a protective manner in models of liver inflammation and injury using mice with deletion of STAT3 in myeloid cells (48, 49). Our findings in the LPS-induced septic shock model are different, with STAT3 activation exerting profound inflammatory responses. However, it should be noted that deletion of SOCS3 from myeloid cells likely has effects other than STAT3 activation, since SOCS3 also functions as an E3 ubiquitin ligase (50). Thus, the regulatory functions of SOCS3 and STAT3 in various inflammatory models are likely to be different.
In conclusion, our results identify SOCS3 as a modulator of macrophage polarization, and show that SOCS3 deficiency skews macrophages towards the M1 phenotype. The beneficial role of SOCS3 in myeloid lineage cells has been documented in restricting inflammatory responses in a variety of animal models for multiple sclerosis, arthritis, allograft rejection, lung injury, atherosclerosis and septic shock (18, 51-53). Therefore, strategies to regulate SOCS3 expression in myeloid cells can be exploited to restrict the duration of M1 polarization, and subsequent effects on inflammation and Th1/Th17 differentiation.
Acknowledgments
The authors thank Dr. Warren Alexander for the SOCS3fl/fl mice, and members of the Benveniste laboratory for helpful discussions.
Footnotes
This work was funded in part by National Institutes of Health Grants NS45290 (E.N.B.), National Multiple Sclerosis Society Collaborative Research Center Grant (CA 1059-A-13) (E.N.B.), and a CFAR Developmental Grant (H.Q.), P30 AI27767. Y.L. is supported by the NMSS Collaborative Research Center Grant. We thank the Rheumatic Diseases Core Center (RDCC) (P30 Flow Core, AR48311) for advice and technical assistance.
References
- 1.Lawrence T, Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol. 2011;11:750–761. doi: 10.1038/nri3088. [DOI] [PubMed] [Google Scholar]
- 2.Murray PJ, Wynn TA. Obstacles and opportunities for understanding macrophage polarization. J Leukoc Biol. 2011;89:557–563. doi: 10.1189/jlb.0710409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11:723–737. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mantovani A, Locati M. Orchestration of macrophage polarization. Blood. 2009;114:3135–3136. doi: 10.1182/blood-2009-07-231795. [DOI] [PubMed] [Google Scholar]
- 5.Khallou-Laschet J, Varthaman A, Fornasa G, Compain C, Gaston AT, Clement M, Dussiot M, Levillain O, Graff-Dubois S, Nicoletti A, Caligiuri G. Macrophage plasticity in experimental atherosclerosis. PLoS One. 2010;5:e8852. doi: 10.1371/journal.pone.0008852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Benoit M, Desnues B, Mege JL. Macrophage polarization in bacterial infections. J Immunol. 2008;181:3733–3739. doi: 10.4049/jimmunol.181.6.3733. [DOI] [PubMed] [Google Scholar]
- 7.Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32:593–604. doi: 10.1016/j.immuni.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 8.Galli SJ, Borregaard N, Wynn TA. Phenotypic and functional plasticity of cells of innate immunity: macrophages, mast cells and neutrophils. Nat Immunol. 2011;12:1035–1044. doi: 10.1038/ni.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krausgruber T, Blazek K, Smallie T, Alzabin S, Lockstone H, Sahgal N, Hussell T, Feldmann M, Udalova IA. IRF5 promotes inflammatory macrophage polarization and T(H)1-T(H)17 responses. Nat Immunol. 2011;12:231–238. doi: 10.1038/ni.1990. [DOI] [PubMed] [Google Scholar]
- 10.Ishii M, Wen H, Corsa CA, Liu T, Coelho AL, Allen RM, Carson WFt, Cavassani KA, Li X, Lukacs NW, Hogaboam CM, Dou Y, Kunkel SL. Epigenetic regulation of the alternatively activated macrophage phenotype. Blood. 2009;114:3244–3254. doi: 10.1182/blood-2009-04-217620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Satoh T, Takeuchi O, Vandenbon A, Yasuda K, Tanaka Y, Kumagai Y, Miyake T, Matsushita K, Okazaki T, Saitoh T, Honma K, Matsuyama T, Yui K, Tsujimura T, Standley DM, Nakanishi K, Nakai K, Akira S. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat Immunol. 2010;11:936–944. doi: 10.1038/ni.1920. [DOI] [PubMed] [Google Scholar]
