

Abstract
The autophagic process is the only known mechanism for mitochondrial turnover and it has been speculated that dysfunction of autophagy may result in mitochondrial error and cellular stress. Emerging investigations have provided new understanding of how autophagy of mitochondria (also known as mitophagy) is associated with cellular oxidative stress and its impact on neuro-degeneration. This impaired autophagic function may be considered as a possible mechanism in the pathogenesis of several neurodegenerative disorders including: Parkinson's disease (PD), Alzheimer's disease (AD), multiple sclerosis (MS), amyotrophic lateral sclerosis (ALS) and Huntington disease (HD). It can be suggested that autophagy dysfunction along with oxidative stress are considered main events in neurodegenerative disorders. New therapeutic approaches have now begun to target mitochondria as a potential drug target. This review discusses evidence supporting the notion that oxidative stress and autophagy are intimately associated with neurodegenerative disease pathogenesis. This review also explores new approaches that can prevent mitochondrial dysfunction, improve neurodegenerative etiology, and also offer possible cures to the aforementioned neurodegenerative diseases.
Keywords: Autophagy, Mitophagy, Neurodegeneration, Oxidative stress
Introduction
Autophagy is an evolutionary conserved pathway that engulfs dysfunctional organelles and/or misfolded proteins in a double membrane compartment known as an autophagosome [1]. The formation of the autophagosome drives a progressive process involving a regulation of signal which triggers the formation of a phagophore. When the membrane edges of this phagophore fuse, a autophagosome is formed [2]. Autophagosomes are thought to play an important role in the pathogenesis of a number of common neurodegenerative disorders including: Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD) and Amyotrophic lateral sclerosis (ALS) [3]. Autophagomsomes are now recognized as a potentially important contributing factor to the pathogenesis of neurodegenerative disorders. Autophagy systems cause increased oxidative stress and free radical formation, impaired bioenergetics and mitochondrial dysfunction, disruption of neuronal Golgi apparatus transport, and impaired molecular chaperones [4]. Neurons are metabolically active cells with high energy demands that are mainly dependent on mitochondrion which is confirmed by the link between diseases of mitochrondria and their common neurodegenerative component [5]. In recent years, a growing number of collective data has inferred the role of mitochondria in the pathogenesis of neurodegenerative disorders and apoptotic processes [6]. Reactive oxygen species (ROS) concentration is mediated by mitochondrial antioxidants such as manganese superoxide dismutase (SOD) and glutathione peroxidase (GPX) which are normal byproducts of the mitochondrial respiratory chain. In addition to the generation of ROS, mitochondria are also involved with calcium homeostasis, apoptosis and lipid peroxidation which are central features of the majority of neurodegenerative disorders [7,8].
Recent reports have highlighted that neurons are reliant particularly on the dynamic properties of mitochondria. They engage in repeated cycles of fusion and fission, which serve to inter mix the population of mitochondria [9]. In addition, mitochondria are actively recruited to subcellular sites, such as the axonal and dendritic processes of neurons. Finally, the quality and health of the mitochondrial population is maintained through mitophagy, a form of autophagy, in which defective mitochondria are selectively degraded. In this review we will focus on the involvement of mitochondria, autophagy and oxidative stress in common neurodegenerative disorders (Fig.1.) and offer possible therapeutic approaches for cures.
Figure 1.

Figure showing the autophagy associated common neurodegenerative disease and their molecule.
Autophagy
Autophagy is characterized by the presence of autophagic vacuoles, autophagosomes, and acts as an arbiter of neuronal survival and death decisions amongst neurodegenerative diseases [10, 11, 12, 13]. The increased autophagy in the brains of patients with neurodegenerative disorders suggest that it contributes to the pathogenesis of these neurodegenerative diseases by causing cell death [14, 15, 16, 17, 18]. Autophagy also plays an important role in neuroprotection as well as in neurodegeneration as supported by in vitro and in vivo models [19, 20, 21].
Micro Autophagy
Microautophagy is somewhat similar to macroautophagy and it is reported that the lysosomes directly engulf cytoplasm by passing the need for autophagosome and autophagolysosomal formation [22]. Microautophagy is active in the resting state and is responsible for the appropriate removal of selective organelles and the continuous turnover of intracellular constituents. Interestingly, microautphagy is not activated by nutritional deprivation or stress.
Macro Aautophagy
Macro autophagy is the most abundant type of autophagy and it is conserved from yeast to mammals. Non-selective autophagy is induced by withholding essential nutrients and selective autophagy ensues to clear unwanted or damaged organelles, including mitochondria [23]. In this process an isolation membrane (known as phagophore) surrounds a portion of the cytoplasm or an organelle forming a double membranous structure called the autophagosome. Mitochondria are removed by a form of macroautophagy (called mitophagy) in which the core machinery of bulk macroautophagy is harnessed for the selective clearance of mitochondrion [24].
Mitophagy
The term mitophagy came to describe the selective degradation of mitochondria by autophagy [25]. Reactive oxygen species (ROS), particularly superoxide anion (O2−), hydrogen peroxide (H2O2), and hydroxyl radical (OH−) are toxic byproducts of normal oxidative phosphorylation. Damaged mitochondria release high levels of Ca2+ and cytochrome c into the cytosol and thereby trigger apoptosis [26, 27] and neurodegenerative diseases [28]. Mitochondria have their own proteolytic system whose function is to maintain health, both internally and externally by interacting with other mitochrondria, by degrading unfolded membrane proteins on the inner and outer mitochondrial membrane [29, 30, 31]. In addition to proteolytic and proteosomal degradation recent evidence points to a lysosomal pathway in which vesicles bud from mitochondrial tubules, sequester selected mitochondrial cargos, and then deliver those mitochondrial components to the lysosome for degradation [32]. Mitophagy pathways account for degradation of mitochondrial proteins and the specific control of mitochondrial morphology has a significant impact on mitochondrial function [33]. Mitochondrial fusion was suggested as a route for the rapid exchange of metabolites, mitochondrial DNA (mtDNA) and membrane components [34]. Fission, however, is thought to facilitate the segregation of mtDNA and isolation of mitochondria from the network to allow their degradation [35]. Through this, mitochondrial fission and fusion influences nearly all aspects of mitochondrial function, including respiration, calcium buffering, and apoptosis [36].
Mitochondria and Redox Stress
Neurons and astrocytes, the two major types of brain cells, require more than 20% of the total consumption of oxygen [37]. Oxidative stress is a condition in which the balance between production of ROS and level of antioxidants is significantly altered, leaving high levels of ROS which eventually damages cells. ROS contribute to the development of neurodegeneration by modulating the function of biomolecules [38, 39]. ROS may target several different substrates in the cell causing protein, DNA, RNA oxidation, or lipid peroxidation. Overproduction of ROS in mitochondria or in other sources can also cause changes in the antioxidant system which leads to imbalance and induces oxidative stress and neurodegeneration [40].
