

Abstract
Many tumors are believed to be maintained by a small number of cancer stem-like cells where cure is thought to require eradication of this cell population. In this study, we investigated the dynamics of acute promyelocytic leukemia (APL) before and during therapy with regard to disease initiation, progression and therapeutic response. This investigation employed a mathematical model of hematopoiesis and a dataset derived from the North American Intergroup Study INT0129. The known phenotypic constraints of APL could be explained by a combination of differentiation blockade of PML-RARα positive cells and suppression of normal hematopoiesis. ATRA neutralizes the differentiation block and decreases the proliferation rate of leukemic stem cells in vivo. Prolonged ATRA treatment after chemotherapy can cure APL patients by eliminating the stem-like cell population over the course of approximately one year. To our knowledge, this study offers the first estimate of the average duration of therapy that is required to eliminate stem-like cancer cells from a human tumor, with the potential for the refinement of treatment strategies to better manage human malignancy.
Major Finding
By combining a mathematical model of hematopoiesis with data from a large randomized trial of acute promyelocytic leukemia, this study offers the first determination of the average duration of therapy required to eliminate all stem-like cells in a human tumor.
Quick Guide to Equations and Assumptions
Our multi-compartment model of hematopoiesis has been described elsewhere, but we describe briefly here for clarity (1-6) (Figure 1). Cells in a given compartment i with probability εi differentiate and produce two cells that migrate to the next downstream compartment (i + 1) or self-renew and increase compartment i by one cell with probability 1 − εi. Here, ‘compartments’ are not understood as physical spaces but as an accounting tool to keep track of each cell's replication and differentiation. Proliferation rates ri are intrinsic for each compartment and with ri < ri+1. Parameters describing cells in non-stem cell compartments i > 0 are fixed, by allometric scaling arguments (1) and data from human hematopoiesis and are given by , with γh = 1.26. Thus, the rate of proliferation increases exponentially with the compartment (subscripts refer to the compartment, whereas superscripts refer to healthy (h) or cancerous (c) cells). In the stem cell compartment i = 0, N0 = 400, and days (7-10). Undisturbed, the above system reaches a steady state, where cell numbers fluctuate around an average cell count. In the absence of disease, the steady state for each compartment is given by
Figure 1.
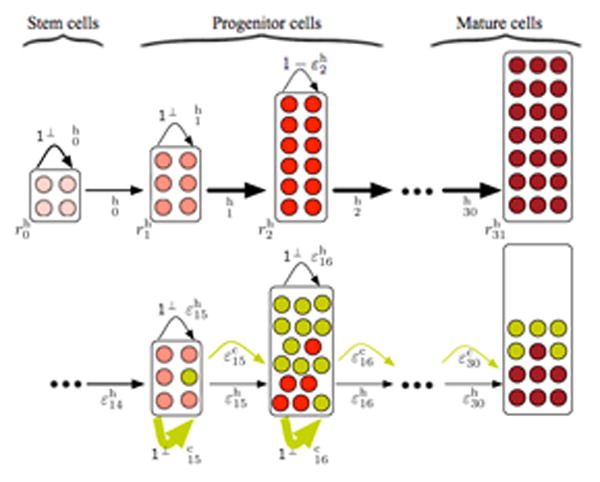
Schematics of our hierarchical hematopoiesis model. At the top, undisturbed healthy hematopoiesis. At the root of our hierarchical structure (compartment 0) is the stem cell compartment with a few slowly dividing stem cells (light red, proliferation rate ) that can self renew with probability and differentiate with probability . Cells undergo several differentiations with probabilities and proliferate with rates until they reach the mature cell state (compartment 31 in our case, dark red). At the bottom, hematopoiesis is disturbed by leukemic cells (dark yellow). The leukemia driving mutation occurs in compartment 15. The self-renewal capacity ( ) of the leukemic cells is significantly increased and malignant cells accumulate in bone marrow compartments. Concomitantly, the proliferation of healthy cells (red) is suppressed by cancer cells and thus the healthy cell count decreases in compartments downstream of compartment 15.
Under this parameter setting we need 32 compartments to represent hematopoiesis with a daily output of ∼ 3.5 × 1011 cells (1-6). This can be viewed as an almost continuous differentiation process of cells (Figure 1).
Because of the high cell turnover rates, the dynamics of normal cells in such hierarchies can be captured by a linear system of differential equations (5, 6). An arbitrary number of mutations can be described analytically by similar equations, if mutated cells proliferate independently (5, 6).
We model the dynamics of normal and leukemic cell lineages by such systems of differential equations. The equations for normal cells follow from the influx and output of healthy cells in each compartment and are derived as follows. Cells in compartment i increase in number due to self-renewal at rate and decrease due to differentiation of cells into compartment i + 1 at rate , leading to a change in number arising from processes within the compartment of . In addition, there is an influx from upstream compartment i − 1, at rate , see Figure 1 for a graphical representation. Collecting all terms give
Here l denotes the compartment where the cancer driving mutation occurs i.e. the leukemic stem-like cells. Compartments upstream of l are not affected by this mutation and proliferate independently (Figure 1). However, proliferation of healthy cells in and downstream of compartment l is potentially inhibited by the leukemic cells . This interference is modeled by Hill-functions (11). In the absence of malignant cells, homeostasis is normal, but proliferation of healthy cells is suppressed with increasing numbers of leukemic cells (12).
The dynamics of leukemic cells can be described by the following set of equations
In the absence of therapy, the number of leukemic cells in the compartment of origin grows exponentially, leading to an even faster growth in downstream compartments. Leukemic cells proliferate independently of healthy cells as evidenced by observations in animal models of disease (13). The leukemia-driving cell(s) occupy compartment l (Figure 1, lower panel). Thus leukemic cells can only be found in and downstream of compartment l. In addition the leukemic cell proliferation properties differ significantly from those of healthy cells.
