

Abstract
Alkylating agents are a frontline therapy for the treatment of several aggressive cancers including pediatric glioblastoma, a lethal tumor in children. Unfortunately, many tumors are resistant to this therapy. We sought to identify ways of sensitizing tumor cells to alkylating agents while leaving normal cells unharmed; increasing therapeutic response while minimizing toxicity. Using a siRNA screen targeting over 240 DNA damage response genes, we identified novel sensitizers to alkylating agents. In particular the base excision repair (BER) pathway, including 3-methylpurine-DNA glycosylase (MPG), as well as ataxia telangiectasia mutated (ATM) were identified in our screen. Interestingly, we identified MPG as a direct novel substrate of ATM. ATM-mediated phosphorylation of MPG was required for enhanced MPG function. Importantly, combined inhibition or loss of MPG and ATM resulted in increased alkylating agent-induced cytotoxicity in vitro and prolonged survival in vivo. The discovery of the ATM-MPG axis will lead to improved treatment of alkylating agent-resistant tumors.
Introduction
Identifying biological targets and pathways that lead to treatment resistance presents a major hurdle to developing effective cancer therapies. Cancer cells acquire or intrinsically harbor resistance to many frontline treatments(1). A major mechanism of cancer cell resistance is through activation of DNA repair pathways which reverse the cytotoxicity of many clinically used DNA-damaging agents, including alkylating agents. Therefore, targeting DNA repair pathways on which tumor cells, and not normal cells, are dependent could potentially overcome treatment resistance in many cancers.
In adults, the most common primary brain tumor is glioblastoma (GBM). Large clinical trials have demonstrated a survival benefit for GBM patients with the addition of the alkylating agent temozolamide (TMZ) over radiation alone(2, 3). TMZ is an oral alkylating agent that readily crosses the blood brain barrier making it an attractive drug to use clinically and has become part of the standard of care for adult GBM(4, 5). Interestingly, not all patients benefit from TMZ and current data suggests that GBM patients with promoter methylation and lack of O6-methylguanine–DNA methyltransferase (MGMT) expression do better than patients with an unmethylated MGMT promoter and MGMT expression. Alkylating agents, including TMZ, work by causing DNA damage – namely alkylation of O6-guanine, N7-guanine and N3-adenine residues on DNA. However, DNA repair mechanisms exist within cells that can reverse this damage. MGMT repairs the alkylated O6-guanine lesion(6) explaining why its expression may mediate resistance to TMZ. Importantly, many patients whose tumors do not express MGMT are nevertheless resistant to TMZ with the mechanism underlying this resistance unclear.
In children with GBM the use of TMZ is more controversial. A recent trial by the Children’s Oncology group demonstrated no survival benefit for patients receiving adjuvant TMZ over those receiving radiation alone(7). Thirty percent of high-grade gliomas in this study expressed MGMT and this was associated with worse progression-free survival. What may have been mediating resistance in the remaining cases is unclear. In other studies of pediatric GBM, TMZ appears to have little or no effect despite a lack of MGMT expression in many cases(8-10). Overall the mechanisms of treatment resistance in pediatric GBM are poorly understood. Recent data have highlighted the genetic differences between pediatric and adult GBM suggesting that novel strategies are required to identify effective therapeutics for children with GBM (11-13). We hypothesized that alternate, non-MGMT dependent, mechanisms of DNA repair and treatment resistance could explain why pediatric GBM fails to respond to alkylating agents. To identify molecular mechanisms of therapeutic resistance, we performed a siRNA screen targeting DNA damage response genes in the presence of sub-lethal doses of TMZ. Our results demonstrate that the MPG mediated base-excision repair (BER) pathway plays a major role in promoting resistance to TMZ in pediatric GBM and that loss of MPG results in an accumulation of unrepaired cytotoxic N3 methyladenine residues, which results in stalled DNA replication and cell death(14, 15). Furthermore, we demonstrate that the BER pathway specifically mediated by 3-methylpurine-DNA glycosylase (MPG) can be regulated by ataxia telangiectasia mutated (ATM) through direct phosphorylation of MPG. MPG is a DNA glycosylase responsible for initial recognition of the damaged DNA caused by TMZ, specifically alkylated N7 guanine and alkylated N3 adenine residues(16-18). By linking ATM signaling with MPG, we identified a novel axis that tumor cells may utilize to repair damage cause by alkylating agents. Importantly, targeting BER and ATM increases sensitivity to TMZ and prolongs survival in and in vivo model of pediatric GBM. By painting a broader landscape of the role of DNA damage response pathways in pediatric GBM than was previously appreciated, this study suggests novel therapeutic combinations to overcome treatment resistance.
Results
A siRNA screen identifies the base excision repair pathway as the major TMZ-sensitizer in pediatric GBM
Pediatric GBM lines were screened for TMZ sensitivity by calculating the half maximal inhibitory concentration (IC50) at days 3 and 7. Cells with an IC50> 100 uM were considered resistant as such doses are not physiologically achievable in TMZ-treated patients (19) (20, 21). Five out of six pediatric GBM lines showed TMZ resistance, and IC50s were comparable to adult GBM lines (Figure S1a). To identify the mechanism mediating this resistance, we performed a siRNA screen targeting over 240 DNA response genes in two of the TMZ resistant pediatric GBM cell lines, SJG2 and KNS42. Cells were incubated with or without TMZ (100 uM) in biological triplicates. Cell viability was assessed 72h post siRNA transfection. Data were normalized using the z-score method and significant TMZ sensitizers were determined using z-score cut off values of less than −1.65 (p < 0.05 in all three biological replicates; Figure 1a-b and Figure S1b-c).
Figure 1. An siRNA screen identifies base excision repair pathway members as sensitizers to TMZ in pediatric GBM.

a-b. SJG2 and KNS42 cells were transfected with a 240 DNA siRNA pool library and 24 h post transfections, cells were cultured with or without TMZ (100uM). Cell viability was assessed by the almarBlue cell viability assay 72 h post siRNA transfection. Data were normalized using the standard z-score method by correcting the raw data for plate to plate variation. Significance of potential TMZ sensitizers were determined using z-score cut off values of less than −1.65 (dotted line), which corresponded to a p value of 0.05 in all three biological replicates (rep1-3).
c. Venn diagram of genes common and unique to both cell lines from the siRNA screen.
d. Heat map of cell viability to validate target genes in TMZ and non-TMZ conditions using SJG2, KNS42, NHAs and normal neural stem cells (NSC). TMZ dose used was 100 uM and viability was assessed using almarBlue assay on day 3. *p<0.05 using ANOVA followed by a post-hoc Dunnett’s test. Each heatmap box represents the average of three independent experiments.
e. Immunoblotting to evaluate the protein expression levels of base excision repair pathway members in pediatric glioblastoma cell lines.
f. Immunofluorescence of MPG in SJG2 cells showing nuclear expression of the protein. Scale bar = 16um.
g. Pearson correlation plots of MPG (black) and MGMT (red) protein expression quantified by chemi luminescence densitometry versus IC50 values following 7 days of TMZ in pediatric GBM cell lines from supplemental figure 1a. Strong correlation is seen with MPG protein levels (r= 0.78 at 7 days) but not with MGMT levels (r= 0.03). All experiments were performed in triplicate, error bars represent standard error of the mean.
We identified 24 genes whose knockdown resulted in decreased cell viability in response to TMZ, 13 of which were common to both cell lines (Figure 1c). Interestingly this analysis revealed enrichment of genes involved in the base excision repair pathway (MPG, APEX1, APEX2 XRCC1, POLB), ATM (a master regulator of DNA repair) and LIG4 (involved in double strand break repair (DSBR) and non homologous end joining (NHEJ)). We confirmed knockdown at the RNA level (Figure S1d) and protein level for our top candidates (MPG, APEX1, LIG4 and ATM) (Figure S1e-h). We next rescreened our top candidates in pediatric GBM cells as well as fetal normal human astrocytes (NHA) and fetal neural stem cells (NSC) with our initial concentration of 100 uM at day 3 to test for toxicity (Figure 1d). We also rescreened at a lower dose (25 uM) of TMZ to evaluate the effect of gene knockdown at a low, clinically achievable, dose over a longer period of time (day 7), Figure S2A. ATF, TDG and DCLRE1 were eliminated as candidates as they were toxic to NHAs and NSCs and loss of these genes, even in the absence of TMZ, caused significant reduction in viability (Figure 1d and Figure S2a).