Mitochondrial dysfunction and increased oxidative damage are often associated with AD, PD, ALS and HD and several other neurodegenerative disorders suggesting that oxidative stress may play an important role in the pathophysiology of these diseases [41]. It has become dogma that mitochondria are a major source of ROS and also a target of ROS. In pathological conditions this organelle actually produces less-free radicals than other cytosolic enzymes such as NADPH oxidase [42]. However mitochondria (electron transport chain-ETC), in contrast to other cellular producers of ROS, generate free radicals constantly. Mitochondria, which harbor the bulk of oxidative pathways, are packed with various redox carriers that can potentially leak single electrons to oxygen and convert it into a superoxide anion [43]. ROS in mitochondria can also be generated by several enzymes including aconitase and α-ketoglutarate dehydrogenase complex [44, 45, 46]. The production of superoxide by the ETC in mitochondria is dependent on the value of mitochondrial membrane potential. In addition to being generated during cellular metabolism in mitochondria, ROS can be produced in response to different environmental stimuli such as growth factors, inflammatory cytokines, chemical oxidants and chemotherapeutics. Dependent on cell types ROS have been found to function as signaling molecules in cell proliferation and cellular senescence [47], or cell death [48]. The observation of increased autophagy in the brains of patients with AD, PD and HD suggests that autophagy contributes to the pathogenesis of these neurodegenerative diseases; a possible cause is increased cell death [49] because of the constant production of free radicals in mitochondria and changes in the antioxidant system which leads to imbalance, oxidative stress, and neurodegeneration [40].
The excessive Ca2+ uptake into mitochondria can be lethal to neurons. Intracellular stress such as Ca2+ overload and oxidative stress leads to cell death [50]. This causes mitochondria to swell and leads to rupture of the outer mitochondrial membrane releasing proteins from the intra membrane space, e.g. cytochrome c, into the cytosol [50]. This results in mitochondrial depolarization, uncoupling of oxidative phosphorylation, and overproduction of ROS to the cytosol which eventually leads to cell death [51]. Physiological mitochondrial Ca2+ concentrations do not induce mitochondrial transition pore (MTP) opening, but will work in synergy with pro-apoptotic stimuli [52].
Alzheimer’s disease
Mitochondria are significantly reduced in various types of cells obtained from patients with Alzheimer’s disease (AD) and their dysfunction has also been associated with the pathophysiology of AD [53]. The most consistent defect in AD is the deficiency in cytochrome c oxidase which is in the family of mitochondrial electron transport enzymes [54]. This deficiency leads to an increase in ROS production and a disturbance in energy metabolism [7]. AD brains also show evidence of ROS mediated-injury as in there are increased levels of malondyaldehyde and 4-hydroxynonenal in the brains and cerebrospinal fluid of AD patients compared to controls [55, 56]. The amyloid β-protein (Aβ) is involved in mitochondrial function and pathology of AD [57] and is also responsible for mitochondrial mutations in AD development. In AD there is massive accumulation of autophagosomes within large swellings along dystrophic and degenerating neuritis which is primarily due to deficits in the maturation of autophagosomes and their retrograde transport towards the neuronal cell body [49].
In general autophagy is considered to be activated in AD primarily because of impaired clearance of autophagosomes that contain both amyloid precursor protein (APP) and its processing enzymes thereby increasing the propensity to generate toxic Aβ peptides [58]. Normally, most Aβ formed during autophagy are degraded within lysosomes via macroautophagy, but in disease, Aβ accumulates within the large pool of autophagosomes in dystrophic neurites and becomes a major intracellular reservoir of toxic peptides in AD brains [59]. Accumulation of autophagosomes in neurons contributes to Aβ generation within plaques and with that, increased autophagy [60]. Consistently oxidative stress induced autophagy of accumulated amyloid β-protein in AD causes permeabilization of the lysosomal membrane, pyramidal neuronal loss, neuronal cell death and a further neuronal subjection to autophagic degradation which summate into causing neurodegeneration [36, 26].
Parkinson ’s disease
Parkinson’s disease (PD) is the second most common progressive disorder of the central nervous system which is characterized prominently by a loss of dopaminergic neurons in the substantia nigra and formation of intraneuronal protein aggregates α-synuclein [61, 62, 63]. PD is associated with multifactorial causes having features of autophagosome-like structures and a pattern of neurodegeneration [64, 65, 66]. The first evidence in support of a significant role of autophagy in PD came from the demonstration that α-synuclein, which is a major constituent of Lewy bodies found in PD, is degraded by macroautophagy [30]. Besides the degradation of α-synuclein the autophagic pathway is also involved in the turnover of mitochondria. Mutations in genes such as PARKIN and PINK1 are known to cause autosomal recessive forms of PD and have been implicated in the control of mitochondrial morphology and function [67]. Recently PARKIN was found to facilitate macroautophagy of impaired mitochondria [68]. In support of a role for autophagy in the clearance of defective mitochondria in PD, knockdown of PINK1 expression induces mitochondrial fragmentation, followed by activation of autophagy/mitophagy [69, 70]. Furthermore, PARKIN, whose loss of function mutation causes early onset PD has been found to promote autophagy of depolarized mitochondria [71, 68] suggesting that a failure to eliminate damaged mitochondria by mutant PARKIN is responsible for the pathogenesis of PD.
Altered mitochondrial respiration and increased production of ROS causes a loss of dopaminergic neurons in human and animal models which strongly suggests a link between oxidative stress, mitochondrial dysfunction, and PD pathogenesis [72, 73]. It is reported that there is an increase in common deletions of mitochondrial DNA in the surviving dopaminergic neurons in PD substantia nigra. These DNA deletions are believed to be the result of oxidative stress [74, 75]. Protein oxidative damage in the form of protein carbonyls is evident in PD brain compared to controls and there is some evidence to suggest a role for nitration and nitrosylation of certain proteins due to reactive nitrogen species in PD brain [76]. Consistent with this concept, a significant decrease in the activity of complex I in the electron transport chain is observed in the substantia nigra from PD patients [72]. Genetic studies of PINK1 and PARKIN further support the role of mitophagy in pathogenesis of PD [77, 78].
Huntington’s Disease
Huntington’s disease (HD) is another hereditary neurodegenerative disorder that affects muscle coordination and leads to cognitive decline and dementia. HD is caused by an autosomal dominant mutation in the Huntingtin (HTT) gene [79, 80]. Mutant HTT is known to induce endosomal and lysosomal activities [81]. Morphologic defects of mitochondria, such as reduced mitochondrial movement and alterations in mitochondrial ultrastructures have been observed in patients with HD or in transgenic HD mouse models [33, 82]. In addition, expression of mutant HTT leads to impaired energy metabolism, abnormal Ca2+ signaling and mitochondrial membrane potential [83], and drastic changes in mitochondrial ultrastructures [84, 85, 86]. Postmortem brains of patients with HD have and increase in endosomal and/or lysosomal organelles and their multivesicular bodies express characteristic features of autophagy [87]. It is recently proposed that mutant HTT conveys its neurotoxicity by evoking defects in mitochondrial dynamics, mitochondrial fission and fusion, and organelle trafficking which in turn result in bioenergetic failure and HD associated neuronal dysfunction [33, 88].
