

Abstract
Objective
To compare the complexity of the amniotic fluid supernatant cell-free fetal transcriptome as described by RNA-Sequencing (RNA-Seq) and gene expression microarrays.
Methods
Cell-free fetal RNA from the amniotic fluid supernatant of five euploid mid-trimester samples was divided and prepared in tandem for analysis by either the Affymetrix HG-U133 Plus 2.0 Gene Chip microarray or Illumina HiSeq. Transcriptomes were assembled and compared on the basis of presence of signal, rank-order gene expression, and pathway enrichment using Ingenuity Pathway Analysis (IPA). RNA-Seq data were also examined for evidence of alternative splicing.
Results
Within individual samples, gene expression was strongly correlated (R=0.43-0.57). Fewer expressed genes were observed using RNA-Seq than gene expression microarrays (4,158 versus 8,842). Most of the top pathways in the “Physiological Systems Development and Function” IPA category were shared between platforms, although RNA-Seq yielded more significant p-values. Using RNA-Seq, examples of known alternative splicing were detected in several genes including H19 and IGF2.
Conclusions
In this pilot study, we found that expression microarrays gave a broader view of overall gene expression, while RNA-Seq demonstrated alternative splicing and specific pathways relevant to the developing fetus. The degraded nature of cell-free fetal RNA presented technical challenges for the RNA-Seq approach.
Introduction
During fetal development, amniotic fluid (AF) is in direct contact with several fetal tissues, including oropharynx, lung, gastrointestinal tract, and skin. Cells and cell-free transcripts from these tissues can be found within AF. Centrifugation of amniotic fluid allows extraction of cells for use by genetic diagnostic laboratories, leaving the cff RNA suspended in the AFS, which is routinely discarded. Our laboratory has shown that a complex fetal transcriptome can be derived from AFS cff RNA. The fetal transcriptome changes with gestational age and the ongoing development of the fetus.1,2,3 This transcriptome includes transcripts for which expression is limited to the central nervous system.4 Additionally, prior work from our laboratory has demonstrated that distinct molecular phenotypes exist for fetuses affected with a variety of conditions, including trisomies 18 and 21 and twin-twin transfusion syndrome.5,6,7
Until recently, characterization of the human fetal transcriptome has relied exclusively on expression microarray-based approaches. The Affymetrix GeneChip Human Genome U133 Plus 2.0 microarray queries expression of over 47,000 coding and noncoding transcripts representing over 20,000 genes. In recent years, massively parallel sequencing (MPS) has been increasingly utilized in both basic research and clinical applications. Among the most widely-used options is the Illumina HiSeq platform for RNA-Seq, which employs sequencing by synthesis to quantify the abundance of RNA transcripts in a given sample. Unlike a standard gene expression microarray, RNA-Seq allows for an unbiased approach to survey gene expression, with the potential to create a census of both known and novel RNA transcripts and transcript isoforms. Importantly, RNA-Seq allows for greater dynamic range than an expression microarray, with an ability to detect transcripts of low abundance, provided there is sufficient depth of sequencing. Also, there is no limit to the maximum expression level that may be detected, which allows for better differentiation among highly expressed transcripts than microarrays, for which signal intensity plateaus, erasing any nuances of differential expression at high levels.
Several prior studies comparing the two technologies have found RNA-Seq to be similar to or more sensitive than expression microarrays when used for differential gene expression analysis of cellular RNA, especially for highly expressed genes.8,9,10,11 In other studies, this conclusion was reiterated, with the caveat that for low expressed genes, microarrays had decreased technical variation compared to RNA-Seq.12,13 In the current study, rather than examining differential gene expression in cellular RNA, we focused on a unique sample type, AFS cff RNA, to profile the fetal transcriptome.
The fetal transcriptome, as derived from AFS cff RNA, presents a snapshot of fetal development, informing only on the processes that are active up to and during the gestational age at which the amniotic fluid is collected.14 Use of this material provides both distinct benefits and challenges. AFS cff RNA provides information on the active development of multiple tissues in the living human fetus, but simultaneously presents a technical challenge due to its dilute and degraded nature.