Next, we screened a series of pediatric GBM lines for endogenous expression of our top candidates. All six pediatric GBM lines demonstrated expression of APEX1, LIG4 and PARP1. MPG was not expressed in SF188 and RES259, both of which are sensitive to TMZ, while SJG2 cells expressed high levels of MPG and are TMZ resistant (Figure 1e). SJG2 and KNS42 cells expressed nuclear MPG and SF188 cells had no expression of MPG (Figure 1f and Figure S2b). In addition to high nuclear MPG, SJG2 cells expressed high MPG glycosylase activity whereas SF188 cells had no detectable MPG activity (Figure S2c). Furthermore, MPG protein expression measured in our pGBM cell lines correlated with the IC50 of TMZ measured on day 3 and day 7 (Figure 1g and Figure S2d). MGMT protein expression did not correlate with the IC50 values of TMZ in pediatric GBM cells and was not expressed in TMZ resistant pediatric GBM cells used in the screen (Figure 1e-g and Figure S2d). In addition to TMZ, MPG protein expression correlated with IC50 values for other alkylating agents including bis-chloroethylnitrosourea (BCNU/carmustine) and methyl methanesulfonate (MMS) (Figure S2e-f).
MPG is strongly expressed in pediatric GBM patients
To ensure the clinical relevance of our findings, we investigated the expression of MPG, base excision repair (BER) proteins (APEX1, APEX2, XRCC1), ATM and LIG4 in pediatric GBM operative samples compared to normal brain. Immunohistochemical (IHC) analysis of tissue microarrays containing 72 clinically annotated supratentorial pediatric GBMs from two independent centres demonstrated high nuclear staining in 48/72 (67%), cytoplasmic staining in 5/72 (7%) and negative staining in 19/72 (26%) patients’ tumors (Figure 2a). Kaplan-Meier survival analysis of the London data set (King’s College, UK) demonstrated patients with negative MPG had better overall survival (Figure 2b, p=0.02). For the entire Toronto dataset, which consisted of 41 heterogeneously treated patients, MPG negative status showed a trend toward survival benefit but was not statistically significant (p=0.14, Figure S2g). Interestingly, on pairwise analysis of the 16 patients in the SickKids cohort treated with an alkylating agent (TMZ or CCNU), MPG negative patients had a significantly better median survival than MPG positive patients (median survival was 3.5 years in the MPG negative group (n=4) vs 1.7 years in the MPG positive group (n=12), *p<0.038). However, these numbers are small and a prospectively TMZ-treated cohort from a clinical trial is needed to fully evaluate the benefit of using MPG and other targets from our screen as predictors of TMZ resistance.
Figure 2. Base excision repair genes are up-regulated in pGBM and adult gliomas.
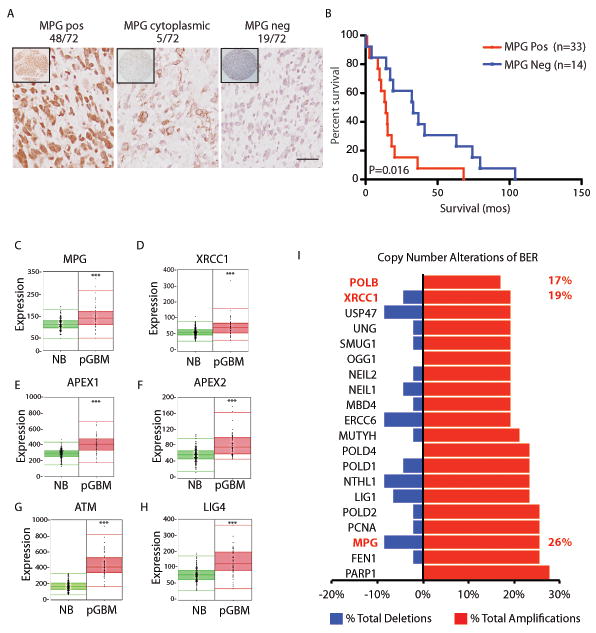
a. Immunohistochemistry analysis of 72 pediatric GBM samples demonstrating strong nuclear, cytoplasmic or negative staining. Scale bar =20um.
b. Kaplan Meir survival curve analysis of patients staining positive for MPG or negative. Log rank p=0.016.
c–h. RNA gene-expression analysis of base excision repair proteins downstream of MPG in pediatric GBM (pGBM, n=53) compared to normal human brain (NB, n=172). ***P<0.001.
i. Copy number alterations identify 20/40 BER DNA repair genes are significantly altered in 47 pediatric high grade gliomas. Gains were established as 2.5 more copies of tumor DNA per gene compared to matched normal controls and losses were established as 1.5 copies or less of tumor DNA compared to normal control tissue both corrected at a false discovery rate (q-value) of <5%.
We next explored gene-expression of MPG and several other components of BER at the mRNA expression level in pediatric GBM compared to normal brain in an additional gene-expression dataset of pediatric GBM (22, 23) (Figure 2c-f). We confirmed that BER proteins: MPG, APEX1, APEX2 and XRCC1 were significantly up-regulated compared to normal brain. ATM and LIG4 were also up-regulated in pediatric GBM compared to normal human brain (Figure 2g-h).
We next explored copy number alterations for all genes with a DNA repair gene ontology involved in BER (GO term: GO:0006284) on 47 of our genomically characterized high grade pediatric gliomas (13, 24). Analysis of 40 BER DNA repair genes revealed significant amplifications in 20/40 BER genes with MPG amplifications among the top 3 most amplified BER genes (Figure 2i, *p<0.05). In summary, the base-excision repair pathway, including MPG, is highly up-regulated at the RNA level, exhibits copy number gains and has high protein expression in pediatric high grade gliomas including pediatric GBM.
MPG modulates resistance to alkylating agents in pediatric GBM
To further characterize the role of MPG in TMZ resistance we generated stable loss of MPG in SJG2 and KNS42 cells by shRNA mediated gene silencing (Figure 3a). TMZ (50-500 uM) treatment led to a significant decrease in cell number in SJG2 and KNS42 cells with MPG knockdown versus scramble control cells (Figure 3b). In addition to reduced cell count, TMZ treatment of SJG2 and KNS42 with MPG knockdown compared to shRNA control cells led to increased DNA damage (increased γH2AX), and reduced DNA repair (increased alkaline comet tail length), Figure 3c and Figure S3a-d. Increased DNA damage and reduced DNA repair led to increased apoptosis in MPG knockdown cells compared to shRNA control cells as measured by activated Caspase 3/7 (Figure 3d, *p<0.05) and expression of cleaved PARP (Figure S3e). A colony forming unit assay (CFU) measured over 14 days demonstrated that both lines with MPG knockdown had reduced colony growth when grown in 100 uM TMZ (Figure 3e) or other alkylating agents (200 uM BCNU or 10 uM MMS, Figure S3f-g) compared to controls.
Figure 3. MPG loss sensitizes pGBM cells to alkylating agents.

a. Immunoblotting confirming MPG protein knockdown in pediatric GBM cell lines SJG2 and KNS42 following transfection with pooled stable clones generated from two unique MPG shRNA constructs.
b. Cell count of pediatric GBM cell lines SJG2 and KNS42 expressing MPG shRNA or shRNA controls (con) exposed to increasing doses of TMZ (0-500 uM). Cell counts were performed 72h post TMZ treatment.
c. Immunofluorescence of gamma H2AX in SJG2 cells expressing MPG shRNA or control (con) shRNA. Scale bar =16um.
d. Activated Cleaved caspase 3/7 assay of SJG2 and KNS42 cells treated with TMZ post 24h treatment. **p<0.01,***p<0.001
e. Colony forming unit (CFU) assay in SJG2 and KNS42 cells cultured with or without 100 uM TMZ. CFUs were counted after 14 days. **p<0.01 knockdown versus control cells.
f. Immunoblotting of MPG and LIG4 following siRNA treatment showing effective protein knockdown.
g-h. Plot of cell viability of SJG2 (g) and KNS42 (h) cells transfected with MPG, LIG4 or dual siRNA following exposure to varying concentrations of TMZ. Viability was measured using the almarBlue viability assay and quantified after 7 days. Dual siRNA knockdowns were also compared to single knockdowns to evaluate additive effects of double knockdown, *p<0.05,**p<0.01,***p<0.001.