Amyotrophic Lateral Sclerosis Disease
Amyotrophic lateral sclerosis (ALS) is characterized by extensive loss of motor neurons functioning in the spinal cord and brain stem, atrophy of ventral roots, degeneration of upper motor neurons in the motor cortex and corticospinal tract, somatic and axonal inclusions of aberrant neurofilament proteins, and reactive astrocytosis [89]. Mitochondrial dysfunction may cause motor neuron death by predisposing them to calcium-mediated excitotoxicity by increasing generation of ROS and by initiating the intrinsic apoptotic pathway [90]. The specific mechanism of ALS still needs to be investigated, but the findings are significant because they implicate cells other than neuron cells in neurodegeneration [91]. But accumulating evidence suggests that mitochondrial dysfunction is involved in the pathogenesis of ALS [92]. It has been reported that abundant autophagosomes and associated increases in autophagy proteins and their activation is detrimental for the survival of motor neurons [93]. However, other studies have found an increase in the LC3II macroautophagy marker protein and a decreased ratio of phosphorylated mTOR-positive motor neurons shows defective autophagy accompanied with motor neuron loss in ALS [94]. Mutant SOD1 was found to be preferentially associated with mitochondria and the subsequent impaired function suggests that axonal transport of mitochondria along microtubules is disrupted in ALS [95]. Furthermore, new evidence suggests that mitochondrial fission and fusion as well as mitophagy clearance may also be affected by mutant SOD1 [96]. A key to answering the question of whether mitochondrial dysfunction plays a crucial role in motor neuron degeneration are therapeutic approaches aimed at modulating mitochondrial function and protecting mitochondria from the pro-apoptotic effect of mutant SOD1. A better understanding of the etiology of ALS is needed to develop effective therapies for the treatment of this fatal neurodegenerative disease.
Multiple Sclerosis
Multiple sclerosis (MS) is a chronic, unpredictable, and often disabling disease that attacks the central nervous system [97]. In MS disease the neurological function starts gradually deteriorating from the onset of the disease [98]. Currently available therapeutic agents are not effective in preventing or reducing the relentless accumulation of neurological deficits during the progressive phase of MS [99]. Although the pathological substrate of disease progression is regarded as axonal degeneration, recent evidence identifies axonal dysfunction as an additional and possibly important contributor to the neurological disability during the progressive phase of MS [100]. The mitochondrial changes in axons lacking healthy myelin sheaths as well as redistribution of sodium channels suggest that demyelinated axons would be more vulnerable to energy deficits than myelinated axons [99]. The axons with dysfunctional Na+/K+ ATPase or without Na+/K+ ATPase will no longer be able to efflux sodium, maintain resting membrane potential, or conduct nerve impulses [100]. A recent study identified the lack of Na+/K+ ATPase in approximately half of chronically demyelinated axons in MS [101]. A dysfunction of mitochondria in lesions as well as in the normal-appearing white and grey matter is increasingly recognized in MS and could be an important determinant of axonal dysfunction and degeneration.
Prevention of Mitochondrial Dysfunction
The alterations of mitochondrial function lead to a plethora of diseases. However, targeting mitochondrial linked pathways and using different approaches can revert or prevent mitochondrial dysfunction, these approaches are: minimizing ROS generation, cellular antioxidant pathways, calcium flux, electron transport chain, anti-apoptoic mechanisms, and mitochondrial permeability transition pore (mPTP) (Figure.2). Several studies have reported therapeutic molecules that could be used as potential mitochondrial dysfunction reverting agents, these are: CDDO-ethyl amide, CDDO-trifluroethylamide,pioglitazone, rosiglitazone, resveratrol, 5-aminoimidazole-4-carboxamide ribonucleoside (AICAR), coenzyme Q10, and bezafibrate. A compiled table of these molecules along with their mode of action, structure, trade name, and references has been given (Table-1). Exploring these agents along with other novel agents/approaches could help in preventing mitochondrial dysfunction and in improving neurodegenerative etiology.
Figure 2.

Figure showing the different target of drugs, molecules and chemical compounds in Mitochondria. These drugs and chemical compound are also used for study of molecular mechanism of various neurodegenerative disease.
Table.1.
Table showing the different chemical compounds, drugs and their targets in mitochndria. These drugs and chemical compounds targeting the oxidative stress pathways, neuroinflammatory pathways, some of targeting electron transport chain. These drugs and compounds are widely used for the study of neurodegenerative disease.
| S. No. |
Compounds | Mechanism | Structure | Trade name |
References |
|---|---|---|---|---|---|
| 1 | CDDO-Ethyl amide | Target Nrf2/ARE (NF-E2-related factor 2/antioxidant response element) signaling pathway | 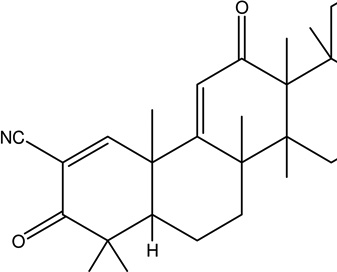 |
Not assigned | [102, 103] |
| 2 | CDDO-Trifluroethylamide | Target Nrf2/ARE (NF-E2-related factor 2/antioxidant response element) signaling pathway, attenuates mitochondria l dysfunction | 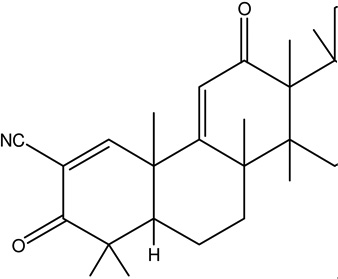 |
Not assigned | [102, 103] |
| 3 | Pioglitazone | Activate peroxisome proliferator-activated receptors (PPARs), provide neuroprotection, attenuates mitochondria l dysfunction by improving mitochondria l DNA (mtDNA) content, levels of mtDNA and nuclearencoded electron transport chain subunit proteins, increased oxygen consumption, and elevated complex I and complex IV V max activities | 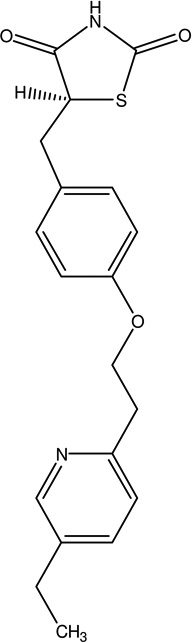 |
Actos Piomed Pioneer | [104, 105] |