Model constraints
Phenotype: Since PML-RARα expression reduces differentiation and enhances self-renewal of cells (13-17), we imposed that εc < εh = 0.85. Generally, APL is associated with pancytopenia due to a reduction in bone marrow output. Therefore, we adjusted our parameters to reduce the cells in compartment 31 to 10 – 20% of normal while ensuring that the intramedullary compartments are hypercellular. ATRA reverses the differentiation block and therefore εc → 0.85. In vitro, ATRA slows down the rate of replication of leukemic cells by at least a factor of 0.61 (18). While chemotherapy kills the majority of cancer cells, the proliferation properties of surviving cancer cells remain unchanged.
Time to diagnosis and origin of the disease: The disease must start from a single leukemic stem cell in keeping with the clonal origin of cancer. Using data from Guibal et al (13), we inferred that the minimum time for diagnosis in mice is ∼ 120 days. Using our previously described allometric scaling relationship (19, 20) comparing timescales between mice and men, where T and M refer to the time to diagnosis and the species specific adult mass respectively, we could determine that the minimum time from the appearance of the first leukemic stem cell in humans to diagnosis is > 872 days. An animal model of APL suggests that the disease may originate in a CFU-GM cell (13), which in our model would reside in compartments 13 - 15. A limited study of three patients also suggests that CD34+CD38- cells do not have the t(15q22;17q12) typical of the disease (21).
Clonal burden in bone marrow: The average tumor burden of leukemic stem cells at the time of diagnosis is ∼1% of the tumor population present in the bone marrow (22), and the leukemic cells represent 65 – 98% of the marrow cellularity (23).
The average time for the bone marrow to appear normal after therapy with ATRA is 38 days (range 25 – 90 days) (23).
Patients treated with ATRA alone for induction often have an increase in their leukocyte count that peaks between 12 and 14 days after initiation of therapy (23).
Time to relapse: If patients are treated with ATRA alone until they have a morphologic remission, they relapse on average after ∼110 days (45 to 300) (24, 25).
ATRA does not alter the dynamic properties of normal hematopoietic cells.
Introduction
The cancer stem cell (CSC) hypothesis states that at the root of most (perhaps all) tumors, there is a population of cancer stem cells that is not only able to renew itself but also gives rise to the bulk of the tumor cell population (26-29). These CSC are essential for the origin and the continued growth and maintenance of the tumor. As a consequence, these cells are an important target of therapy since it is thought that eradication of such cells is necessary for a potential cure of the tumor. Various models have been developed that address this hypothesis from a theoretical perspective and there is increasing evidence for its support from animal models of cancer. Although CSC were initially isolated from patients with acute leukemia (26-29), they have now been identified in virtually all types of tumors. It is therefore important to understand the dynamics of these cells under therapy and whether they can be eradicated leading to cure of the disease and long term survival of patients. Here, we utilize data from a clinical trial of therapy for acute promyelocytic leukemia (APL) to understand the dynamics of leukemic stem cells under therapy. We use a mathematical/computational model of hematopoiesis together with quantitative data from the North American Intergroup Study INT0129 (30, 31) to determine the probability that the leukemic stem cells are eradicated at a certain time under this treatment regimen. APL was chosen for a number of reasons: (i) the disease is well defined with most patients having the translocation t(15q22;17q12) leading to PML-RARα oncogene activation (14-16), (ii) the tumor burden can be quantitated using Q-RT-PCR (31, 32), (iii) targeted therapy in the form of all-trans retinoic acid (ATRA) is available and highly effective (33, 34), (iv) the availability of serial quantitative data on disease burden from a large randomized clinical trial that allows us to investigate the effects of ATRA treatment and chemotherapy separately (31, 32, 35), and (v) the availability of a mathematical/computational model of hematopoiesis that has already been utilized to understand the dynamics of mutations in other hematologic disorders (1-6). In the following, we provide a brief summary of the clinical trial, a description of the mathematical model together with the justification of the constraints used to determine the model parameters, followed by a description of the data fitting and presentation of results. Details of the mathematical model as well as the clinical trial are provided in the methods section. In summary, our mathematical model contains a fixed number of compartments that represent different stages of cell differentiation. At each stage, cells proliferate with a fixed rate ri and differentiate into the next downstream compartment with probability εi. This general framework allows us to describe normal hematopoiesis as well as the initiation and progression of different types of leukemia, defined by changes in proliferation and differentiation parameters of malignant cells. As a result, we provide an estimate of the timescale with which a leukemic stem cell population is eliminated in humans.
Materials and Methods
Clinical trial INT0129
The North American Intergroup trial of all-trans retinoic acid (ATRA) in acute promyelocytic leukemia (APL) was initially reported in 1997 with subsequent updates (30, 36, 37). Patients were recruited from centers affiliated with the Cancer and Leukemia Group B, the Southwest Oncology Group and the Eastern Cooperative Oncology Group. Briefly, patients with newly diagnosed APL were randomly assigned to induction therapy either with combination chemotherapy (CA): daunorubicin (45mg/m2 daily on days 1 – 3) and cytosine arabinoside (100 mg/m2 by continuous infusion for 7 days) (N=191) or ATRA alone (45mg/m2 orally in two divided doses daily) that could be given for up to 90 days (N=188). Pediatric patients less than 3 years of age were treated with the same protocol, but with appropriate dose modifications. Subsequently, patients who achieved a complete remission with induction therapy received two cycles of consolidation: the first cycle was identical to the initial induction chemotherapy regimen (CA) while the second cycle consisted of high dose cytosine arabinoside (2g/m2 every 12 hours for 4 days) with daunorubicin 45mg/m2 daily on days 1 and 2). The patients were subsequently randomized to observation or maintenance therapy with ATRA (45mg/m2 orally in two divided doses daily) for up to 1 year. Fifty four patients who received induction with ATRA were randomized to maintenance with ATRA while 56 patients initially treated with chemotherapy went on ATRA maintenance after the second randomization. Patients had serial measurement of the PML-RARα oncogene as previously described (31, 32, 35). Our mathematical/computational model was fitted to this serial data, after normalization of PML-RARα to GAPDH with the pretreatment copy number value of PML-RARα/GAPDH normalized to 1. Serial collection of blood samples for Q-RT-PCR quantification was not mandatory for the trial and as a result the data set is incomplete in this respect.