Conversely, we re-expressed MPG in two low/negative MPG lines, RES259 and SF188 (Figure S4a). Stable expression of MPG increased resistance to TMZ at doses from 50-250 uM as measured by cell count (Figure S4b). Stable expressing MPG cells in the presence of TMZ had reduced DNA damage as measured by comet tail assay, γH2AX staining, increased CFU number, and decreased apoptosis as measured by activated caspase 3/7 and cleaved PARP (Figure S4c-h).
Targeting multiple BER pathway members and non-BER pathway remembers increases sensitivity to alkylating agents in pediatric GBM
We next tested whether targeting MPG with genes involved in non-BER pathways or multiple targeting of BER might lead to an additive sensitivity to TMZ. LIG4 is involved in double strand break repair (DSBR) and non-homologous break repair (NHEJ). Knockdown of LIG4, as confirmed by western blotting (Figure 3f), led to significantly reduced cell survival. The effect was additive with MPG loss at several doses of TMZ in SJG2 cells (Figure 3g-h, *p<0.05). Combining MPG knockdown with knockdown of ATM, a master controller for cell response to DNA damage and of genome stability, also led to an additive sensitivity to TMZ in SJG2 cells (Figure S5a-b, *p<0.05). Furthermore, knockdown of APEX1 or POLB, downstream BER effector proteins, led to a significant decrease in cell viability in response to TMZ vs scrambled siRNA controls (Figure S5a,c-d, *p<0.05). Combined loss of MPG with APEX1 but not POLB led to a further additive reduction in cell viability (Figure S5c-d, *p<0.05). Lastly, as MGMT is critical for repairing O6-methyl guanine damaged bases induced by TMZ, we next wanted to determine whether targeting MPG mediated BER in MGMT expressing cells could sensitize them to TMZ. siRNA mediated knockdown of either MGMT or MPG in RES186 and UW479 cells (normally express both MPG and MGMT) led to reduced cell viability when treated with several doses of TMZ (Figure S5e-g, *p<0.05). Combined loss of MPG and MGMT resulted in an additive reduction of cell viability (Figure S5f-g, *p<0.05).
In summary, targeting MPG mediated BER with non-BER pathways regulated by LIG4, MGMT and ATM or additional downstream BER proteins, including APEX1, results in additive cytotoxicity when cells are treated with TMZ.
MPG is phosphorylated and activated by ATM
Having determined that MPG modulates response to alkylating agents, we next attempted to elucidate novel regulators of MPG. Using an in silico kinase screen of MPG we identified several phosphorylation motifs (Table S1)(25). Interestingly, we identified ataxia telangiectasia mutated (ATM), a major effector of the DNA damage response, as a putative kinase of MPG. ATM itself was also a top validated target in our siRNA screen (Figure 1 and Figure S5). In silico analysis demonstrated that the ATM/ATR (ataxia telangiectasia and Rad3 related) phosphorylation site SQ (serine-glutamine, at serine 172) on MPG is highly conserved across several species (Figure 4a). We observed significant constitutively active phospho-ATM in our adult (T98G) and pediatric GBM cells (SJG2, KNS42 and RES259), which was further enhanced in the presence of TMZ (Figure 4b and Figure S6a for densitometric quantification). ATR, on the other hand, was only phosphorylated in the presence of TMZ and was not identified as a sensitizer to TMZ in our siRNA screen thus we proceeded to further investigate ATM.
Figure 4. MPG is a substrate of ATM and phosphorylation of MPG is essential for function.
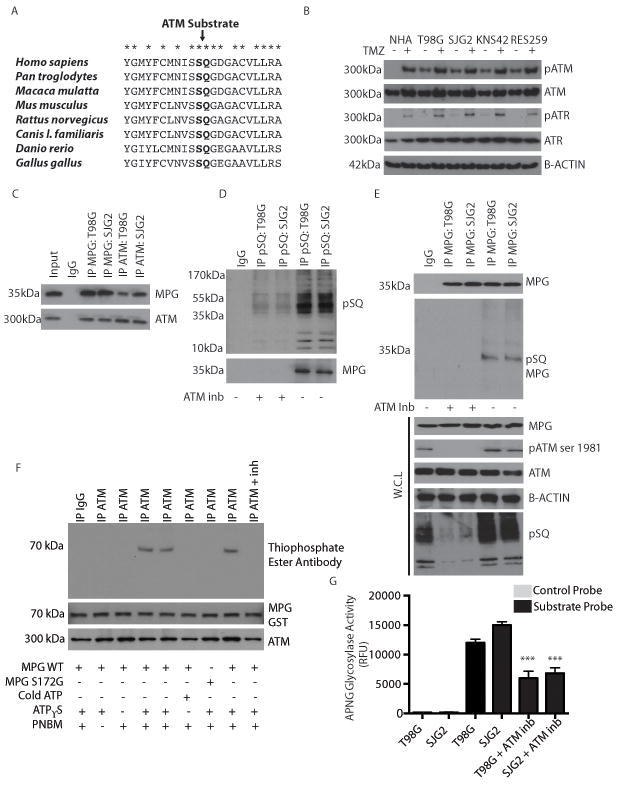
a. Primary amino acid sequence of MPG across species demonstrates that the pSQ ATM substrate residue is evolutionarily conserved.
b. Immunoblotting of ATM, phospho-ATM (pATM), ATR and phospho-ATR (pATR) in adult GBM (T98G) and pediatric GBM (SJG2, KNS42 and RES259) cell lines in the presence (+) and absence (-) of TMZ. Cells were treated with TMZ for 48h.
c. Immunoblots demonstrating co-immunoprecipitation of ATM and MPG in adult (T98G) and pediatric (SJG2) GBM cell lines. Both proteins are detected following immunoprecipitation (IP) of either ATM or MPG.
d. Immunoblots demonstrating detection of MPG following immunoprecipitation with a phospho-(serine/ threonine) (pSQ) ATM/ATR substrate antibody in both adult (T98G) and pediatric (SJG2) GBM cell lines in the absence (-) but not in the presence (+) of an ATM inhibitor (ATM inb, ku55933 used at 5uM).
e. Denaturing immunoprecipitation of MPG and immunoblotting of pSQ substrate specific antibody in presence and absence of ATM inhibitor to demonstrate MPG is phosphorylated at the SQ residue. W.C.L=whole cell lysate. ATM inhibitor ku55933 was used at 5uM
f. In vitro kinase ATM substrate assay. Immunoblots for thiophosphate ester to detect phosphate incorporation into MPG from a thiol labelled ATP analog in presence of ATM kinase. In vitro kinase assay was performed with wild-type MPG (MPG WT), mutant MPG (MPG S172G) in the presence (+) or absence (-) of cold ATP, ATPγS, PNBM or with an ATM inhibitor (ku55933, 5uM) demonstrating MPG is directly phosphorylated by ATM kinase at serine 172.
g. Fresh lysed whole cell lysates from e, were used for the MPG molecular beacon assay to confirm that inhibition of ATM resulted in reduced MPG glycosylase activity.
We next explored whether ATM and MPG were associated and if MPG was a direct target of ATM. Endogenous immunoprecipitations (IPs) of MPG from T98G cells (a well characterized model of TMZ resistance) and SJG2 cells were enriched for ATM compared to IgG controls while reverse IPs of ATM were enriched for MPG (Figure 4c). Interestingly, following treatment of the cells with an ATM inhibitor (ku55933) MPG was no longer enriched in IPs of phospho-SQ (ATM substrate site)(Figure 4d), suggesting the ATM phosphorylation is necessary for the interaction. In keeping with this, phospho-MPG was detected using the ATM phospho-specific SQ antibody after specific immunoprecipitation of MPG from a protein interaction dissociative buffer to enrich only for MPG protein in the absence, but not in the presence, of an ATM inhibitor (ku55933) (Figure 4e).