| 4 | Rosiglitazone | Activate PPAR gamma activators, attenuates mitochondria l dysfunction. | 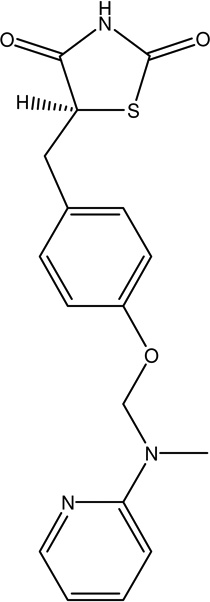 |
NA | [106] |
| 5. | Resveratrol | Antioxidant, anti-inflammatory, and metalchelating effects, prevent mitochondria l dysfunction | 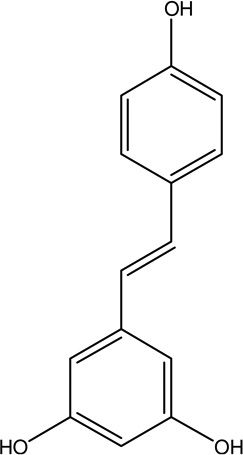 |
Not assigned | [107]. |
| 6. | 5-aminoimidazo le-4-carboxamide ribonucleoside (AICAR) | Block proinflammatory cytokines, inhibit ROS generation, deplete glutathione, prevent oxidative stress | 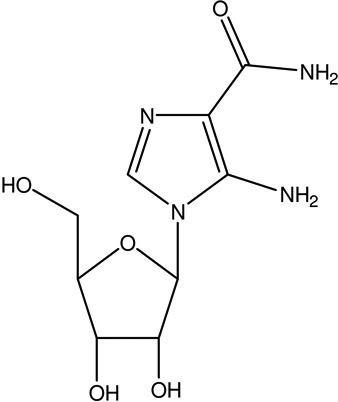 |
Huapin | [108]. |
| 7 | Coenzyme Q10 | Improve respiratory chain dysfunction, oxidative stress and neurodegeneration | 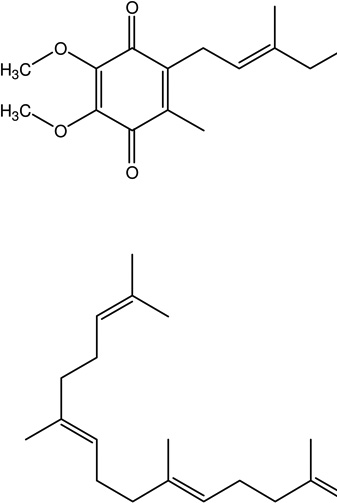 |
Rite Aid Coenzyme Q-10 | [109, 110] |
| 8. | Bezafibrate | Anti-inflammatory, neuroprotective role, attenuate astrogliosis, improve peroxisome proliferator-activated receptor (PPAR)-γ- | 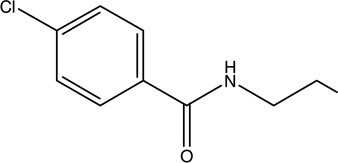 |
Bezalip Beza–XL Fenolip Globez | [111, 112] |
Future direction and conclusions
It is evident from recent findings that mitochondrial abnormalities are actively engaged in the pathogenesis of neurodegenerative diseases. Evidence from the collected data suggested that mitochondrial-DNA mutations, autophagy, and oxidative stress are contributory factors for multiple neurodegenerative diseases. Autophagic dysfunction and oxidative stress occur early in all major neurodegenerative diseases and there is strong evidence that this dysfunction has a causal role in disease pathogenesis. Autophagy represents a major route for degradation of toxic proteins and dysfunctional organelles. Alterations in autophagy processes play a significant role in the pathogenesis of many neurodegenerative diseases. Since the autophagy pathway may be compromised in various neurodegenerative disorders, understanding the autophagy pathways in each neurodegenerative disorder could explain differences in the course of these pathologies and will be essential to developing targeted therapeutic approaches for each disease. In our opinion a better understanding of the cellular response to oxidative stress, mitochondrial dysfunction, and its relation to autophagy will lead to new therapeutic approaches for the prevention and amelioration of neurodegenerative diseases. The future challenges are to devise models to better understand the common pathways and relative contribution of mitochondrial dysfunction to the pathogenesis of neurodegenerative disorders as well as to identify therapeutic approaches that target mitophagy and its consequences.
Acknowledgments
This work was supported by National Institutes of Health grants HL107640-NT and NS-051568 to SCT.
Abbreviations
- AD
Alzhiemers disease
- PD
Parkinson disease
- ALS
Amyotrophic lateral sclerosis
- HD
Huntington’s disease
- ROS
Reactive oxygen species
- MS
multiple sclerosis
Footnotes
Conflict of interest
Authors having no conflict of interest
References
- 1.Shacka JJ, Roth KA, Zhang J. The autophagy–lysosomal degradation pathway: role in neurodegenerative disease and therapy. Front Biosci. 2008;13:718–736. doi: 10.2741/2714. [DOI] [PubMed] [Google Scholar]
- 2.Cherra SJ, 3rd, Chu CT. Autophagy in neuroprotection and neurodegeneration: A question of balance. Future Neurol. 2008;3:309–323. doi: 10.2217/14796708.3.3.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chinnery PF, Schon EA. Mitochondria. J Neurol Neurosurg Psychiatry. 2003;74:1188–1199. doi: 10.1136/jnnp.74.9.1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jellinger KA. Recent advances in our understanding of neurodegeneration. J Neural Transm. 2009;9:1111–1162. doi: 10.1007/s00702-009-0240-y. [DOI] [PubMed] [Google Scholar]
- 5.Hashimoto M, Rockenstein E, Crews L, Masliah E. Role of protein aggregation in mitochondrial dysfunction and neurodegeneration in Alzheimer's and Parkinson's diseases. NeuroMol. Med. 2003;4:21–36. doi: 10.1385/NMM:4:1-2:21. [DOI] [PubMed] [Google Scholar]
- 6.Cardaioli E, Sicurelli F, Carluccio MA, Gallus GN, Da Pozzo P, Mondelli M, Margollicci MA, Micheli V, Federico A, Dotti MT. A new thymidine phosphorylase mutation causing elongation of the protein underlies mitochondrial neurogastrointestinal encephalomyopathy. J Neurol. 2012;259:172–174. doi: 10.1007/s00415-011-6113-y. [DOI] [PubMed] [Google Scholar]
- 7.Kamat PK, Tota S, Shukla R, Ali S, Najmi AK, Nath C. Mitochondrial dysfunction: a crucial event in okadaic acid (ICV) induced memory impairment and apoptotic cell death in rat brain. Pharmacol Biochem Behav. 2011;100:311–319. doi: 10.1016/j.pbb.2011.08.019. [DOI] [PubMed] [Google Scholar]
- 8.Gavin PD, Prescott M, Devenish RJ. F1F0-ATP synthase complex interactions in vivo can occur in the absence of the dimer specific subunit. J Bioenerg Biomembr. 2005;37:55–66. doi: 10.1007/s10863-005-4128-8. [DOI] [PubMed] [Google Scholar]
- 9.Kim I, Rodríguez-Enríquez S, Lemasters JJ. Selective degradation of mitochondria by mitophagy. Arch. Biochem. Biophys. 2007;462:245–253. doi: 10.1016/j.abb.2007.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jing K, Lim K. Why is autophagy important in human diseases? Exp Mol Med. 2012;44:69–72. doi: 10.3858/emm.2012.44.2.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gottlieb RA, Carreira RS. Autophagy in health and disease. Am J Physiol Cell Physiol. 2010;299:C203–C210. doi: 10.1152/ajpcell.00097.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Komatsu M, Waguri S, Chiba T, Murata S, Iwata JI, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 13.Banerjee R, Bea,l MF, Thomas B. Autophagy in neurodegenerative disorders: pathogenic roles and therapeutic implications. Trends Neurosci. 2010;33:541–549. doi: 10.1016/j.tins.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wong E, Cuervo AM. Autophagy gone awry in neurodegenerative diseases. Nat Neurosci. 2010;13:805–811. doi: 10.1038/nn.2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moreira PI, Santos RX, Zhu X, Lee HG, Smith MA, Casadesus G, Perry G. Autophagy in Alzheimer's disease. Expert Rev Neurother. 2010;10:1209–1218. doi: 10.1586/ern.10.84. [DOI] [PubMed] [Google Scholar]