Parameter estimation
We use the initial condition of 1 leukemic stem cell in compartment l with the described constraints to numerically solve the deterministic equations from above for each compartment using standard numerical procedures implemented in Mathematica. Throughout this process, we assume that the effect of ATRA on leukemic cells is constant. Each numerical solution gives the disease scenario arising from a given set of model parameters. In our case we had to determine four key parameters: (i) the proliferation rate and (ii) differentiation probability in the compartment of the cancer initiating cell ( and ) and (iii) the proliferation rate and (iv) differentiation probability of cancer cells in the bone marrow ( and ). We then performed an extensive parameter search, varying these four parameters within a wide range compatible with the phenotypic constrains of the disease (i.e. and as well as and ). We compared the properties of the modeled disease scenario to the restrictive phenotypic constrains of the disease (points (i)-(vii)) described above. If the model prediction deviated in a single point from the phenotypic constraints, the parameter set was discarded. This resulted in a restricted set of parameters for the disease in the absence of therapy.
We assumed that therapy with either ATRA or chemotherapy was started as soon as a diagnosis was made. The proliferation and differentiation parameters of the leukemic cells were altered to fit the available response data in case of ATRA therapy. Chemotherapy was implemented as a single catastrophic event, in which the majority of cancer and healthy cells are killed, but the proliferation properties of the surviving cancer cells remain unchanged. Healthy cells are not affected by ATRA therapy and thus their dynamic properties are only indirectly altered by the response of the leukemic cells to therapy.
Stochastic simulations
We inferred the distribution of extinction times of leukemic stem-like cells by performing stochastic simulations implemented by a Gillespie algorithm (38), based on an agent-based representation of our hierarchical organization (5). We used parameter estimations from our deterministic fitting procedure and therefore parameters were fixed during all stochastic simulations. We performed in total 104 independent stochastic simulations and recorded the time until all cancer stem-like cells went extinct, leading to a distribution of cancer stem-like extinction times.
Results
Dynamics of untreated APL
Initially, we had to determine the time course and dynamic properties of the leukemic stem and progenitor cells taking into account the constraints that we identified from the literature, in particular the known biology of the disease (13-17, 21, 23) (Figure 1). Given that PML-RARα expression leads to a block in differentiation, we had to impose a lower probability for the differentiation of leukemic cells compared to healthy cells in our model. The leukemic stem cell in APL may arise in a CFU-GM cell (13, 21), and therefore l = 15 was chosen as the founding compartment of APL in our hierarchical model, based on our prior results (1, 39). We estimated that the minimum time between the onset of the first leukemic stem cell and disease was 872 days (13, 19, 20), where disease was defined as a reduction in bone marrow output to ∼20% of normal, leading to cytopenias that are typical for this leukemia. At the same time, the bone marrow compartments will be expanded and the marrow appears hypercellular. The parameter estimates that led to the best fit to the data are presented in Table 1.
Table 1.
Parameter estimate for APL stem and progenitor cells before therapy. Here i = 15 represents the compartment of the founding APL cancer stem-like cell. Compartments 16 ≤ i ≤ 25 represent the bone marrow cell load and compartments 26 ≤ i ≤ 31 differentiated cells that can be found in the bloodstream. The parameters denote the differentiation probabilities of cancerous cells in compartment i and the parameters the increase in proliferation rate of cancerous cells per compartment.
| Parameter | i = 15 | 16 ≤ i ≤ 25 | 26 ≤ i ≤ 31 | |
|---|---|---|---|---|
|
|
1.34 | 1.12 | 1.12 | |
|
|
0.45 | 0.07 | 0.85 |
Our fits suggest that the leukemic stem cells replicate faster and self-renew with a higher probability than normal CFU-GM cells in the same compartment. Even based on their higher self-renewal capacity alone, they have a considerable fitness advantage compared to the normal progenitor cells. If we count the number of offspring cells produced by a mutant cell in a given compartment and compare it to the number of offspring of a normal cell, we obtain for relative fitness in our mathematical model (fj) (40),
This translates into a relative fitness advantage of 6.9 compared to normal CFU-GM (17). Moreover, our fitting suggests that the leukemic progenitor cells (downstream of the leukemic stem cells) have an extremely high fitness advantage (based on the virtual absence of normal cells in the circulation), estimated at 75 compared to their normal counterparts (normalized to 1) due to the block in differentiation (and enhanced self-renewal). These estimates provide a vivid explanation of the rapid disease progression and early high lethality associated with this disease prior to the advent of ATRA therapy.
Dynamics of disease under chemotherapy treatment
We implement chemotherapy treatment as a single catastrophic event, in which the majority of both leukemic and healthy cells are killed instantly. The proliferation parameters of surviving cells remain unchanged after chemotherapy treatment. Therefore, we only need to infer a single parameter (the fraction of killed cells under chemotherapy) to determine the dynamics of APL patients under chemotherapy from our mathematical model. We vary the fraction of killed cells and fit the resulting dynamics to the serial Q-RT-PCR data of the fraction of patients treated with chemotherapy in the INT0129 trial. Our best parameter estimates, see Figures 2 and 3, suggest that only 0.3% of all cancer cells survived, compatible with more than 2 log kill of leukemic cells. However, this also implies that approximately 105 leukemic stem cells survive and relapse is to be expected. This prediction is confirmed by observations from the follow up of the INT0129 trial were the risk of relapse of chemotherapy treated patients (without ATRA maintenance) was high. Note, that the characteristic peak of bone marrow output, 10-12 days after ATRA treatment, does not occur with chemotherapy (Figure 2A versus C).