To complement our endogenous IPs, we transfected MPG-MYC tagged and ATM-FLAG tagged constructs into T98G and SJG2 GBM cells and observed exogenous ATM and MPG associated with each other (Figure S6b), similar to our endogenous IP results. Phosphorylation of our MPG-FLAG construct was ablated in cells treated with an ATM inhibitor (Figure S6c). Furthermore, a site directed mutant MPG-FLAG in which serine 172 (the putative ATM phosphorylation site) was replaced with glycine by site directed mutagenesis, was no longer phosphorylatable by ATM (Figure S6c). Phospho-MPG and phospho-ATM were only observed in normal human astrocytes when subjected to TMZ or radiation induced DNA damage (Figure S6d). Using an ATP-γ-thiol analog we performed an in vitro kinase assay with MPG purified using the Glutathione S-transferases (GST) tag system and immunoprecipitated ATM kinase to test whether ATM directly phosphorylates MPG. ATM phosphorylates MPG at serine 172 (Figure 4f). Removal of ATP-γ-thiol, use of cold ATP, performing the reaction in the presence of an ATM inhibitor or use of a S172G MPG-GST mutant causes loss of phospho-serine on MPG (Figure 4f). Inhibition of ATM by ku55933 (Figure 4e), directly reduced MPG glycosylase activity (Figure 4g, p<0.05) in both T98G and SJG2 cells. Treatment of pediatric GBM primary cultures (pGBM 462 and pGBM477) with ku55933 also significantly reduced MPG glycosylase activity (Figure S6e-f, *p<0.05). In summary, our results show that ATM and MPG are directly associated in vitro and in vivo, phosphorylation of MPG at serine 172 is dependent on ATM and that of loss of phospho ATM reduces MPG glycosylase activity.
Phosphorylation of MPG is required for optimal its function in DNA repair
We next generated wildtype MPG, catalytic-dead MPG (R182A), phospho-dead MPG (S172G) and phospho-mimetic (S172D) mutant constructs (Figure 5a). Transient transfection of these constructs in RES259 cells (TMZ sensitive cells) demonstrated that wild type MPG transfected cells but not MPG S172G or MPG R182A cells significantly increased MPG glycosylase activity, increased DNA damage repair (as measured by comet tail assay) and cell viability and significantly reduced apoptosis when compared to empty vector controls (Figure 5b-e). MPG S172D, a phospho-mimetic, was able to partially restore wild type function (Figure 5b-e). To test whether ATM-mediated MPG phosphorylation occurs in clinical pediatric GBM patient samples, we immunoprecipitated (IP) ATM/ATR substrates from 5 frozen, treatment naïve, pediatric GBM patient samples using a pSQ ATM/ATR substrate antibody. In 3/5 cases MPG protein was detected in ATM/ATR substrate pulldowns (Figure 5f). Furthermore, we performed a reverse IP from these lysates in 1% SDS to specifically enrich for MPG and no binding partners. We were able to immunopreciptated MPG in all 5 clinical samples and 3/5 were phosphorylated as detected by the phosphorylated SQ antibody (Figure 5g). In summary, phosphorylated MPG was detected in 60% (3/5) of our clinical pediatric GBM samples.
Figure 5. Phosphorylation of MPG is required for optimal its function in DNA repair.
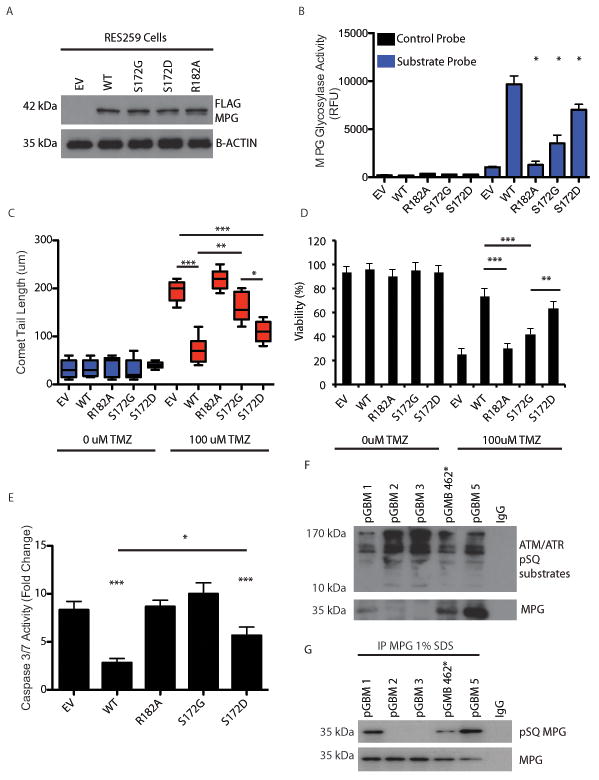
a. Immunoblot using anti-Flag antibody demonstrating robust expression of MPG flag tagged constructs in pediatric GBM cell line RES259. EV: empty vector, WT: wild type, S172G: MPG mutant in which the serine at residue 172 is replaced by glycine, S172D: MPG mutant in which the serine at residue 172 is replaced by aspartic acid, R182A: MPG mutant in which the arginine at residue 182 is replaced by alanine, B-actin: beta-actin.
b. Quantification of molecular beacon MPG activity assay after transfection of empty vector (EV), wild-type (WT) or mutant MPG constructs (R182A, S172G or S172D) in presence of TMZ (100uM).
c. Quantification (b) of comet tail assay (a) after transfection of empty vector (EV), wild-type (WT) or mutant MPG constructs (R182A or S172G) in presence (+) or absence (-) of TMZ (100uM).
d. Cell viability assay after transfection of wildtype or mutant MPG constructs in the presence or absence of TMZ at 48h.
e. Activated cleaved caspase 3/7 assay after transfection of wildtype or mutant MPG constructs in presence of TMZ (100uM).
f. Immunoprecipitation of ATM/ATR pSQ substrates in 5 frozen pGBM operative samples. Immuno precipitates were additionally probed for MPG and detected in 3/5 samples. IgG was used as a negative control.
g. Immunoprecipitation of MPG is 1% SDS followed by western blotting of pSQ from the 5 samples used in f to demonstrate that MPG is phosphorylated at the pSQ residue in clinical samples.
*P<0.05,**P<0.01***P<0.001. All experiments were performed in triplicate with mean and SEM reported where appropriate.
Combined loss of MPG and ATM cooperate to sensitize pediatric GBM cells to TMZ in vivo
To determine the effect of single or dual inhibition of ATM and MPG we generated stable knockdowns using shRNA in SJG2 cells lines (Figure 6a). The cells were injected into the frontal lobe of immunocompromised mice. After confirming tumor by MRI, mice were treated with TMZ (65mg/kg/5days) or vehicle (1% DMSO dissolved in PBS). There were no survival differences between control (shRNA scramble), single or dual knockdowns of ATM and/or MPG (Figure 6b) and the control PBS-treated mice. Similarly, there was no effect of TMZ treatment on mice harboring tumor cells with normal MPG and/or ATM levels (scramble shRNA control) confirming the resistance of SJG2 cells to TMZ in vivo (Figure 6c). Importantly, mice treated with TMZ harboring tumor cells with loss of MPG or ATM had significantly better survival than control mice. Further, mice harboring tumors with dual knockdown of ATM and MPG had the greatest survival when treated with TMZ (Figure 6c). Extended life correlated with reduced tumor cell proliferation as measured by Ki67 staining (Figure 6d) and knockdown of ATM and MPG was maintained at the time of sacrifice as measured by IHC analysis (Figure S7a).
Figure 6. Combined loss of MPG and ATM sensitizes pGBM cells to TMZ in vivo.

a. Immunoblotting of SJG2 cells demonstrating effective MPG, ATM or dual knockdown.
b. Kaplan Meir survival curve analysis of intracranial injected SJG2 cells expressing control shRNA, MPG, ATM or dual knockdown into NOD-SCID mice.
c. Kaplan Meir survival curve analysis of intracranial injected SJG2 cells expressing control shRNA, MPG, ATM or dual knockdown into NOD-SCID mice treated with TMZ (65 mg/kg/5 days).
d. Quantification of ki67 staining in mice (n=3 mice per group) from c.
e. Kaplan Meir survival curve analysis of intracranial injected SJG2 into NOD-SCID mice treated with vehicle, TMZ (65mg/kg/5days), Methoxyamine (MA 100mg/kg/5days) and both TMZ+MA. Mice were treated upon confirmation of tumour by T2-MRI.
f. Quantification of ki67 staining in mice (n=3 mice per group) from e.
g. Kaplan Meir survival curve analysis of an orthotopic patient derived xenograft (PDX) mouse model from pGBM 462 cells. 20 mice were injected with 5 per each treatment arm: Vehicle, TMZ (65mg/kg/2 weeks), Methoxyamine (MA 100mg/kg/2 weeks) and both TMZ+MA (65mg/kg TMZ and MA 100mg/kg for 2 weeks). Mice were treated upon confirmation of tumour by T2-MRI.
h. Hematoxylin and eosin stain (H&E stain) of represented tumors mice from each treatment arm confirming high grade glioma/GBM morphology. Scale bar, Large insets at 500um and small magnified insets at 25um. N denotes normal mouse brain and T denotes xenograft tumor growth.