- 16.Yang YP, Liang ZQ, Gu ZL, Qin ZH. Molecular mechanism and regulation of autophagy. Acta Pharmacol Sin. 2005;26:1421–1434. doi: 10.1111/j.1745-7254.2005.00235.x. [DOI] [PubMed] [Google Scholar]
- 17.Ravikumar B, Stewart A, Kita H, Kato K, Duden R, Rubinsztein DC. Raised intracellular glucose concentrations reduce aggregation and cell death caused by mutant huntingtin exon 1 by decreasing mTOR phosphorylation and inducing autophagy. Hum Mol Genet. 2003;12:985–994. doi: 10.1093/hmg/ddg109. [DOI] [PubMed] [Google Scholar]
- 18.Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem. 2003;278:25009–25013. doi: 10.1074/jbc.M300227200. [DOI] [PubMed] [Google Scholar]
- 19.Liu B, Cheng Y, Zhang B, Bian HJ, Bao JK. Polygonatum cyrtonema lectin induces apoptosis and autophagy in human melanoma A375 cells through a mitochondria-mediated ROS-p38-p53 pathway. Cancer Lett. 2009;275:54–60. doi: 10.1016/j.canlet.2008.09.042. [DOI] [PubMed] [Google Scholar]
- 20.Chu CT, Plowey ED, Dagda RK, Hickey RW, Cherra SJ, 3rd, Clark RS. Autophagy in neurite injury and neurodegeneration: in vitro and in vivo models. Methods Enzymol. 2009;453:217–249. doi: 10.1016/S0076-6879(08)04011-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bossy B, Perkins G, Bossy-Wetzel E. Clearing the brain's cobwebs: the role of autophagy in neuroprotection. Curr Neuropharmacol. 2008;6:97–101. doi: 10.2174/157015908784533897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Filosto M, Scarpelli M, Cotelli MS, Vielmi V, Todeschini A, Gregorelli V, Tonin P, Tomelleri G, Padovani A. The role of mitochondria in neurodegenerative diseases. J Neurol. 2011;258:1763–1774. doi: 10.1007/s00415-011-6104-z. [DOI] [PubMed] [Google Scholar]
- 23.Khan MU, Cheema Y, Shahbaz AU, Ahokas RA, Sun Y, Gerling IC, Bhattacharya SK, Weber KT. Mitochondria play a central role in nonischemic cardiomyocyte necrosis: common to acute and chronic stressor states. Pflugers Arch. 2012;464:123–131. doi: 10.1007/s00424-012-1079-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zheng YZ, Berg KB, Foster LJ. Mitochondria do not contain lipid rafts, and lipid rafts do not contain mitochondrial proteins. J Lipid Res. 2009;50:988–998. doi: 10.1194/jlr.M800658-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Santos JM, Kowluru RA. Role of mitochondria biogenesis in the metabolic memory associated with the continued progression of diabetic retinopathy and its regulation by lipoic acid. Invest Ophthalmol Vis Sci. 2011;52:8791–8798. doi: 10.1167/iovs.11-8203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Parsons MJ, Green DR. Mitochondria and apoptosis: a quick take on a long view. F1000 Biol Rep. 2009;1:17. doi: 10.3410/B1-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Palikaras K, Tavernarakis N. Mitophagy in neurodegeneration and aging. Front Genet. 2012;3:297. doi: 10.3389/fgene.2012.00297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hagen T, Lagace CJ, Modica-Napolitano JS, Aprille JR. Permeability transition in rat liver mitochondria is modulated by the ATP-Mg/Pi carrier. Am J Physiol Gastrointest Liver Physiol. 2003;285:G274–G281. doi: 10.1152/ajpgi.00052.2003. [DOI] [PubMed] [Google Scholar]
- 29.Wallace DC. Mitochondria and cancer: Warburg addressed. Cold Spring Harb Symp Quant Biol. 2005;70:363–374. doi: 10.1101/sqb.2005.70.035. [DOI] [PubMed] [Google Scholar]
- 30.Karbowski M. Mitochondria on guard: role of mitochondrial fusion and fission in the regulation of apoptosis. Adv Exp Med Biol. 2010;687:131–142. doi: 10.1007/978-1-4419-6706-0_8. [DOI] [PubMed] [Google Scholar]
- 31.Langer T, Kaser M, Klanner C, Leonhard K. AAA proteases of mitochondria: quality control of membrane proteins and regulatory functions during mitochondrial biogenesis. Biochem Soc Trans. 2001;29:431–436. doi: 10.1042/bst0290431. [DOI] [PubMed] [Google Scholar]
- 32.Soubannier V, McLelland GL, Zunino R, Braschi E, Rippstein P, Fon EA, McBride HM. A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr Biol. 2012;22:135–141. doi: 10.1016/j.cub.2011.11.057. [DOI] [PubMed] [Google Scholar]
- 33.Miwa S, St-Pierre J, Partridge L, Brand MD. Superoxide and hydrogen peroxide production by Drosophila mitochondria. Free Radic Biol Med. 2003;35:938–948. doi: 10.1016/s0891-5849(03)00464-7. [DOI] [PubMed] [Google Scholar]
- 34.Twig G, Graf SA, Wikstrom JD, Mohamed H, Haigh SE, Elorza A, Deutsch M, Zurgil N, Reynolds N, Shirihai OS. Tagging and tracking individual networks within a complex mitochondrial web with photoactivatable GFP. Am J Physiol Cell Physiol. 2006;291:C176–C184. doi: 10.1152/ajpcell.00348.2005. [DOI] [PubMed] [Google Scholar]
- 35.Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE, Shirihai OS. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Frieden M, Arnaudeau S, Castelbou C, Demaurex N. Subplasmalemmal mitochondria modulate the activity of plasma membrane Ca2+-ATPases. J Biol Chem. 2005;280:43198–43208. doi: 10.1074/jbc.M510279200. [DOI] [PubMed] [Google Scholar]
- 37.Gruber J, Fong S, Chen CB, Yoong S, Pastorin G, Schaffer S, Cheah I, Halliwell B. Mitochondria-targeted antioxidants and metabolic modulators as pharmacological interventions to slow ageing. Biotechnol Adv. 2012 doi: 10.1016/j.biotechadv.2012.09.005. [DOI] [PubMed] [Google Scholar]
- 38.Chen J, Tang XQ, Zhi JL, Cui Y, Yu HM, Tang EH, Sun SN, Feng JQ, Chen PX. Curcumin protects PC12 cells against 1-methyl-4-phenylpyridinium ion-induced apoptosis by bcl-2-mitochondria-ROS-iNOS pathway. Apoptosis. 2006;11:943–953. doi: 10.1007/s10495-006-6715-5. [DOI] [PubMed] [Google Scholar]
- 39.Tang L, Zhang Y, Jobson HE, Li J, Stephenson KK, Wade KL, Fahey JW. Potent activation of mitochondria-mediated apoptosis and arrest in S and M phases of cancer cells by a broccoli sprout extract. Mol Cancer Ther. 2006;5:935–944. doi: 10.1158/1535-7163.MCT-05-0476. [DOI] [PubMed] [Google Scholar]
- 40.Lemasters JJ. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 2005;8:3–5. doi: 10.1089/rej.2005.8.3. [DOI] [PubMed] [Google Scholar]