- 12.Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol. 2007;7:454–465. doi: 10.1038/nri2093. [DOI] [PubMed] [Google Scholar]
- 13.Qin H, Roberts KL, Niyongere SA, Cong Y, Elson CO, Benveniste EN. Molecular mechanism of lipopolysaccharide-Induced SOCS-3 gene expression in macrophages and microglia. J Immunol. 2007;179:5966–5976. doi: 10.4049/jimmunol.179.9.5966. [DOI] [PubMed] [Google Scholar]
- 14.Qin H, Niyongere SA, Lee SJ, Baker BJ, Benveniste EN. Expression and functional significance of SOCS-1 and SOCS-3 in astrocytes. J Immunol. 2008;181:3167–3176. doi: 10.4049/jimmunol.181.5.3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qin H, Wang L, Feng T, Elson CO, Niyongere SA, Lee SJ, Reynolds SL, Weaver CT, Roarty K, Serra R, Benveniste EN, Cong Y. TGF-β promotes Th17 cell development through inhibition of SOCS3. J Immunol. 2009;183:97–105. doi: 10.4049/jimmunol.0801986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sommer U, Schmid C, Sobota RM, Lehmann U, Stevenson NJ, Johnston JA, Schaper F, Heinrich PC, Haan S. Mechanisms of SOCS3 phosphorylation upon IL-6 stimulation: Contributions of SRC- and receptor tyrosine kinases. J Biol Chem. 2005;280:31478–31488. doi: 10.1074/jbc.M506008200. [DOI] [PubMed] [Google Scholar]
- 17.Babon JJ, Kershaw NJ, Murphy JM, Varghese LN, Laktyushin A, Young SN, Lucet IS, Norton RS, Nicola NA. Suppression of cytokine signaling by SOCS3: characterization of the mode of inhibition and the basis of its specificity. Immunity. 2012;36:239–250. doi: 10.1016/j.immuni.2011.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qin H, Yeh WI, De Sarno P, Holdbrooks AT, Liu Y, Muldowney MT, Reynolds SL, Yanagisawa LL, Fox THI, Park K, Harrington LE, Raman C, Benveniste EN. Signal transducer and activator of transcription-3/suppressor of cytokine signaling-3 (STAT3/SOCS3) axis in myeloid cells regulates neuroinflammation. Proc Natl Acad Sci USA. 2012;109:5004–5009. doi: 10.1073/pnas.1117218109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kang Z, Altuntas CZ, Gulen MF, Liu C, Giltiay N, Qin H, Liu L, Qian W, Ransohoff RM, Bergmann C, Stohlman S, Tuohy VK, Li X. Astrocyte-restricted ablation of Interleukin-17-induced Act1-mediated signaling ameliorates autoimmune encephalomyelitis. Immunity. 2010;32:414–425. doi: 10.1016/j.immuni.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Croker BA, Krebs DL, Zhang JG, Wormald S, Willson TA, Stanley EG, Robb L, Greenhalgh CJ, Förster I, Clausen BE, Nicola NA, Metcalf D, Hilton DJ, Roberts AW, Alexander WS. SOCS3 negatively regulates IL-6 signaling in vivo. Nat Immunol. 2003;4:540–545. doi: 10.1038/ni931. [DOI] [PubMed] [Google Scholar]
- 21.Akhtar LN, Qin H, Muldowney MT, Yanagisawa LL, Kutsch O, Clemens JE, Benveniste EN. Suppressor of cytokine signaling 3 inhibits antiviral IFN-β signaling to enhance HIV-1 replication in macrophages. J Immunol. 2010;185:2393–2404. doi: 10.4049/jimmunol.0903563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feng T, Wang L, Schoeb TR, Elson CO, Cong Y. Microbiota innate stimulation is a prerequisite for T cell spontaneous proliferation and induction of experimental colitis. J Exp Med. 2010;207:1321–1332. doi: 10.1084/jem.20092253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma X, Reynolds SL, Baker BJ, Li X, Benveniste EN, Qin H. IL-17 enhancement of the IL-6 signaling cascade in astrocytes. J Immunol. 2010;184:4898–4906. doi: 10.4049/jimmunol.1000142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Greenhill CJ, Rose-John S, Lissilaa R, Ferlin W, Ernst M, Hertzog PJ, Mansell A, Jenkins BJ. IL-6 trans-signaling modulates TLR4-dependent inflammatory responses via STAT3. J Immunol. 2011;186:1199–1208. doi: 10.4049/jimmunol.1002971. [DOI] [PubMed] [Google Scholar]
- 25.Mylonas KJ, Nair MG, Prieto-Lafuente L, Paape D, Allen JE. Alternatively activated macrophages elicited by helminth infection can be reprogrammed to enable microbial killing. J Immunol. 2009;182:3084–3094. doi: 10.4049/jimmunol.0803463. [DOI] [PubMed] [Google Scholar]