Methoxyamine, an inhibitor of base excision repair, sensitizes pediatric GBM to TMZ in vitro and in vivo
Methoxyamine (MA) is a potent inhibitor of base excision repair and works by binding and inhibiting further repair of abasic sites generated by MPG. To complement our genetic manipulation of the BER pathway through knockdown of ATM and MPG, we hypothesized that MA could also overcome TMZ resistance of pediatric GBM. Methoxyamine (MA) was able to sensitize pediatric (SJG2) GBM cells in vitro to TMZ and induce apoptosis (Figure S7b-c). Knockdown of MPG, ATM or both in SJG2 cells increased cell death when treated with TMZ alone but abolished the ability of MA to further sensitize cells to TMZ compared to control siRNA treated cells, suggesting that MA’s effect maybe mediated through MPG and ATM (Figure S7c). Similar results were observed in adult GBM cell lines U87 and T98G (data not shown). To test the in vivo effect of MA we implanted SJG2 pediatric GBM cells into the forebrains of immunocompromised mice. After MRI confirmation of tumor, mice were randomized into four treatment arms: vehicle, TMZ (65mg/kg), MA (100mg/kg) or TMZ+MA (65mg/kg TMZ +100mg/kg MA) for two weeks. Compared to vehicle treated cells, only the dual treated mice had a significant increase in survival (~ doubling of overall survival, p<0.001, Figure 6e). Increased survival correlated with decreased tumor cell proliferation (p<0.01, Figure 6f). To complement our cell line work we tested primary low passage adult (G179) and pediatric (pGBM 462 generated from samples in Figure 5f) GBM patient derived cultures established from newly resected tumors maintained in defined neural stem cell media. These cells expressed both ATM and MPG (Figure S7d). Methoxyamine sensitized primary adult GBM cultures to TMZ treatment leading to reduced cell viability (Figure S7e). Dual loss of ATM and MPG, but not single loss, reduced cell viability in pGBM462 (Figure S8a), which was further enhanced by TMZ (Figure S8b). To complement our gene silencing of MPG and ATM, dual treatment of TMZ and MA but not vehicle or single agents reduced pGBM462 cell viability (Figure S8c) and increased apoptosis (cleaved caspase activity, Figure S9a). Surprisingly, single knockdowns of ATM, MPG or dual knockdowns did not reduce viability in normal neural fetal stem cells in the absence or presence of TMZ (Figure S8d-e). Similarly, dual treatment of MA and TMZ did not reduce viability (Figure S8f) or increase apoptosis (cleaved caspase activity, Figure S9a) in normal fetal neural stem cells compared to controls.
We next generated a patient derived xenograft (PDX) model of pediatric GBM from our primary culture of pGBM462 cells as another in vivo orthotopic model to complement our in vivo work using SJG2 cells. Mice were treated with vehicle, TMZ, MA and dual treatments (TMZ+MA) for two weeks. Mice receiving dual treatment but not vehicle or single treatments had a significant increase in survival of approximately 40% with H&E confirming our xenograft model retained the features of pGBM including hyper-cellularity and an invasive edge (Figure 6g-h, *p<0.01).
Functional redundancy in DNA repair pathways protects normal cells from damage when targeting the ATM-MPG axis
In both our siRNA screen and subsequent functional work we observed that normal cells including glial cells and neural stem cells were resistant to depletion of the ATM-MPG axis. We hypothesized that normal CNS cells have several intact DNA repair pathways and therefore functional redundancy protects them, whereas cancer cells by definition have alterations, mutations, and deletions in their DNA repair machinery making them susceptible. To test this we generated MPG stable knockdown normal human astrocytes by shRNA and knocked down ATM by transient siRNA (Figure 7a). In keeping with our siRNA knockdown data (Figure 1d) we did not observe any significant decrease in viability in either single or dual knockdown cells compared to controls when treated with TMZ (Figure 7b). Similarly, cell viability of normal human astrocytes was not affected by treatment with TMZ and MA compared to controls (Figure S9b).
Figure 7. Model of ATM-MPG mediated therapeutic resistance in pGBM.

a. Western blot analysis demonstrating MPG knockdown by retroviral shRNA transduction and ATM knockdown by siRNA at 72h. Control cells were transduced with shRNA control retrovirus and treated with siRNA scramble controls.
b. Control or double MPG and ATM knockdown astrocytes were treated with 100uM TMZ and assessed for viability at 72h. ns = non significance.
c. Control or double MPG and ATM knockdown astrocytes were targeted by siRNA for DNA damage response genes redundant to either ATM or MPG and treated with 100uM TMZ and assessed for viability at 72h. All experiments were performed in triplicate with the standard error of the mean reported.
d. Summary model of targeting the base excision and ATM pathways for sensitization to alkylating agents.
To address the issue of functional redundancy, we silenced additional genes redundant to ATM or MPG by siRNA to generate triple knockdowns. Loss of genes redundant to ATM, namely ATR/TP53 and CHEK2, in addition to ATM and MPG, significantly reduced normal human astrocyte cell viability in the presence of TMZ (Figure 7c, column 4 of heatmap). Furthermore, loss of additional genes in the BER pathway downstream of MPG, namely APEX1/PARP1/XRCC1 or MGMT (which repairs other DNA lesions induced by TMZ) significantly reduced normal human astrocyte cell viability when combined with ATM and MPG loss in the presence, but not the absence, of TMZ (Figure 7c, column 4). Single loss of ATR, PARP1, CHEK2, XRCC1, MGMT and APEX1 did not result in reduced viability when treated with or without TMZ (Figure 7c, columns 1-2). Interestingly, loss of ATR/CHEK2/TP53 but not BRCA1 in combination with TMZ and MA significantly reduced cell viability of normal astrocytes (Figure S9c). Additionally, loss of APEX1, PARP1 and XRCC1 genes downstream of MPG also reduced cell viability when combined with TMZ and MA compared to controls (Figure S9c). Lastly, loss of MGMT, which repairs cytotoxic lesions distinct from those repaired by MPG also reduced cell viability when combined with TMZ and MA (Figure S9c). In summary, normal human astrocytes have significant redundant DNA repair pathways protecting them from alkylating agent induced cell death.
Discussion
Pediatric GBM is a devastating disease with poor survival. Our current approaches have thus far failed to effectively treat these tumors. This may be due, in part, to a flaw in our therapeutic strategies. Many pediatric GBM treatments are based on adult data and it is now clear that pediatric and adult GBM are distinct entities (12, 26). Pediatric GBM, in most cases, fails to respond to alkylating agent therapy leading us to hypothesize that they may express pathways that mediate this resistance. Finding effective sensitizers to TMZ would allow for more effective treatment and use of less toxic doses(27). Furthermore, using monotherapies to treat pediatric GBM may be futile and multimodality therapies, which show synergism and low toxicity would be advantageous. Here, using a siRNA screening approach, we identified multiple members of the BER pathway mediating alkylating agent resistance in pediatric GBM. The average plasma concentration of TMZ in adult GBM patients is ~72uM, and is thought to be similar in pediatric patients(28, 29). We performed our initial screen at 100uM TMZ, thus it is possible that we may have not identified the full spectrum of mediators of TMZ resistance(19). Nevertheless, when tested with 25uM TMZ, a clinically achievable, low dose of TMZ, inhibition of the BER pathway continued to sensitize pediatric GBM to TMZ therapy and the combination of BER-inhibition with TMZ led to increased survival of mice bearing pediatric GBM tumors.