Figure 2.

Clinical data and model predictions, sub-divided in different treatment classes. Panel a shows the treatment progression of patients with ATRA therapy for up to 90 days (triangles) and patients with continued ATRA therapy for up to 1 year (dots) and our best model fit (line, R2 = 0.43, see Methods and Materials section for details on the fitting procedure) for the first 250 days after treatment initiation. Panel b the complete available data set for ATRA treated patients (follow up of up to 1600 days after treatment initiation). The initial treatment response cannot be distinguished for both groups. Yet the relapse probability for patients without maintenance therapy is higher and they tend to relapse faster. Panel c Group of patients treated with chemotherapy only (cubes) and our model prediction for this treatment scenario (line, (R2 = 0.38). First, the significant initial peak in tumor burden that is typical for ATRA treatment is absent in this case. Second, also there is a good initial response, chemotherapy is likely not able to kill all cancer stem cells. Therefore relapse occurs often and relatively quickly. If chemotherapy is followed by ATRA maintenance therapy (diamonds), tumor burden tends to diminish. Panel d shows the complete available data set for chemotherapy with and without ATRA maintenance.
Figure 3.
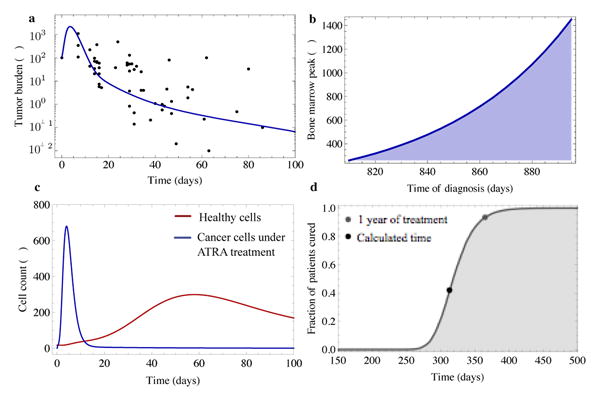
a Response of APL cells to ATRA treatment inferred from the North American Intergroup trial (dots) and our model predictions (line, R2 = 0.54, model parameter as in Tables 1 and 2). A peak in tumor burden (here after 7-10 days) of ATRA treatment is typical of APL and is caused by the rapid differentiation of APL progenitor cells. B Dependence of the height of the bone marrow peak on the time of disease diagnosis. Later diagnosis results in a higher bone marrow cancer cell load and thus in a higher peak after treatment. c Cell count of healthy and leukemic cells under ATRA treatment inferred from the model (parameters as in a). The peak of leukemia cells occurs approximately 7 days after treatment initiation. The healthy cell count peaks approximately 50 days after treatment initiation and declines to normal levels afterwards. d Estimated probability of patients being cured (grey line) under continuous ATRA treatment inferred from stochastic individual based computer simulations (see methods section for details, parameters as in a). The black dot corresponds to the deterministic extinction time (here after 312 days) of the leukemic cancer stem-like cells given by equation (3). Because of the inherent stochastic nature of the proliferation properties of the cancer stem-like cells only 42% of patients are cured after 312 days of ATRA treatment and we expect 58% of the patients with remaining APL stem-like cells in the bone marrow. After one year of ATRA treatment (grey dot) approximately 92% of patients have no remaining cancer stem-like cells left.
Dynamics of disease under therapy with ATRA
ATRA alters the behavior of leukemic cells by (i) inducing differentiation and (ii) slows down the rate of replication of leukemic stem cells. Fitting of our model to serial RT-PCR data from the INT0129 trial (Figure 2 and 3A) provides an estimate for the leukemic cell parameters under therapy as reported in Table 2. We consider that therapy starts immediately after diagnosis or 870 days from the appearance of the first leukemic stem cell. We assume that the differentiation probabilities return to normal under ATRA therapy. The proliferation rates within the bone marrow, , are fixed by the time until the bone marrow output peaks. The proliferation rate of cancer stem cells, , has only a minor influence on this dynamics due to the exponential growth characteristics of hematopoiesis, and is thus difficult to estimate from the data. However, it is a crucial parameter to assess the probability of relapse. For the best parameter estimate, the rate of replication of the leukemic stem cells returns to normal compared to other CFU-GM cells . At the same time, the differentiation block of the cells is removed . Zhu et al. showed that the doubling time of NB4 cells treated with ATRA increased in vitro from 25.2 hours to 41.26 (slowed the cells by a factor of 0.6) (18). In our in vivo model, there is also such a slowdown effect. Our proliferation rates in compartment i scale via ri = (γ)ir0.
Table 2.
Parameters estimates for APL cells in response to ATRA therapy. Here and denote the differentiation probabilities in compartment i of healthy cells and cancerous under ATRA treatment respectively. The parameters and represent the relative increase of the proliferation rate per compartment for healthy cells and cancer cells under ATRA treatment.
| Parameter | i = 15 | 16 ≤ i ≤ 25 | 26 ≤ i ≤ 31 | |
|---|---|---|---|---|
|
|
1.26 | 1.26 | 1.26 | |
|
|
1.26 | 1.44 | 1.44 | |
|
|
0.85 | 0.85 | 0.85 | |
|
|
0.85 | 0.85 | 0.85 |
Thus a decrease in doubling times of cancer cells under ATRA treatment by a factor of 0.6 in the experiment corresponds to ≈ 0.4 in our theoretical model based on the scaling of replication rates. With this in mind, our relative reduction in leukemic stem cell replication (γc = 0.34 → γATRA = 1.26) is in qualitative agreement with the finding of Zhu et al. We note that the replication rate in vivo would be expected to be slower than what is observed in vitro (41).