- 26.Stout RD, Jiang C, Matta B, Tietzel I, Watkins SK, Suttles J. Macrophages sequentially change their functional phenotype in response to changes in microenvironmental influences. J Immunol. 2005;175:342–349. doi: 10.4049/jimmunol.175.1.342. [DOI] [PubMed] [Google Scholar]
- 27.Liao X, Sharma N, Kapadia F, Zhou G, Lu Y, Hong H, Paruchuri K, Mahabeleshwar GH, Dalmas E, Venteclef N, Flask CA, Kim J, Doreian BW, Lu KQ, Kaestner KH, Hamik A, Clement K, Jain MK. Kruppel-like factor 4 regulates macrophage polarization. J Clin Invest. 2011;121:2736–2749. doi: 10.1172/JCI45444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sierra-Filardi E, Puig-Kroger A, Blanco FJ, Nieto C, Bragado R, Palomero MI, Bernabeu C, Vega MA, Corbi AL. Activin A skews macrophage polarization by promoting a proinflammatory phenotype and inhibiting the acquisition of anti-inflammatory macrophage markers. Blood. 2011;117:5092–5101. doi: 10.1182/blood-2010-09-306993. [DOI] [PubMed] [Google Scholar]
- 29.Barton GM, Medzhitov R. Toll-like receptor signaling pathways. Science. 2003;300:1524–1525. doi: 10.1126/science.1085536. [DOI] [PubMed] [Google Scholar]
- 30.Qin H, Wilson CA, Lee SJ, Zhao X, Benveniste EN. LPS induces CD40 gene expression through the activation of NF-κB and STAT-1α in macrophages and microglia. Blood. 2005;106:3114–3122. doi: 10.1182/blood-2005-02-0759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van der Does AM, Beekhuizen H, Ravensbergen B, Vos T, Ottenhoff TH, van Dissel JT, Drijfhout JW, Hiemstra PS, Nibbering PH. LL-37 directs macrophage differentiation toward macrophages with a proinflammatory signature. J Immunol. 2010;185:1442–1449. doi: 10.4049/jimmunol.1000376. [DOI] [PubMed] [Google Scholar]
- 32.Cassol E, Cassetta L, Rizzi C, Alfano M, Poli G. M1 and M2a polarization of human monocyte-derived macrophages inhibits HIV-1 replication by distinct mechanisms. J Immunol. 2009;182:6237–6246. doi: 10.4049/jimmunol.0803447. [DOI] [PubMed] [Google Scholar]
- 33.Shaykhiev R, Krause A, Salit J, Strulovici-Barel Y, Harvey BG, O'Connor TP, Crystal RG. Smoking-dependent reprogramming of alveolar macrophage polarization: implication for pathogenesis of chronic obstructive pulmonary disease. J Immunol. 2009;183:2867–2883. doi: 10.4049/jimmunol.0900473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Greenhill CJ, Gould J, Ernst M, Hertzog PJ, Mansell A, Jenkins BJ. LPS hypersensitivity of gp130 mutant mice is independent of elevated haemopoietic TLR4 signaling. Immunol Cell Biol. 2011;56:1–5. doi: 10.1038/icb.2011.56. [DOI] [PubMed] [Google Scholar]
- 35.Refaie S, Gagnon S, Gagnon H, Desjardins R, D'Anjou F, D'Orleans-Juste P, Zhu X, Steiner DF, Seidah NG, Lazure C, Salzet M, Day R. Disruption of PC1/3 expression in mice causes innate immune defects and uncontrolled cytokine secretion. J Biol Chem. 2012 doi: 10.1074/jbc.M111.323220. 2012 Mar 6. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yasukawa H, Ohishi M, Mori H, Murakami M, Chinen T, Aki D, Hanada T, Takeda K, Akira S, Hoshijima M, Hirano T, Chien KR, Yoshimura A. IL-6 induces an anti-inflammatory response in the absence of SOCS3 in macrophages. Nat Immunol. 2003;4:551–556. doi: 10.1038/ni938. [DOI] [PubMed] [Google Scholar]
- 37.Lehmann U, Schmitz J, Weissenbach M, Sobota RM, Hortner M, Friederichs K, Behrmann I, Tsiaris W, Sasaki A, Schneider-Mergener J, Yoshimura A, Neel BG, Heinrich PC, Schaper F. SHP2 and SOCS3 contribute to Tyr-759-dependent attenuation of interleukin-6 signaling through gp130. J Biol Chem. 2003;278:661–671. doi: 10.1074/jbc.M210552200. [DOI] [PubMed] [Google Scholar]
- 38.Palmer DC, Restifo NP. Suppressors of cytokine signaling (SOCS) in T cell differentiation, maturation, and function. Trends Immunol. 2009;30:592–602. doi: 10.1016/j.it.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Horn PJ, Peterson CL. Chromatin higher order folding–wrapping up transcription. Science. 2002;297:1824–1827. doi: 10.1126/science.1074200. [DOI] [PubMed] [Google Scholar]