Modulation of the MPG mediated BER pathway has been shown to promote both sensitivity and resistance to alkylating agents, demonstrating how tissue specificity and cancer type must be carefully evaluated before targeting the BER pathway(30-34). We observed that modulation of MPG, the first step of TMZ induced BER, can promote sensitivity to TMZ when lost, or resistance when re-expressed in pediatric GBM. A similar finding has been seen in adult GBM in cases where MGMT, a common mechanism of TMZ resistance in these tumors, could not explain resistance to TMZ in vitro and in vivo (30, 35). However, forced over-expression of MPG in pediatric GBM cells already expressing MPG may be toxic as it may lead to accumulation of BER toxic intermediates that downstream BER enzymes cannot quickly resolve (31, 36).
The crystal structure of MPG has been identified, but thus far there are no effective small molecule inhibitors of this glycosylase (17). MPG contains more disordered regions in the active site that are needed for substrate specificity, thus it may be difficult to develop structure-based rational drug design targeting MPG(37, 38). However, if MPG cannot be targeted directly, the BER pathway may be therapeutically targeted through use of methoxyamine (MA), a small molecule inhibitor that binds to 3’ hydroxyl groups that are left behind by MPG following excision of the damaged base and thus inhibits BER activity (5, 39, 40). Of great interest is that loss of MPG or other BER pathway members in normal astrocytes and neural stem cells did not promote cytotoxicity. This suggests that targeting BER should have limited toxicity to normal brain. We did not directly test whether targeting BER may be toxic to non-CNS tissues, although mice did not suffer systemic side-effects during their treatment in this study. Redundant and proficient DNA repair pathways in normal cells may limit their toxicity while allowing targeting of tumor cells. In accordance with this, we observed that loss of additional DNA repair proteins in normal humans astrocytes, including genes in the ATM/TP53/CHEK2 pathway or BER pathway (APEX1,PARP1,XRCC1), sensitized these cells to TMZ and led to reduced cell viability when MPG and ATM were also targeted either by siRNA knockdown or by treatment with MA. Similar screening strategies were used to identify PARP inhibitors as effective therapeutics in several cancers where tumor cells with BRCA1/2 mutations were sensitive to PARP inhibition but not normal tissue (41, 42). Of interest pediatric GBMs frequently harbor mutations in DNA repair proteins, TP53 and ATRX (11, 13) suggesting that targeting of the ATM-MPG axis is of therapeutic relevance. However, it should be noted that patients with underlying germline conditions where DNA repair proteins are mutated such as Li-Fraumeni (mutations in TP53), or Seckel syndrome (mutations in Ataxia telangiectasia and Rad3 (ATR)) maybe not benefit from targeting MPG and ATM as it may result in severe toxicity to normal tissues or even secondary malignancies.
Cellular responses to DNA damage are mediated by a number of master protein kinases and at the core of these signaling pathways are ATM (ataxia telangiectasia mutated) and ATR (ATM and Rad3-related). Constitutively active ATM may arise through persistent double stranded breaks or oxidative stress observed in many cancers, including pediatric GBM, and can initiate cross talk between DNA repair pathways(43-46). Interestingly, serine 172, the target of ATM-mediated MPG phosphorylation, is in between tyrosine 162 and arginine 182, both critical amino acids for MPG substrate specificity and catalytic domain function(17). Phosphorylation of serine 172 may be important for maintaining the domain structure or function of MPG mediated base excision. Tumor cells may become dependent on this pathway for survival in the face of cellular stress making it a prime therapeutic target. Loss or inhibition of ATM is known to sensitize tumor cells to TMZ but the mechanism is unknown. Our results suggest that ATM inhibitors, at least in part, may lead to TMZ sensitization through BER pathway inhibition. ATM-mediated phosphorylation of MPG and its associated increased activity would be blocked by ATM-inhibitors leading to reduced base excision repair. Combination with downstream BER-inhibitors, such as methoxyamine, leads to even greater TMZ sensitivity in pediatric GBM (Figure 7d) and potentially other cancers showing alkylating agent resistance (47). In addition to identifying a novel function for ATM, our study demonstrates the role of BER in pediatric GBM resistance to alkylating agents and a direct targetable pathway that may lead to more effective treatments.
Materials and Methods
Human samples and Study approval
Pediatric GBM biopsy samples and matching clinical data were obtained from the Hospital for Sick Children following approval by the institutional Research Ethics Board(48). All samples were de-identified prior to analysis. 35/41 patients were given chemotherapy with 16/35 having received TMZ. Additional GBM tissue microarrays were obtained from London Cancer Institute (Dr Chris Jones) with a material transfer agreement.
In vivo mouse work
Mice were maintained in accordance with UHN institutional animal protocols. Stereotactic guided intracranial injections in NOD-SCID mice were performed by injecting 250,000 SJG2 cells stably expressing control shRNA, MPG shRNA, ATM shRNA or dual MPG/ATM shRNA. Injections were made into the frontal cortex (Coordinates were as follows X=-1.0, Y=1.5, Z=2.4, with Bregma serving as the 0 point for X and Y). Mice were treated with vehicle (PBS) or temozolomide 65mg/kg daily for 5 days. For methoxyamine experiments, mice were injected with parental SJG2 cells as described above. Two weeks after tumour confirmation by MRI, mice were given temozolomide, methoxyamine, both or vehicle by oral gavage (65mg/kg TMZ daily for 2 weeks (weekends off) and/or methoxyamine 100mg/kg daily for 2 weeks (weekends off)) and sacrificed upon signs of sickness. T2-MRI imaging was used to confirm tumor formation as previously published. PDX pGBM 462 model was derived by generating a primary culture from a pGBM patient. 1 × 105 cells were injected and treatment commenced on day 10. Mice were given temozolomide, methoxyamine, both or vehicle by oral gavage (65mg/kg TMZ daily for 2 weeks (weekends off) and/or methoxyamine 100mg/kg daily for 2 weeks (weekends off)) and sacrificed upon signs of sickness Animal use protocols (AUPs) were approved by the University Health Network (UHN) animal care committee (ACC) under AUP 884.14 and AUP1191.13.
siRNA screen
2500 SJG2 pediatric GBM and KNS42 pediatric GBM cell lines were plated in black 96 well plates. 12h post plating, SJG2 and KNS42 cells were transfected with a 240 DNA siRNA pool library (Thermo scientific Cat#G-006005-025) and 24h post transfections, cells were cultured with TMZ (100uM). Cell viability was assessed by the almarBlue cell viability assay (Resazurin viability dye, Invitrogen, Cat# DAL1025) 72h post siRNA transfection (48h post TMZ). Data were normalized using the standard z-score method by correcting the raw data for plate to plate variation(49, 50). Significance of potential TMZ sensitizers was determined using z-score cut off values of less than −1.65, which corresponded to a p value of 0.05 in all three biological replicates.
Tissue culture and drugs
T98G cell lines were obtained from American Type Culture Collection (ATCC, Manassas, VA). SF188, KNS42, RES186, RES259, UW479 cells were obtained from Dr. Chris Jones (London, Institute of Cancer Research) (27, 51). All cells have been extensively characterized previously, and were authenticated by short tandem repeat (STR) profiling in 2011. The cells were grown in DMEM F12 containing 10% FBS (DMEM/FBS) at 37°C in a 95% air/5% CO2 atmosphere. Pediatric primary cultures and fetal normal neural stem cell lines maintained in stem like media were provided by Dr. Peter Dirks and cultured as previously demonstrated(35, 52). Cells were exposed to varying concentrations and durations of TMZ (Sigma), methyl methanesulfonate (MMS, Sigma, St. Louis, MO, USA cat#M4016), N,N’-Bis(2-chloroethyl)-N-nitrosourea (BCNU, Sigma, St. Louis, MO, USA), methoxyamine (Sigma Cat# 226904), and ATM inhibitor ku55933 (Selleck chemicals, Houston TX, Cat# S1092) as described in the results and figure legends.
Copy number analysis and gene-expression dataset analysis
Gene-expression for pGBM and normal brain was obtained and processed from the following datasets(22, 23). Using the R2 software, we analyzed candidate gene expression levels in primary high-grade glioma samples (WHO grade III, n=16; WHO grade IV, n=37), and compared gene expression patterns to normal brain tissues (n=172). The data is deposited in the Gene-expression Omnibus (GEO) with the following accession numbers: gse19578, gse11882.