In this proof-of-principle pilot experiment, we sought to compare the information obtained on fetal development as provided by expression microarray and RNA-Seq. For each of five amniotic fluid supernatant samples, RNA was extracted and prepared in tandem for analysis by both approaches (Figure 1). For each platform, we defined an amniotic fluid core transcriptome (AFCT), which was comprised of the set of genes for which transcripts were detected in all of the five samples. These two AFCTs were compared on the basis of number of genes observed, relative expression levels of these genes, and active molecular pathways identified. The ultimate goal of this work was to address whether the AFCT as described by RNA-Seq captured the same complexity as the AFCT captured by microarray.
Figure 1.
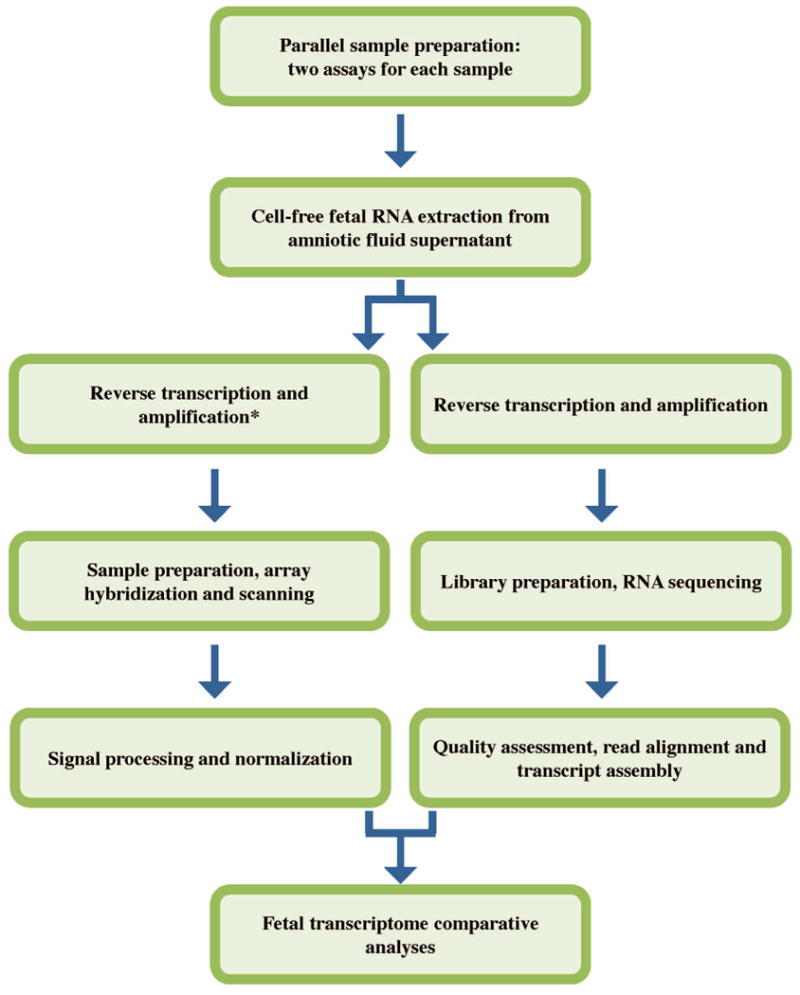
Materials and Methods
Ethical approval
The procedures employed in this study were reviewed and approved by the Tufts Medical Center Institutional Review Board.
Extraction of RNA from amniotic fluid
This study used five samples of amniotic fluid from second-trimester euploid male or female fetuses free from known structural anomalies. Cff RNA was extracted from five individual second trimester amniotic fluid supernatant samples as previously described.15 RNA quality was checked by qRT-PCR for GAPDH gene expression (Applied Biosystems) and the Agilent 2100 Bioanlyzer Total RNA Pico chip (Agilent Technologies). The extracted RNA was then divided in half, with one half dedicated for preparation on the microarray pipeline, and one half for preparation on the RNA-Seq pipeline.
Preparation for and analysis by expression microarray
Full methods are available online (Supplementary Methods). cDNA generation and fragmentation, microarray processing and signal normalization were performed as previously described.5 Array data for the five samples used in this study were previously published as part of a larger study that included sixteen samples.7 Raw microarray CEL files, along with normalized expression values, for the subset of five samples used in this experiment are publicly available at NCBI's Gene Expression Omnibus16 using the GEO Series accession number GSE49893. RefSeq annotation as assigned by Ingenuity Pathway Analysis (IPA, Ingenuity® Systems, www.ingenuity.com) was used (Content version 17199142).