- 40.Murphy KM, Stockinger B. Effector T cell plasticity: flexibility in the face of changing circumstances. Nat Immunol. 2010;11:674–680. doi: 10.1038/ni.1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ramgolam VS, Markovic-Plese S. Regulation of suppressors of cytokine signaling as a therapeutic approach in autoimmune diseases, with an emphasis on multiple sclerosis. J Signal Transduct. 2011;2011:635721. doi: 10.1155/2011/635721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grutkoski PS, Chen Y, Chung CS, Ayala A. Sepsis-induced SOCS-3 expression is immunologically restricted to phagocytes. J Leukoc Biol. 2003;74:916–922. doi: 10.1189/jlb.0303108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fang M, Dai H, Yu G, Gong F. Gene delivery of SOCS3 protects mice from lethal endotoxic shock. Cell Mol Immunol. 2005;2:373–377. [PubMed] [Google Scholar]
- 44.Shouda T, Yoshida T, Hanada T, Wakioka T, Oishi M, Miyoshi K, Komiya S, Kosai K, Hanakawa Y, Hashimoto K, Nagata K, Yoshimura A. Induction of the cytokine signal regulator SOCS3/CIS3 as a therapeutic strategy for treating inflammatory arthritis. J Clin Invest. 2001;108:1781–1788. doi: 10.1172/JCI13568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jo D, Liu D, Yao S, Collins RD, Hawiger J. Intracellular protein therapy with SOCS3 inhibits inflammation and apoptosis. Nat Med. 2005;11:892–898. doi: 10.1038/nm1269. [DOI] [PubMed] [Google Scholar]
- 46.Hilberath JN, Carlo T, Pfeffer MA, Croze RH, Hastrup F, Levy BD. Resolution of Toll-like receptor 4-mediated acute lung injury is linked to eicosanoids and suppressor of cytokine signaling 3. FASEB J. 2011;25:1827–1835. doi: 10.1096/fj.10-169896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mignon A, Rouquet N, Fabre M, Martin S, Pages JC, Dhainaut JF, Kahn A, Briand P, Joulin V. LPS challenge in D-galactosamine-sensitized mice accounts for caspase-dependent fulminant hepatitis, not for septic shock. Am J Respir Crit Care Med. 1999;159:1308–1315. doi: 10.1164/ajrccm.159.4.9712012. [DOI] [PubMed] [Google Scholar]
- 48.Lafdil F, Wang H, Park O, Zhang W, Moritoki Y, Yin S, Fu XY, Gershwin ME, Lian ZX, Gao B. Myeloid STAT3 inhibits T cell-mediated hepatitis by regulating T helper 1 cytokine and interleukin-17 production. Gastroenterology. 2009;137:2125–2135. doi: 10.1053/j.gastro.2009.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Horiguchi N, Lafdil F, Miller AM, Park O, Wang H, Rajesh M, Mukhopadhyay P, Fu XY, Pacher P, Gao B. Dissociation between liver inflammation and hepatocellular damage induced by carbon tetrachloride in myeloid cell-specific signal transducer and activator of transcription 3 gene knockout mice. Hepatology. 2010;51:1724–1734. doi: 10.1002/hep.23532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Babon JJ, Sabo JK, Soetopo A, Yao S, Bailey MF, Zhang JG, Nicola NA, Norton RS. The SOCS box domain of SOCS3: Structure and interaction with the elonginBC-cullin5 ubiquitin ligase. J Mol Biol. 2008;381:928–940. doi: 10.1016/j.jmb.2008.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li Y, Chu N, Rostami A, Zhang GX. Dendritic cells transduced with SOCS-3 exhibit a tolerogenic/DC2 phenotype that directs type 2 Th cell differentiation in vitro and in vivo. J Immunol. 2006;177:1679–1688. doi: 10.4049/jimmunol.177.3.1679. [DOI] [PubMed] [Google Scholar]
- 52.Ronn SG, Borjesson A, Bruun C, Heding PE, Frobose H, Mandrup-Poulsen T, Karlsen AE, Rasschaert J, Sandler S, Billestrup N. Suppressor of cytokine signalling-3 expression inhibits cytokine-mediated destruction of primary mouse and rat pancreatic islets and delays allograft rejection. Diabetologia. 2008;51:1873–1882. doi: 10.1007/s00125-008-1090-0. [DOI] [PubMed] [Google Scholar]
- 53.Chen YH, Hsieh SC, Chen WY, Li KJ, Wu CH, Wu PC, Tsai CY, Yu CL. Spontaneous resolution of acute gouty arthritis is associated with rapid induction of the anti-inflammatory factors TGFbeta1, IL-10 and soluble TNF receptors and the intracellular cytokine negative regulators CIS and SOCS3. Ann Rheum Dis. 2011;70:1655–1663. doi: 10.1136/ard.2010.145821. [DOI] [PubMed] [Google Scholar]