DNA from forty-three paediatric high-grade astrocytomas and 4 high-grade glioma cell lines were hybridized to Affymetrix 250K Nsp arrays (Santa Clara, CA) as previously reported (13, 24). Sample preparation, which included DNA extraction, digestion and labelling, was performed as directed by the manufacturer’s protocol. CEL data was analyzed using in Partek Genomics Suite version 6.6 (Partek Incorporated, St. Louis, MO). Paired samples were normalized to their matched non-neoplastic control. Unpaired sample copy number was normalized to HapMap. Genomic segmentation analysis tool was used at default parameters. A total of 22,721 genes among all 47 samples had significant copy number alterations; gains (2.5 copies +) or losses (1.5 copies -) compared to controls (p<0.05). Genes associated with DNA repair pathways (432 genes) were selected for by gene ontology terms - GO:0006281 (“DNA repair”) and under child terms for SSBR, NER, HR, BER, MMR. Of these, 377 were represented in the segmentation data. We next extracted all genes involved in base excision repair (n=40) (BER) and significant alteration was identified. Frequency of samples with gains or deletions was enumerated for each BER gene. The data has been deposited into geo with the following accession numbers: GSE18828, GSE59678.
Statistical analysis
All experiments were performed in triplicate with mean and standard error of the mean reported where appropriate. Analysis of variance (ANOVA) was conducted for multi-group comparisons followed by a post-hoc Dunnett’s test (groups compared to one control group) or post-hoc Tukey’s test (to identify differences among sub-groups). Where appropriate, direct comparisons were conducted using an unpaired two-tailed Student’s t-test. *Significance was p<0.05, **Significance was p<0.01, ***Significance was p<0.001.
Supplementary Material
Significance.
Inhibition of ATM and MPG mediated BER cooperate to sensitize tumor cells to alkylating agents, impairing tumor growth in vitro and in vivo with no toxicity to normal cells providing an ideal therapeutic window.
Acknowledgments
This study was funded by Canadian Institutes of Health Research grants (CIHR MOP 102513 and 115004) to C. Hawkins. RWS was supported by a grant from the National Institute of Health (NIH): CA148629. MR was funded by the Dr. Mildred Scheel foundation (German Cancer Aid). We would like to thank Dr. Geoff Margison at the Paterson Institute for Cancer Research for input and evaluation of the manuscript. For MRI images we would like to thank Dr. Warren Foltz and the Spatio-Temporal Targeting and Amplification of Radiation Response (STTARR) program and its affiliated funding agencies. The MPG-GST construct was a generous gift from Dr. Mitsuyoshi Nakao (Institute of Molecular Embryology and Genetics Kumamoto University). We would also like to thank Dr. Michael Kastan for the ATM construct (Duke University, School of Medicine). Both MPG-GST and ATM constructs were obtained by a materials transfer agreement (MTA).
Footnotes
Please see Supplemental data for Expanded details of Materials and Methods
The authors have no conflicts or disclosures to declare
References
- 1.Fu D, Calvo JA, Samson LD. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nature reviews Cancer. 2012;12:104–20. doi: 10.1038/nrc3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 3.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–96. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 4.Denny BJ, Wheelhouse RT, Stevens MF, Tsang LL, Slack JA. NMR and molecular modeling investigation of the mechanism of activation of the antitumor drug temozolomide and its interaction with DNA. Biochemistry. 1994;33:9045–51. doi: 10.1021/bi00197a003. [DOI] [PubMed] [Google Scholar]
- 5.Hang B, Singer B, Margison GP, Elder RH. Targeted deletion of alkylpurine-DNA-N-glycosylase in mice eliminates repair of 1,N6-ethenoadenine and hypoxanthine but not of 3,N4-ethenocytosine or 8-oxoguanine. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:12869–74. doi: 10.1073/pnas.94.24.12869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weller M, Stupp R, Reifenberger G, Brandes AA, van den Bent MJ, Wick W, et al. MGMT promoter methylation in malignant gliomas: ready for personalized medicine? Nat Rev Neurol. 2010;6:39–51. doi: 10.1038/nrneurol.2009.197. [DOI] [PubMed] [Google Scholar]
- 7.Cohen KJ, Pollack IF, Zhou T, Buxton A, Holmes EJ, Burger PC, et al. Temozolomide in the treatment of high-grade gliomas in children: a report from the Children’s Oncology Group. Neuro-oncology. 2011;13:317–23. doi: 10.1093/neuonc/noq191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Broniscer A, Chintagumpala M, Fouladi M, Krasin MJ, Kocak M, Bowers DC, et al. Temozolomide after radiotherapy for newly diagnosed high-grade glioma and unfavorable low-grade glioma in children. Journal of neuro-oncology. 2006;76:313–9. doi: 10.1007/s11060-005-7409-5. [DOI] [PubMed] [Google Scholar]
- 9.Lashford LS, Thiesse P, Jouvet A, Jaspan T, Couanet D, Griffiths PD, et al. Temozolomide in malignant gliomas of childhood: a United Kingdom Children’s Cancer Study Group and French Society for Pediatric Oncology Intergroup Study. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2002;20:4684–91. doi: 10.1200/JCO.2002.08.141. [DOI] [PubMed] [Google Scholar]
- 10.Ruggiero A, Cefalo G, Garre ML, Massimino M, Colosimo C, Attina G, et al. Phase II trial of temozolomide in children with recurrent high-grade glioma. Journal of neuro-oncology. 2006;77:89–94. doi: 10.1007/s11060-005-9011-2. [DOI] [PubMed] [Google Scholar]
- 11.Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482:226–31. doi: 10.1038/nature10833. [DOI] [PubMed] [Google Scholar]
- 12.Sturm D, Witt H, Hovestadt V, Khuong-Quang DA, Jones DT, Konermann C, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer cell. 2012;22:425–37. doi: 10.1016/j.ccr.2012.08.024. [DOI] [PubMed] [Google Scholar]
- 13.Khuong-Quang DA, Buczkowicz P, Rakopoulos P, Liu XY, Fontebasso AM, Bouffet E, et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta neuropathologica. 2012;124:439–47. doi: 10.1007/s00401-012-0998-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fronza G, Gold B. The biological effects of N3-methyladenine. J Cell Biochem. 2004;91:250–7. doi: 10.1002/jcb.10698. [DOI] [PubMed] [Google Scholar]
- 15.Iyer P, Srinivasan A, Singh SK, Mascara GP, Zayitova S, Sidone B, et al. Synthesis and characterization of DNA minor groove binding alkylating agents. Chemical research in toxicology. 2013;26:156–68. doi: 10.1021/tx300437x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Engelward BP, Dreslin A, Christensen J, Huszar D, Kurahara C, Samson L. Repair-deficient 3-methyladenine DNA glycosylase homozygous mutant mouse cells have increased sensitivity to alkylation-induced chromosome damage and cell killing. The EMBO journal. 1996;15:945–52. [PMC free article] [PubMed] [Google Scholar]
- 17.Lau AY, Scharer OD, Samson L, Verdine GL, Ellenberger T. Crystal structure of a human alkylbase-DNA repair enzyme complexed to DNA: mechanisms for nucleotide flipping and base excision. Cell. 1998;95:249–58. doi: 10.1016/s0092-8674(00)81755-9. [DOI] [PubMed] [Google Scholar]
- 18.Ye N, Holmquist GP, O’Connor TR. Heterogeneous repair of N-methylpurines at the nucleotide level in normal human cells. J Mol Biol. 1998;284:269–85. doi: 10.1006/jmbi.1998.2138. [DOI] [PubMed] [Google Scholar]
- 19.Ostermann S, Csajka C, Buclin T, Leyvraz S, Lejeune F, Decosterd LA, et al. Plasma and cerebrospinal fluid population pharmacokinetics of temozolomide in malignant glioma patients. Clinical cancer research : an official journal of the American Association for Cancer Research. 2004;10:3728–36. doi: 10.1158/1078-0432.CCR-03-0807. [DOI] [PubMed] [Google Scholar]
- 20.Baker SD, Wirth M, Statkevich P, Reidenberg P, Alton K, Sartorius SE, et al. Absorption, metabolism, and excretion of 14C-temozolomide following oral administration to patients with advanced cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 1999;5:309–17. [PubMed] [Google Scholar]
- 21.Hammond LA, Eckardt JR, Baker SD, Eckhardt SG, Dugan M, Forral K, et al. Phase I and pharmacokinetic study of temozolomide on a daily-for-5-days schedule in patients with advanced solid malignancies. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 1999;17:2604–13. doi: 10.1200/JCO.1999.17.8.2604. [DOI] [PubMed] [Google Scholar]