In addition, ATRA therapy affects more downstream progenitor cells correcting their differentiation block back to normal . Fitting also suggests that ATRA increases the rate of replication of downstream leukemic progenitors compared to normal cells. The latter prediction is compatible with the observation of a rapid increase in the neutrophil count in patients treated with ATRA alone (Figure 2 and 3C).
In Figure 4, we provide a comparison of individual fits of the model to patient specific data for a patient induced with ATRA (A) and another patient randomized to chemotherapy only induction (B). The model fits especially well the ATRA treated patient.
Figure 4.

Panel a Disease dynamics for a single patient treated with ATRA (R2 = 0.95). Panel b Disease dynamics of a single patient treated with chemotherapy (R2 = 0.61). The initial response to ATRA treatment is well described by our model prediction (blue line). However, this patient did not receive maintenance therapy and finally relapse 861 days after the initial treatment. In contrast, the patient treated with chemotherapy has an initial response to therapy, but progresses in between cycles of chemotherapy. This variation could be caused by stochastic effects of cell proliferation together with a varying number of surviving cancer stem-like cells per treatment.
Extinction time of leukemic stem cells
Our model is in line with in vivo mouse experiments (13) that prior to the initiation of therapy, the leukemic stem cell population (i = 15), increases exponentially (Figure 3B). Thus at the time of diagnosis (Tdiag), we expect to find cells, where
| (1) |
For our parameters, we obtain 4 × 107 leukemic stem cells at the time of diagnosis in compartment 15 (CFU-GM), where the mutation originates (19,20). Under ATRA therapy, the number of leukemic stem cells decreases exponentially such that
| (2) |
Therefore, the average extinction time for the leukemic stemlike-cells (i.e. for the elimination of all leukemic stem-like cells) is given by:
| (3) |
On average the time for clonal extinction under ATRA treatment is ∼312 days (0.36 × 870 days). However, this time increases exponentially with the number of cells at diagnosis, and therefore, continued therapy with ATRA is a prerequisite to cure this disease. These results are compatible with clinical observations and justify the need for maintenance therapy for ∼ 1 year that seems to lead to a cure in many patients as treated in the INT0129 trial (Figure 3D).
Relapse
Relapse of seemingly successful treated patients is a common phenomenon in acute leukemia. Presumably, this relapse is caused by the remaining cancer stem and progenitor cells after therapy as well as the selection of mutant cells resistant to therapy through a variety of mechanisms, including mutations in the LBD domain of RARα, increased ATRA catabolism, abnormal trafficking of ATRA to the nucleus, the presence of cytoplasmic retinoic acid binding protein and overexpression of BP1 (42, 43). Thus the question of whether and when treatment eradicates all cancer stem cells is critical. As intrinsic cell properties such as the exact time of cell proliferation or differentiation are stochastic, one naturally expects the actual extinction time of the leukemic stem cell pool to differ in patients (44, 45). To obtain the distribution of these extinction times under ATRA treatment we implemented a computational representation of the mathematical model (see Methods) by utilizing standard Gillespie algorithms (38) and ran exact individual based stochastic simulations on this model. We assumed that the initial decline from approximately 4 × 107 to 105 leukemic stem cells under ATRA treatment follows the deterministic equations. We performed 104 independent realizations of the stochastic simulations initialized with 105 leukemic stem cells under ATRA treatment that use the parameter estimations from Table 1 and 2 and recorded the extinction times of the leukemic stem cell pool. The probability of leukemic stem cell eradication under ATRA therapy that lasted less than 200 days is negligible (Figure 3D and Figure 5). Only 42% of patients would be expected to be cured after 312 days (deterministic extinction time), imposing a high risk of relapse for over half of the patients. However, after one year of ATRA treatment up to 92% of patients are free of leukemic stem cells according to this model and therefore ‘cured’ of their disease.
Figure 5.
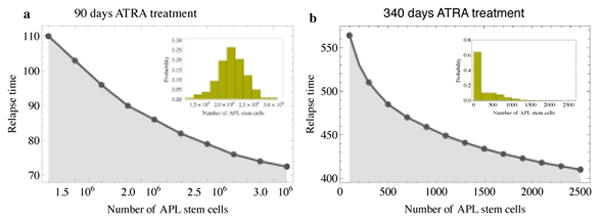
The insets show the distribution of remaining APL stem cells for (a) 90 days of continued ATRA treatment and (b) 340 days of continued ATRA treatment due to stochastic individual based simulations with the parameters from Table 1 and 2. After 340 days of ATRA treatment almost 60% of patients are expected to be cancer stem cell free and thus potentially cured. The main panels shows the expected relapse time of patients where elimination of cancer stem cells was not achieved, depending on the size of the remaining pool of cancer stem cells. These relapse times correspond well with observations in clinical studies.
Discussion
If we hope to better treat cancer, with emerging therapeutic approaches, a detailed understanding of cancer initiation, cancer progression and response to treatment is essential. Here, we combine a mathematical/computational approach with clinical trial data of treatment response of APL patients to ATRA therapy and/or chemotherapy. This approach allows us to model leukemia progression from the occurrence of the first leukemic stem cell until the potential elimination of the last cancer stem cell under treatment and thus provides a detailed understanding of all phases of APL under two different treatment regimes. At least one murine model of APL suggests that the disease originates in a CFU-GM cell (13, 22) and thus within the lower half (here compartment 15) of our hierarchical model. We acknowledge that there is still some disagreement on the true origin of the leukemic stem cell in APL (21, 46, 47), partly based on the animal model used (46, 48) and it is likely that other mutations in addition to the t(15q22;17q12) are required for APL to develop (49). Although the cancer stem cell hypothesis is increasingly accepted, and cancer initiating (stem) cells have been isolated from many tumors, the field is still somewhat controversial (27-29). There are several possible explanations for the divergent results observed vis-à-vis the presence, frequency, surface marker expression and functional properties of these putative cells including (i) the animal model used for engraftment that provides the complex microenvironment for cells to survive and grow, (ii) genetic/epigenetic heterogeneity between tumors, (iii) stage of the tumor, and others (47-49). However the presence of cellular hierarchies within acute leukemia is less controversial and a critical component of our modeling approach.