- 41.Lin MT, Beal MF. Alzheimer's APP mangles mitochondria. Nat Med. 2006;12:1241–1243. doi: 10.1038/nm1106-1241. [DOI] [PubMed] [Google Scholar]
- 42.Frazziano G, Champion HC, Pagano PJ. NADPH oxidase-derived ROS and the regulation of pulmonary vessel tone. Am J Physiol Heart Circ Physiol. 2012;302:H2166–H2177. doi: 10.1152/ajpheart.00780.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Takahashi E, Endoh H, Doi K. Intracellular gradients of O2 supply to mitochondria in actively respiring single cardiomyocyte of rats. Am J Physiol. 1999;276:H718–H724. doi: 10.1152/ajpheart.1999.276.2.H718. [DOI] [PubMed] [Google Scholar]
- 44.McKenna MC. Glutamate dehydrogenase in brain mitochondria: do lipid modifications and transient metabolon formation influence enzyme activity? Neurochem Int. 2011;59:525–533. doi: 10.1016/j.neuint.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nordberg J, Arner ES. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Radic Biol Med. 2001;31:1287–1312. doi: 10.1016/s0891-5849(01)00724-9. [DOI] [PubMed] [Google Scholar]
- 46.Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552:335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lundberg IE. New possibilities to achieve increased understanding of disease mechanisms in idiopathic inflammatory myopathies. Curr Opin Rheumatol. 2002;14:639–642. doi: 10.1097/00002281-200211000-00001. [DOI] [PubMed] [Google Scholar]
- 48.Burdon RH, Gill V, Boyd PA, Rahim RA. Hydrogen peroxide and sequence-specific DNA damage in human cells. FEBS Lett. 1996;383:150–154. doi: 10.1016/0014-5793(96)00230-x. [DOI] [PubMed] [Google Scholar]
- 49.Rubinsztein DC, DiFiglia M, Heintz N, Nixon RA, Qin ZH, Ravikumar B, Stefanis L, Tolkovsky A. Autophagy and its possible roles in nervous system diseases, damage and repair. Autophagy. 2005;1:11–22. doi: 10.4161/auto.1.1.1513. [DOI] [PubMed] [Google Scholar]
- 50.Galluzzi L, Blomgren K, Kroemer G. Mitochondrial membrane permeabilization in neuronal injury. Nat Rev Neurosci. 2009;10:481–494. doi: 10.1038/nrn2665. [DOI] [PubMed] [Google Scholar]
- 51.Pinton P, Giorgi C, Siviero R, Zecchini E, Rizzuto R. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene. 2008;27:6407–6418. doi: 10.1038/onc.2008.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zecchini V, Mills IG. Putting chromatin immunoprecipitation into context. J Cell Biochem. 2009;107:19–29. doi: 10.1002/jcb.22080. [DOI] [PubMed] [Google Scholar]
- 53.Rogawski MA, Wenk GL. The neuropharmacological basis for the use of memantine in the treatment of Alzheimer's disease. CNS Drug Rev. 2003;9:275–308. doi: 10.1111/j.1527-3458.2003.tb00254.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cardoso SM, Santana I, Swerdlow RH, Oliveira CR. Mitochondria dysfunction of Alzheimer's disease cybrids enhances Abeta toxicity. J Neurochem. 2004;89:1417–1426. doi: 10.1111/j.1471-4159.2004.02438.x. [DOI] [PubMed] [Google Scholar]
- 55.Butterfield DA, Lauderback CM. Lipid peroxidation and protein oxidation in Alzheimer's disease brain: potential causes and consequences involving amyloid beta-peptide-associated free radical oxidative stress. Free Radic Biol Med. 2002;32:1050–1060. doi: 10.1016/s0891-5849(02)00794-3. [DOI] [PubMed] [Google Scholar]
- 56.Aksenov MY, Aksenova MV, Butterfield DA, Geddes JW, Markesbery WR. Protein oxidation in the brain in Alzheimer's disease. Neuroscience. 2000;103:373–383. doi: 10.1016/s0306-4522(00)00580-7. [DOI] [PubMed] [Google Scholar]
- 57.Maruszak A, Zekanowski C. Mitochondrial dysfunction and Alzheimer's disease. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35:320–330. doi: 10.1016/j.pnpbp.2010.07.004. [DOI] [PubMed] [Google Scholar]
- 58.Butler D, Nixon RA, Bahr BA. Potential compensatory responses through autophagic/lysosomal pathways in neurodegenerative diseases. Autophagy. 2006;2:234–237. doi: 10.4161/auto.2729. [DOI] [PubMed] [Google Scholar]
- 59.Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, Cuervo AM. Extensive involvement of autophagy in Alzheimer disease: an immunoelectron microscopy study. Neuropathol Exp Neurol. 2005;64:113–122. doi: 10.1093/jnen/64.2.113. [DOI] [PubMed] [Google Scholar]
- 60.Nixon RA. Autophagy, amyloidogenesis and Alzheimer disease. J Cell Sci. 2007;120:4081–4091. doi: 10.1242/jcs.019265. [DOI] [PubMed] [Google Scholar]
- 61.Kirik D, Rosenblad C, Burger C, Lundberg C, Johansen TE, Muzyczka N, Mandel RJ, Bjorklund A. Parkinson-like neurodegeneration induced by targeted overexpression of alpha-synuclein in the nigrostriatal system. J Neurosci. 2002;22:2780–2791. doi: 10.1523/JNEUROSCI.22-07-02780.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tian L, Ma L, Kaarela T, Li Z. Neuroimmune crosstalk in the central nervous system and its significance for neurological diseases. J Neuroinflammation. 2012;9:155. doi: 10.1186/1742-2094-9-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Battisti C, Formichi P, Radi E, Federico A. Oxidative-stress-induced apoptosis in PBLs of two patients with Parkinson disease secondary to alpha-synuclein mutation. J Neurol Sci. 2008;267:120–124. doi: 10.1016/j.jns.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 64.Alvarez-Erviti L, Rodriguez-Oroz MC, Cooper JM, Caballero C, Ferrer I, Obeso JA, Schapira AH. Chaperone-mediated autophagy markers in Parkinson disease brains. Arch Neurol. 2010;67:1464–1472. doi: 10.1001/archneurol.2010.198. [DOI] [PubMed] [Google Scholar]
- 65.Rogaeva E, Johnson J, Lang AE, Gulick C, Gwinn-Hardy K, Kawarai T, Sato C, Morgan A, Werner J, Nussbaum R, Petit A, Okun MS, McInerney A, Mandel R, Groen JL, Fernandez HH, Postuma R, Foote KD, Salehi-Rad S, Liang Y, Reimsnider S, Tandon A, Hardy J, St George-Hyslop P, Singleton AB. Analysis of the PINK1 gene in a large cohort of cases with Parkinson disease. Arch Neurol. 2004;61:1898–1904. doi: 10.1001/archneur.61.12.1898. [DOI] [PubMed] [Google Scholar]