- 22.Paugh BS, Qu C, Jones C, Liu Z, Adamowicz-Brice M, Zhang J, et al. Integrated molecular genetic profiling of pediatric high-grade gliomas reveals key differences with the adult disease. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2010;28:3061–8. doi: 10.1200/JCO.2009.26.7252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Berchtold NC, Cribbs DH, Coleman PD, Rogers J, Head E, Kim R, et al. Gene expression changes in the course of normal brain aging are sexually dimorphic. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:15605–10. doi: 10.1073/pnas.0806883105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zarghooni M, Bartels U, Lee E, Buczkowicz P, Morrison A, Huang A, et al. Whole-genome profiling of pediatric diffuse intrinsic pontine gliomas highlights platelet-derived growth factor receptor alpha and poly (ADP-ribose) polymerase as potential therapeutic targets. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2010;28:1337–44. doi: 10.1200/JCO.2009.25.5463. [DOI] [PubMed] [Google Scholar]
- 25.Amanchy R, Periaswamy B, Mathivanan S, Reddy R, Tattikota SG, Pandey A. A curated compendium of phosphorylation motifs. Nature biotechnology. 2007;25:285–6. doi: 10.1038/nbt0307-285. [DOI] [PubMed] [Google Scholar]
- 26.Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gaspar N, Marshall L, Perryman L, Bax DA, Little SE, Viana-Pereira M, et al. MGMT-independent temozolomide resistance in pediatric glioblastoma cells associated with a PI3-kinase-mediated HOX/stem cell gene signature. Cancer research. 2010;70:9243–52. doi: 10.1158/0008-5472.CAN-10-1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gururangan S, Fisher MJ, Allen JC, Herndon JE, 2nd, Quinn JA, Reardon DA, et al. Temozolomide in children with progressive low-grade glioma. Neuro-oncology. 2007;9:161–8. doi: 10.1215/15228517-2006-030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nicholson HS, Krailo M, Ames MM, Seibel NL, Reid JM, Liu-Mares W, et al. Phase I study of temozolomide in children and adolescents with recurrent solid tumors: a report from the Children’s Cancer Group. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 1998;16:3037–43. doi: 10.1200/JCO.1998.16.9.3037. [DOI] [PubMed] [Google Scholar]
- 30.Agnihotri S, Gajadhar AS, Ternamian C, Gorlia T, Diefes KL, Mischel PS, et al. Alkylpurine-DNA-N-glycosylase confers resistance to temozolomide in xenograft models of glioblastoma multiforme and is associated with poor survival in patients. The Journal of clinical investigation. 2012;122:253–66. doi: 10.1172/JCI59334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fishel ML, He Y, Smith ML, Kelley MR. Manipulation of base excision repair to sensitize ovarian cancer cells to alkylating agent temozolomide. Clinical cancer research : an official journal of the American Association for Cancer Research. 2007;13:260–7. doi: 10.1158/1078-0432.CCR-06-1920. [DOI] [PubMed] [Google Scholar]
- 32.Ito M, Ohba S, Gaensler K, Ronen SM, Mukherjee J, Pieper RO. Early Chk1 phosphorylation is driven by temozolomide-induced, DNA double strand break- and mismatch repair-independent DNA damage. PloS one. 2013;8:e62351. doi: 10.1371/journal.pone.0062351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tang JB, Svilar D, Trivedi RN, Wang XH, Goellner EM, Moore B, et al. N-methylpurine DNA glycosylase and DNA polymerase {beta} modulate BER inhibitor potentiation of glioma cells to temozolomide. Neuro-oncology. 2011 doi: 10.1093/neuonc/nor011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goellner EM, Grimme B, Brown AR, Lin YC, Wang XH, Sugrue KF, et al. Overcoming temozolomide resistance in glioblastoma via dual inhibition of NAD+ biosynthesis and base excision repair. Cancer research. 2011;71:2308–17. doi: 10.1158/0008-5472.CAN-10-3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Agnihotri S, Wolf A, Munoz DM, Smith CJ, Gajadhar A, Restrepo A, et al. A GATA4-regulated tumor suppressor network represses formation of malignant human astrocytomas. J Exp Med. 2011;208:689–702. doi: 10.1084/jem.20102099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rinne M, Caldwell D, Kelley MR. Transient adenoviral N-methylpurine DNA glycosylase overexpression imparts chemotherapeutic sensitivity to human breast cancer cells. Molecular cancer therapeutics. 2004;3:955–67. [PubMed] [Google Scholar]
- 37.Setser JW, Lingaraju GM, Davis CA, Samson LD, Drennan CL. Searching for DNA lesions: structural evidence for lower- and higher-affinity DNA binding conformations of human alkyladenine DNA glycosylase. Biochemistry. 2012;51:382–90. doi: 10.1021/bi201484k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hegde ML, Hazra TK, Mitra S. Functions of disordered regions in mammalian early base excision repair proteins. Cellular and molecular life sciences : CMLS. 2010;67:3573–87. doi: 10.1007/s00018-010-0485-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Elder RH, Jansen JG, Weeks RJ, Willington MA, Deans B, Watson AJ, et al. Alkylpurine-DNA-N-glycosylase knockout mice show increased susceptibility to induction of mutations by methyl methanesulfonate. MolCell Biol. 1998;18:5828–37. doi: 10.1128/mcb.18.10.5828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Margison GP, O’Connor PJ. Biological implications of the instability of the N-glycosidic bone of 3-methyldeoxyadenosine in DNA. Biochim Biophys Acta. 1973;331:349–56. doi: 10.1016/0005-2787(73)90021-x. [DOI] [PubMed] [Google Scholar]
- 41.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–7. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 42.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–21. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 43.Somyajit K, Basavaraju S, Scully R, Nagaraju G. ATM- and ATR-mediated phosphorylation of XRCC3 regulates DNA double-strand break-induced checkpoint activation and repair. Molecular and cellular biology. 2013;33:1830–44. doi: 10.1128/MCB.01521-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Willis J, Patel Y, Lentz BL, Yan S. APE2 is required for ATR-Chk1 checkpoint activation in response to oxidative stress. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:10592–7. doi: 10.1073/pnas.1301445110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chou WC, Wang HC, Wong FH, Ding SL, Wu PE, Shieh SY, et al. Chk2-dependent phosphorylation of XRCC1 in the DNA damage response promotes base excision repair. The EMBO journal. 2008;27:3140–50. doi: 10.1038/emboj.2008.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, 3rd, Hurov KE, Luo J, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160–6. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- 47.Eich M, Roos WP, Nikolova T, Kaina B. Contribution of ATM and ATR to the resistance of glioblastoma and malignant melanoma cells to the methylating anticancer drug temozolomide. Molecular cancer therapeutics. 2013 doi: 10.1158/1535-7163.MCT-13-0136. [DOI] [PubMed] [Google Scholar]
- 48.Liang ML, Ma J, Ho M, Solomon L, Bouffet E, Rutka JT, et al. Tyrosine kinase expression in pediatric high grade astrocytoma. Journal of neuro-oncology. 2008;87:247–53. doi: 10.1007/s11060-007-9513-1. [DOI] [PubMed] [Google Scholar]
- 49.Birmingham A, Selfors LM, Forster T, Wrobel D, Kennedy CJ, Shanks E, et al. Statistical methods for analysis of high-throughput RNA interference screens. Nature methods. 2009;6:569–75. doi: 10.1038/nmeth.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Henderson MC, Gonzales IM, Arora S, Choudhary A, Trent JM, Von Hoff DD, et al. High-throughput RNAi screening identifies a role for TNK1 in growth and survival of pancreatic cancer cells. Molecular cancer research : MCR. 2011;9:724–32. doi: 10.1158/1541-7786.MCR-10-0436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bax DA, Little SE, Gaspar N, Perryman L, Marshall L, Viana-Pereira M, et al. Molecular and phenotypic characterisation of paediatric glioma cell lines as models for preclinical drug development. PloS one. 2009;4:e5209. doi: 10.1371/journal.pone.0005209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pollard SM, Yoshikawa K, Clarke ID, Danovi D, Stricker S, Russell R, et al. Glioma stem cell lines expanded in adherent culture have tumor-specific phenotypes and are suitable for chemical and genetic screens. Cell Stem Cell. 2009;4:568–80. doi: 10.1016/j.stem.2009.03.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.