We find that an interaction of leukemic and healthy cells is sufficient to explain the known phenotypic constrains of APL. The bone marrow in APL usually appears hypercellular at diagnosis, but a fraction of patients may have a hypocellular bone marrow at diagnosis. This can be explained by the suppression of healthy cells combined with a differentiation block of APL cells in the bone marrow. Our model of acute promyelocytic leukemia also reveals that bone marrow failure syndromes are not necessarily due to failures of the normal hematopoietic stem cell pool. They can also occur by suppression of the proliferation of normal cells by leukemic cells, for example due to competition for cytokines. This also implies that hematopoiesis returns to normal after the eradication of the malignant cells as is typical for many acute leukemias and is in line with our model as well as recent observations in vivo (12).
Our model suggests that, in addition to the block of differentiation, APL stem-like cells have an increased proliferation rate, and thus they have a significant fitness advantage (6.9 to normal CFU-GM cells with 1.0) compared to normal cells. Despite this fitness advantage the disease progresses slowly initially, as cells accumulate in the bone marrow, but only weakly affect the output of normal hematopoietic cells (17). The output of fully differentiated healthy cells only starts to decrease slowly approximately 600 days after the occurrence of the first leukemic stem cell and diagnosis typically occurs after approximately 870 days (2.4 years). This is substantially shorter than the timescale for the clinical development of chronic myeloid leukemia, multiple myeloma or solid malignancies such as colon cancer (3, 50).
We find that chemotherapy provides a significant initial response but is unlikely to cure the disease, as a substantial fraction of leukemic stem-like cells is expected to survive treatment and leads to relapse of the disease. Indeed we find several patients in the INT0129 trial that underwent three cycles of chemotherapy (per protocol) but relapsed in time intervals of approximately 100-300 days.
The model suggests that ATRA has differential effects on APL stem-like cells compared to leukemic cells further downstream within its hierarchy. ATRA removes the differentiation block and increases the proliferation rate of APL cells in the bone marrow. This leads to a rapid expansion of most APL progenitor cells and causes the typical bone marrow peak after 10-13 days of ATRA treatment. The bone marrow will appear free of APL blasts after approximately 20 days. However, cure requires the extinction of all APL stem-like cells. We find, in line with in vitro studies, that ATRA decreases the proliferation rate of leukemic stem-like cells to 0.4 compared to untreated leukemic stem-like cells and thus slows down the eradication of APL stem-like cells. Thus, despite a fast initial response, continued ATRA therapy is necessary to reduce the risk of relapse. Our model suggests that, in the absence of additional mutations, ATRA therapy for one year would result in a high likelihood of eradication of all leukemic stem-like cells and thus a cure in many patients. This is compatible with long term follow-up of patients on the INT0129 trial where only 3.3% of patients who achieved complete remission experienced a late relapse (defined as occurring >3 years after diagnosis) (37). Our model in its current form does not consider the intrinsic heterogeneity present in most leukemias or the emergence of mutant subclones that may lead to relapse of the disease. Such extensions of the model may be possible in the future but require more detailed knowledge about the structure of the tumor. Regardless, our modeling also illustrates the impact of stochastic effects on the response to treatment, and in part explains why outcomes can be vastly different between patients with similar disease status at diagnosis, even without considerations of tumor evolution. However, our model suggests that continued ATRA treatment can eventually eradicate all leukemic stem-like cells and thus potentially cure APL, and provides for the first time a time scale for the in vivo eradication of leukemic stem-like cells in humans.
Acknowledgments
This study was coordinated by the ECOG-ACRIN Cancer Research Group (Robert L. Comis, MD and Mitchell D. Schnall, MD, PhD, Group Co-Chairs) and supported in part by Public Health Service Grants CA21115, CA14958, CA13650, CA17145, CA86726, and CA56771 from the National Cancer Institute, National Institutes of Health and the Department of Health and Human Services.
Footnotes
Its content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute.