- 66.Yang L, Xia Y, Zhao H, Zhao J, Zhu X. Magnetic resonance imaging of transplanted neural stem cells in Parkinson disease rats. J Huazhong Univ Sci Technolog Med Sci. 2006;26:489–492. doi: 10.1007/s11596-006-0430-x. [DOI] [PubMed] [Google Scholar]
- 67.Banerjee R, Starkov AA, Beal MF, Thomas B. Mitochondrial dysfunction in the limelight of Parkinson's disease pathogenesis. Biochim Biophys Acta. 2009;7:651–663. doi: 10.1016/j.bbadis.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chu CT, Zhu J, Dagda R. Beclin 1-independent pathway of damage-induced mitophagy and autophagic stress: implications for neurodegeneration and cell death. Autophagy. 2007;3:663–666. doi: 10.4161/auto.4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dagda RK, Cherra SJ, 3rd, Kulich SM, Tandon A, Park D, Chu CT. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J Biol Chem. 2009;284:13843–13855. doi: 10.1074/jbc.M808515200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.McBride HM. Parkin mitochondria in the autophagosome. J Cell Biol. 2008;183:757–759. doi: 10.1083/jcb.200810184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Greenamyre JT, Sherer TB, Betarbet R, Panov AV. Complex I and Parkinson's disease. IUBMB Life. 2001;52:135–141. doi: 10.1080/15216540152845939. [DOI] [PubMed] [Google Scholar]
- 73.Seet RC, Lee CY, Lim EC, Tan JJ, Quek AM, Chong WL, Looi WF, Huang SH, Wang H, Chan YH, Halliwell B. Oxidative damage in Parkinson disease: Measurement using accurate biomarkers. Free Radic Biol Med. 2010;48:560–566. doi: 10.1016/j.freeradbiomed.2009.11.026. [DOI] [PubMed] [Google Scholar]
- 74.Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH, Jaros E, Hersheson JS, Betts J, Klopstock T, Taylor RW, Turnbull DM. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet. 2006;38:515–517. doi: 10.1038/ng1769. [DOI] [PubMed] [Google Scholar]
- 75.Dalfo E, Ferrer I. Alpha-synuclein binding to rab3a in multiple system atrophy. Neurosci Lett. 2005;380:170–175. doi: 10.1016/j.neulet.2005.01.034. [DOI] [PubMed] [Google Scholar]
- 76.Murphy BM, Dandy DS, Henry CS. Analysis of oxidative stress biomarkers using a simultaneous competitive/non-competitive micromosaic immunoassay. Anal Chim Acta. 2009;640:1–6. doi: 10.1016/j.aca.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chen CY, Jang JH, Park MH, Hwang SJ, Surh YJ, Park OJ. Attenuation of Abeta-induced apoptosis of plant extract (Saengshik) mediated by the inhibition of mitochondrial dysfunction and antioxidative effect. Ann N Y Acad Sci. 2007;1095:399–411. doi: 10.1196/annals.1397.043. [DOI] [PubMed] [Google Scholar]
- 78.Dodson MW, Guo M. Pink1, Parkin, DJ-1 and mitochondrial dysfunction in Parkinson's disease. Curr Opin Neurobiol. 2007;17:331–337. doi: 10.1016/j.conb.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 79.Pavese N, Gerhard A, Tai YF, Ho AK, Turkheimer F, Barker RA, Brooks DJ, Piccini P. Microglial activation correlates with severity in Huntington disease: a clinical and PET study. Neurology. 2006;66:1638–1643. doi: 10.1212/01.wnl.0000222734.56412.17. [DOI] [PubMed] [Google Scholar]
- 80.Gusella JF, MacDonald ME. Huntington's disease: CAG genetics expands neurobiology. Curr Opin Neurobiol. 1995;5:656–662. doi: 10.1016/0959-4388(95)80072-7. [DOI] [PubMed] [Google Scholar]
- 81.Kegel KB, Kim M, Sapp E, McIntyre C, Castano JG, Aronin N, DiFiglia M. Huntingtin expression stimulates endosomal-lysosomal activity, endosome tubulation, and autophagy. J Neurosci. 2000;20:7268–7278. doi: 10.1523/JNEUROSCI.20-19-07268.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Orr AL, Li S, Wang CE, Li H, Wang J, Rong J, Xu X, Mastroberardino PG, Greenamyre JT, Li XJ. N-terminal mutant huntingtin associates with mitochondria and impairs mitochondrial trafficking. J Neurosci. 2008;28:2783–2792. doi: 10.1523/JNEUROSCI.0106-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Panov AV, Gutekunst CA, Leavitt BR, Hayden MR, Burke JR, Strittmatter WJ, Greenamyre JT. Early mitochondrial calcium defects in Huntington's disease are a direct effect of polyglutamines. Nat Neurosci. 2002;5:731–736. doi: 10.1038/nn884. [DOI] [PubMed] [Google Scholar]
- 84.Modugno N, Curra A, Giovannelli M, Priori A, Squitieri F, Ruggieri S, Manfredi M, Berardelli A. The prolonged cortical silent period in patients with Huntington's disease. Clin Neurophysiol. 2001;112:1470–1474. doi: 10.1016/s1388-2457(01)00599-5. [DOI] [PubMed] [Google Scholar]
- 85.Herishanu YO, Parvari R, Pollack Y, Shelef I, Marom B, Martino T, Cannella M, Squitieri F. Huntington disease in subjects from an Israeli Karaite community carrying alleles of intermediate and expanded CAG repeats in the HTT gene: Huntington disease or phenocopy? J Neurol Sci. 2009;277:143–146. doi: 10.1016/j.jns.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 86.Tellez-Nagel I, Johnson AB, Terry RD. Studies on brain biopsies of patients with Huntington's chorea. J Neuropathol Exp Neurol. 1974;33:308–332. doi: 10.1097/00005072-197404000-00008. [DOI] [PubMed] [Google Scholar]
- 87.Hirano M. VAPB: new genetic clues to the pathogenesis of ALS. Neurology. 2008;70:1161–1162. doi: 10.1212/01.wnl.0000307756.15383.fc. [DOI] [PubMed] [Google Scholar]
- 88.Martinez-Vicente M, Talloczy Z, Wong E, Tang G, Koga H, Kaushik S, de Vries R, Arias E, Harris S, Sulzer D, Cuervo AM. Cargo recognition failure is responsible for inefficient autophagy in Huntington's disease. Nat Neurosci. 2010;13:567–576. doi: 10.1038/nn.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kawamata H, Manfredi G. Mitochondrial dysfunction and intracellular calcium dysregulation in ALS. Mech Ageing Dev. 2010;131:517–526. doi: 10.1016/j.mad.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Julien JP. ALS: astrocytes move in as deadly neighbors. Nat Neurosci. 2007;10:535–537. doi: 10.1038/nn0507-535. [DOI] [PubMed] [Google Scholar]