References
- 1.Dingli D, Traulsen A, Pacheco JM. Compartmental architecture and dynamics of hematopoiesis. PloS one. 2007;2:e345. doi: 10.1371/journal.pone.0000345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Traulsen A, Pacheco JM, Dingli D. On the origin of multiple mutant clones in paroxysmal nocturnal hemoglobinuria. Stem cells (Dayton, Ohio) 2007;25:3081–4. doi: 10.1634/stemcells.2007-0427. [DOI] [PubMed] [Google Scholar]
- 3.Dingli D, Traulsen A, Pacheco JM. Chronic myeloid leukemia: origin, development, response to therapy and relapse. Clin Leukemia. 2008;2:133–9. [Google Scholar]
- 4.Dingli D, Luzzatto L, Pacheco JM. Neutral evolution in paroxysmal nocturnal hemoglobinuria. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:18496–500. doi: 10.1073/pnas.0802749105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Werner B, Dingli D, Lenaerts T, Pacheco JM, Traulsen A. Dynamics of mutant cells in hierarchical organized tissues. PLoS computational biology. 2011;7:e1002290. doi: 10.1371/journal.pcbi.1002290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Werner B, Dingli D, Traulsen A. A deterministic model for the occurrence and dynamics of multiple mutations in hierarchically organized tissues. J R Soc Interface. 2013;10:20130349. doi: 10.1098/rsif.2013.0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buescher ES, Alling DW, Gallin JI. Use of an X-linked human neutrophil marker to estimate timing of lyonization and size of the dividing stem cell pool. J Clin Invest. 1985;76:1581–4. doi: 10.1172/JCI112140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rufer N, Brummendorf TH, Kolvraa S, Bischoff C, Christensen K, Wadsworth L, et al. Telomere fluorescence measurements in granulocytes and T lymphocyte subsets point to a high turnover of hematopoietic stem cells and memory T cells in early childhood. J Exp Med. 1999;190:157–67. doi: 10.1084/jem.190.2.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shepherd BE, Guttorp P, Lansdorp PM, Abkowitz JL. Estimating human hematopoietic stem cell kinetics using granulocyte telomere lengths. Exp Hematol. 2004;32:1040–50. doi: 10.1016/j.exphem.2004.07.023. [DOI] [PubMed] [Google Scholar]
- 10.Shepherd BE, Kiem HP, Lansdorp PM, Dunbar CE, Aubert G, Larochelle A, et al. Hematopoietic stem cell behavior in non-human primates. Blood. 2007;110:1806–13. doi: 10.1182/blood-2007-02-075382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heck HD. Statistical theory of cooperative binding to proteins. The Hill equation and the binding potential. J Am Chem Soc. 1971;93:23–9. doi: 10.1021/ja00730a004. [DOI] [PubMed] [Google Scholar]
- 12.Miraki-Moud F, Anjos-Afonso F, Hodby KA, Griessinger E, Rosignoli G, Lillington D, et al. Acute myeloid leukemia does not deplete normal hematopoietic stem cells but induces cytopenias by impeding their differentiation. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:13576–81. doi: 10.1073/pnas.1301891110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guibal FC, Alberich-Jorda M, Hirai H, Ebralidze A, Levantini E, Di Ruscio A, et al. Identification of a myeloid committed progenitor as the cancer-initiating cell in acute promyelocytic leukemia. Blood. 2009;114:5415–25. doi: 10.1182/blood-2008-10-182071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de The H, Chomienne C, Lanotte M, Degos L, Dejean A. The t(15;17) translocation of acute promyelocytic leukaemia fuses the retinoic acid receptor alpha gene to a novel transcribed locus. Nature. 1990;347:558–61. doi: 10.1038/347558a0. [DOI] [PubMed] [Google Scholar]
- 15.Piazza F, Gurrieri C, Pandolfi PP. The theory of APL. Oncogene. 2001;20:7216–22. doi: 10.1038/sj.onc.1204855. [DOI] [PubMed] [Google Scholar]
- 16.Ablain J, de The H. Revisiting the differentiation paradigm in acute promyelocytic leukemia. Blood. 2011;117:5795–802. doi: 10.1182/blood-2011-02-329367. [DOI] [PubMed] [Google Scholar]
- 17.Welch JS, Yuan W, Ley TJ. PML-RARA can increase hematopoietic self-renewal without causing a myeloproliferative disease in mice. J Clin Invest. 2011;121:1636–45. doi: 10.1172/JCI42953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu J, Shi XG, Chu HY, Tong JH, Wang ZY, Naoe T, et al. Effect of retinoic acid isomers on proliferation, differentiation and PML relocalization in the APL cell line NB4. Leukemia. 1995;9:302–9. [PubMed] [Google Scholar]
- 19.Pacheco JM, Traulsen A, Antal T, Dingli D. Cyclic neutropenia in mammals. American journal of hematology. 2008;83:920–1. doi: 10.1002/ajh.21295. [DOI] [PubMed] [Google Scholar]
- 20.Pacheco JM, Traulsen A, Dingli D. The allometry of chronic myeloid leukemia. J Theor Biol. 2009;259:635–40. doi: 10.1016/j.jtbi.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 21.Turhan AG, Lemoine FM, Debert C, Bonnet ML, Baillou C, Picard F, et al. Highly purified primitive hematopoietic stem cells are PML-RARA negative and generate nonclonal progenitors in acute promyelocytic leukemia. Blood. 1995;85:2154–61. [PubMed] [Google Scholar]
- 22.Nasr R, Guillemin MC, Ferhi O, Soilihi H, Peres L, Berthier C, et al. Eradication of acute promyelocytic leukemia-initiating cells through PML-RARA degradation. Nature medicine. 2008;14:1333–42. doi: 10.1038/nm.1891. [DOI] [PubMed] [Google Scholar]
- 23.Chen ZX, Xue YQ, Zhang R, Tao RF, Xia XM, Li C, et al. A clinical and experimental study on all-trans retinoic acid-treated acute promyelocytic leukemia patients. Blood. 1991;78:1413–9. [PubMed] [Google Scholar]
- 24.Huang ME, Ye YC, Chen SR, Chai JR, Lu JX, Zhoa L, et al. Use of all-trans retinoic acid in the treatment of acute promyelocytic leukemia. Blood. 1988;72:567–72. [PubMed] [Google Scholar]
- 25.Frankel SR, Eardley A, Heller G, Berman E, Miller WH, Jr, Dmitrovsky E, et al. All-trans retinoic acid for acute promyelocytic leukemia. Results of the New York Study. Ann Intern Med. 1994;120:278–86. doi: 10.7326/0003-4819-120-4-199402150-00004. [DOI] [PubMed] [Google Scholar]
- 26.Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645–8. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- 27.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–11. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 28.Rosen JM, Jordan CT. The increasing complexity of the cancer stem cell paradigm. Science (New York, NY. 2009;324:1670–3. doi: 10.1126/science.1171837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kreso A, Dick JE. Evolution of the Cancer Stem Cell Model. Cell Stem Cell. 2014;14:275–91. doi: 10.1016/j.stem.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 30.Tallman MS, Andersen JW, Schiffer CA, Appelbaum FR, Feusner JH, Ogden A, et al. All-trans-retinoic acid in acute promyelocytic leukemia. N Engl J Med. 1997;337:1021–8. doi: 10.1056/NEJM199710093371501. [DOI] [PubMed] [Google Scholar]
- 31.Gallagher RE, Yeap BY, Bi W, Livak KJ, Beaubier N, Rao S, et al. Quantitative real-time RT-PCR analysis of PML-RAR alpha mRNA in acute promyelocytic leukemia: assessment of prognostic significance in adult patients from intergroup protocol 0129. Blood. 2003;101:2521–8. doi: 10.1182/blood-2002-05-1357. [DOI] [PubMed] [Google Scholar]
- 32.Slack JL, Willman CL, Andersen JW, Li YP, Viswanatha DS, Bloomfield CD, et al. Molecular analysis and clinical outcome of adult APL patients with the type V PML-RARalpha isoform: results from intergroup protocol 0129. Blood. 2000;95:398–403. [PubMed] [Google Scholar]
- 33.Tallman MS, Altman JK. How I treat acute promyelocytic leukemia. Blood. 2009;114:5126–35. doi: 10.1182/blood-2009-07-216457. [DOI] [PubMed] [Google Scholar]
- 34.Chomienne C, Balitrand N, Ballerini P, Castaigne S, de The H, Degos L. All-trans retinoic acid modulates the retinoic acid receptor-alpha in promyelocytic cells. J Clin Invest. 1991;88:2150–4. doi: 10.1172/JCI115547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gallagher RE, Willman CL, Slack JL, Andersen JW, Li YP, Viswanatha D, et al. Association of PML-RAR alpha fusion mRNA type with pretreatment hematologic characteristics but not treatment outcome in acute promyelocytic leukemia: an intergroup molecular study. Blood. 1997;90:1656–63. [PubMed] [Google Scholar]
- 36.Tallman MS, Andersen JW, Schiffer CA, Appelbaum FR, Feusner JH, Woods WG, et al. All-trans retinoic acid in acute promyelocytic leukemia: long-term outcome and prognostic factor analysis from the North American Intergroup protocol. Blood. 2002;100:4298–302. doi: 10.1182/blood-2002-02-0632. [DOI] [PubMed] [Google Scholar]
- 37.Douer D, Zickl LN, Schiffer CA, Appelbaum FR, Feusner JH, Shepherd L, et al. All-trans retinoic acid and late relapses in acute promyelocytic leukemia: very long-term follow-up of the North American Intergroup Study I0129. Leuk Res. 2013;37:795–801. doi: 10.1016/j.leukres.2013.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gillespie DT. Exact stochastic simulation of coupled chemical reactions. The Journal of Physical Chemistry. 1977;81:2340–61. [Google Scholar]
- 39.Dingli D, Antal T, Traulsen A, Pacheco JM. Progenitor cell self-renewal and cyclic neutropenia. Cell proliferation. 2009;42:330–8. doi: 10.1111/j.1365-2184.2009.00598.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Traulsen A, Pacheco JM, Dingli D. Reproductive fitness advantage of BCR-ABL expressing leukemia cells. Cancer letters. 2010;294:43–8. doi: 10.1016/j.canlet.2010.01.020. [DOI] [PubMed] [Google Scholar]
- 41.West GB, Woodruff WH, Brown JH. Allometric scaling of metabolic rate from molecules and mitochondria to cells and mammals. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(Suppl 1):2473–8. doi: 10.1073/pnas.012579799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Awwad RT, Do K, Stevenson H, Fu SW, Lo-Coco F, Costello M, et al. Overexpression of BP1, a homeobox gene, is associated with resistance to all-trans retinoic acid in acute promyelocytic leukemia cells. Ann Hematol. 2008;87:195–203. doi: 10.1007/s00277-007-0402-7. [DOI] [PubMed] [Google Scholar]
- 43.Tomita A, Kiyoi H, Naoe T. Mechanisms of action and resistance to all-trans retinoic acid (ATRA) and arsenic trioxide (As2O 3) in acute promyelocytic leukemia. Int J Hematol. 2013;97:717–25. doi: 10.1007/s12185-013-1354-4. [DOI] [PubMed] [Google Scholar]
- 44.Dingli D, Traulsen A, Pacheco JM. Stochastic dynamics of hematopoietic tumor stem cells. Cell cycle (Georgetown, Tex. 2007;6:441–6. doi: 10.4161/cc.6.4.3853. [DOI] [PubMed] [Google Scholar]
- 45.Traulsen A, Lenaerts T, Pacheco JM, Dingli D. On the dynamics of neutral mutations in a mathematical model for a homogeneous stem cell population. J R Soc Interface. 2013;10:20120810. doi: 10.1098/rsif.2012.0810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grimwade D, Enver T. Acute promyelocytic leukemia: where does it stem from? Leukemia. 2004;18:375–84. doi: 10.1038/sj.leu.2403234. [DOI] [PubMed] [Google Scholar]
- 47.Wojiski S, Guibal FC, Kindler T, Lee BH, Jesneck JL, Fabian A, et al. PML-RARalpha initiates leukemia by conferring properties of self-renewal to committed promyelocytic progenitors. Leukemia. 2009;23:1462–71. doi: 10.1038/leu.2009.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wartman LD, Welch JS, Uy GL, Klco JM, Lamprecht T, Varghese N, et al. Expression and function of PML-RARA in the hematopoietic progenitor cells of Ctsg-PML-RARA mice. PloS one. 2012;7:e46529. doi: 10.1371/journal.pone.0046529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wartman LD, Larson DE, Xiang Z, Ding L, Chen K, Lin L, et al. Sequencing a mouse acute promyelocytic leukemia genome reveals genetic events relevant for disease progression. J Clin Invest. 2011;121:1445–55. doi: 10.1172/JCI45284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hobbs JR. Growth rates and responses to treatment in human myelomatosis. Br J Haematol. 1969;16:607–17. doi: 10.1111/j.1365-2141.1969.tb00441.x. [DOI] [PubMed] [Google Scholar]