- 91.Liu B, Tewari AK, Zhang L, Green-Church KB, Zweier JL, Chen YR, He G. Proteomic analysis of protein tyrosine nitration after ischemia reperfusion injury: mitochondria as the major target. Biochim Biophys Acta. 2009;1794:476–485. doi: 10.1016/j.bbapap.2008.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Beckman JS, Estevez AG, Crow JP, Barbeito L. Superoxide dismutase and the death of motoneurons in ALS. Trends Neurosci. 2001;24:S15–S20. doi: 10.1016/s0166-2236(00)01981-0. [DOI] [PubMed] [Google Scholar]
- 93.Okamoto K, Fujita Y, Mizuno Y. Pathology of protein synthesis and degradation systems in ALS. Neuropathology. 2010;30:189–193. doi: 10.1111/j.1440-1789.2009.01088.x. [DOI] [PubMed] [Google Scholar]
- 94.Li B, Liu XY, Li Z, Bu H, Sun MM, Guo YS, Li CY. Effect of ALS IgG on motor neurons in organotypic spinal cord cultures. Can J Neurol Sci. 2008;35:220–225. doi: 10.1017/s0317167100008672. [DOI] [PubMed] [Google Scholar]
- 95.Shi P, Strom AL, Gal J, Zhu H. Effects of ALS-related SOD1 mutants on dynein- and KIF5-mediated retrograde and anterograde axonal transport. Biochim Biophys Acta. 2010;1802:707–716. doi: 10.1016/j.bbadis.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 97.Roxburgh RH, Seaman SR, Masterman T, Hensiek AE, Sawcer SJ, Vukusic S, Achiti I, Confavreux C, Coustans M, le Page E, Edan G, McDonnell GV, Hawkins S, Trojano M, Liguori M, Cocco E, Marrosu MG, Tesser F, Leone MA, Weber A, Zipp F, Miterski B, Epplen JT, Oturai A, Sorensen PS, Celius EG, Lara NT, Montalban X, Villoslada P, Silva AM, Marta M, Leite I, Dubois B, Rubio J, Butzkueven H, Kilpatrick T, Mycko MP, Selmaj KW, Rio ME, Sa M, Salemi G, Savettieri G, Hillert J, Compston DA. Multiple Sclerosis Severity Score: using disability and disease duration to rate disease severity. Neurology. 2005;64:1144–1151. doi: 10.1212/01.WNL.0000156155.19270.F8. [DOI] [PubMed] [Google Scholar]
- 98.Noseworthy J, Kappos L, Daumer M. Competing interests in multiple sclerosis research. Lancet. 2003;361:350–351. doi: 10.1016/S0140-6736(03)12354-9. [DOI] [PubMed] [Google Scholar]
- 99.Waxman SG. Axonal dysfunction in chronic multiple sclerosis: meltdown in the membrane. Ann Neurol. 2008;63:411–413. doi: 10.1002/ana.21361. [DOI] [PubMed] [Google Scholar]
- 100.Mahad D, Ziabreva I, Lassmann H, Turnbull D. Mitochondrial defects in acute multiple sclerosis lesions. Brain. 2008;131:1722–1735. doi: 10.1093/brain/awn105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Young EA, Fowler CD, Kidd GJ, Chang A, Rudick R, Fisher E, Trapp BD. Imaging correlates of decreased axonal Na+/K+ ATPase in chronic multiple sclerosis lesions. Ann Neurol. 2008;63:428–435. doi: 10.1002/ana.21381. [DOI] [PubMed] [Google Scholar]
- 102.Neymotin A, Calingasan NY, Wille E, Naseri N, Petri S, Damiano M, Liby KT, Risingsong R, Sporn M, Beal MF, Kiaei M. Neuroprotective effect of Nrf2/ARE activators, CDDO ethylamide and CDDO trifluoroethylamide, in a mouse model of amyotrophic lateral sclerosis. Free Radic Biol Med. 2011;51:88–96. doi: 10.1016/j.freeradbiomed.2011.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Stack Cliona, Ho Daniel, Will Elizabeth, Noel Y. Calingasan, Charlotte Williams, Karen Liby, Michael Sporn, Magali Dumont, and M. Flint Beal Triterpenoids CDDO-ethyl amide and CDDO-trifluoroethyl amide improve the behavioral phenotype and brain pathology in a transgenic mouse model of Huntington's disease. Free Radic Biol Med. 2010;49:147–158. doi: 10.1016/j.freeradbiomed.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sauerbeck A, Gao J, Readnower R, Liu M, Pauly JR, Bing G, Sullivan PG. Pioglitazone attenuates mitochondrial dysfunction, cognitive impairment, cortical tissue loss, and inflammation following traumatic brain injury. Exp Neurol. 2011;227:128–135. doi: 10.1016/j.expneurol.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ghosh Sangeeta, Patel Nishant, Rahn Douglas, McAllister Jenna, Sadeghi Sina, Horwitz Geoffrey, Berry Diana, Xuan Kai, Wang, Russell H. Swerdlow The Thiazolidinedione Pioglitazone Alters Mitochondrial Function in Human Neuron-Like Cells. Mol Pharmacol. 2007;71:1695–1702. doi: 10.1124/mol.106.033845. [DOI] [PubMed] [Google Scholar]
- 106.Quintanilla RA, Jin YN, Fuenzalida K, Bronfman M, Johnson GV. Rosiglitazone treatment prevents mitochondrial dysfunction in mutant huntingtinexpressing cells: possible role of peroxisome proliferator-activated receptor-gamma (PPARgamma) in the pathogenesis of Huntington disease. J Biol Chem. 2008;283:25628–25637. doi: 10.1074/jbc.M804291200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sun Albert Y. Resveratrol as a Therapeutic Agent for Neurodegenerative Diseases. Mol Neurobiol. 2010;41:375–383. doi: 10.1007/s12035-010-8111-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ayasolla Kamesh R, Giri Shailendra, Singh Avtar K, Singh Inderjit. 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside (AICAR) attenuates the expression of LPS- and Aβ peptide-induced inflammatory mediators in astroglia. Journal of Neuroinflammation. 2005;2:21. doi: 10.1186/1742-2094-2-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Orsucci D, Mancuso M, Ienco EC, LoGerfo A, Siciliano G. Targeting mitochondrial dysfunction and neurodegeneration by means of coenzyme Q10 and its analogues. Curr Med Chem. 2011;18:4053–4064. doi: 10.2174/092986711796957257. [DOI] [PubMed] [Google Scholar]
- 110.Mancuso Michelangelo, Orsucci Daniele, Calsolaro Valeria, Choub Anna, Siciliano Gabriele. Coenzyme Q10 and Neurological Diseases. Pharmaceuticals. 2009;2:134–149. doi: 10.3390/ph2030134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Noe N, Dillon L, Lellek V, Diaz F, Hida A, Moraes CT, Wenz T. Bezafibrate improves mitochondrial function in the CNS of a mouse model of mitochondrial encephalopathy. Mitochondrion. 2012;12:258–259. doi: 10.1016/j.mito.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Johri Ashu, Calingasan Noel Y, Hennessey Thomas M, Sharma Abhijeet, Yang Lichuan, Wille Elizabeth, Chandra Abhishek, Beal M Flint. Pharmacologic activation of mitochondrial biogenesis exerts widespread beneficial effects in a transgenic mouse model of Huntington's disease. Hum. Mol. Genet. 2012;21:1124–1137. doi: 10.1093/hmg/ddr541. [DOI] [PMC free article] [PubMed] [Google Scholar]