Preparation for and analysis by RNA-Seq
Full methods are available online (Supplementary Methods). cDNA was generated using the Ovation RNA-Seq System V2 (NuGEN) and purified with the MinElute Reaction Cleanup Kit (QIAGEN). One paired-end indexed library was sequenced per sample to a length of 50 nucleotides per mate at a depth of 17.7 × 106 − 98.5 × 106 mate-pairs per library using the Illumina HiSeq 2000 instrument. Reads were aligned to the Hg19 UCSC using the Spliced Transcripts Alignment to a Reference (STAR) aligner.17 Cufflinks was used for transcript assembly18 with the Hg19 goldenPath UCSC annotation GTF file. Raw FASTQ files for these experiments, along with processed files, are publicly available at NCBI's Gene Expression Omnibus16 using the GEO Series accession number GSE49893. Genes were considered “present” in a given sample if the fragments per kilobase of transcript per million mapped fragments (FPKM) was ≥1. For the purposes of matching against the microarray data, UCSC gene symbols were translated to RefSeq gene symbols using IPA.
Functional analyses
Traditionally, IPA is used with differential gene expression results, but we have successfully used it in the past to identify active biological pathways in the amniotic fluid core transcriptome.4 AFCTs for each platform were subjected to IPA core analysis, the results of which were analyzed using the “comparison analysis” option. IPA uses a right-tailed Fisher exact test to calculate a p-value corresponding to the probability that a biological function that is not relevant to the input data set is falsely identified as relevant. These p-values were corrected using a Benjamini-Hochberg false discovery rate of 0.05.
Results
Data Quality
Results for each of the five samples on the two platforms are summarized in Table 1. For the gene expression microarray analyses, each sample showed similar scale factors (0.86-1.08) and hybridization rates (42-44%), within a range consistent with expectations for this sample type.5 Examination of the RNA-Seq read quality data showed overrepresentation of Illumina adaptor and Illumina PCR primer sequences in our samples. Many reads contained short fragments of RNA flanked by Illumina sequence within the 50-base read sequence. This overrepresentation varied in magnitude from library to library, affecting between 3% and 84% of reads. Inversely proportional to the degree of Illumina sequence overrepresentation, there was wide variation in the extent of RNA-Seq genomic alignment for each library: between 5% and 71% of reads aligned to the genome. Despite a reduced level of alignment, we obtained usable data from all five Illumina libraries.
Table 1.
Results for each of five samples assessed on two platforms.
| Sample | GA | Sex | Paired-end reads | % Aligned | RNA-Seq genes | Microarray genes | Pearson R |
|---|---|---|---|---|---|---|---|
| 1 | 19+4 | F | 25,893,912 | 32.4 | 7,322 | 11,746 | 0.574 |
| 2 | 18+2 | F | 46,412,870 | 33.8 | 7,687 | 11,721 | 0.551 |
| 3 | 17+2 | M | 98,469,492 | 70.6 | 8,368 | 11,908 | 0.433 |
| 4 | 17+6 | M | 20,072,545 | 25.6 | 7,289 | 12,125 | 0.547 |
| 5 | 19+6 | M | 17,696,918 | 5.0 | 7,140 | 11,324 | 0.532 |
GA, Gestational age in weeks + days at the time of amniocentesis.
Gene Expression
Within individual samples, expression levels were compared between the platforms, and showed a strong, positive correlation (R=0.43-0.57, Figure 2a-e). Comparison of the AFCTs for each platform revealed that fewer genes were observed using RNA-Seq than microarray (4,158 versus 8,842, Figure 3). The majority of genes (89%) in the RNA-Seq AFCT are also detected by microarray. When ranked by microarray expression level, among the genes found within the top 10% of expression values in the microarray AFCT, 84% were also represented in the RNA-Seq AFCT. In contrast, of the 10% of genes that had lowest expression in the microarray AFCT, only 8% were also represented in the RNA-Seq AFCT (Table 2). The reciprocal comparison was also performed: genes were ranked in the RNA-Seq AFCT by expression level and the top and bottom 10% were examined for presence or absence in the microarray AFCT. In this case, we did not see a similar discrepancy between representations of the high and low expressing genes (93.8 % in the top expressers versus 88.2% in the lowest expressers). We observed, however, that in both types of comparisons the highly expressed genes are represented in both AFCTs.
Figure 2.
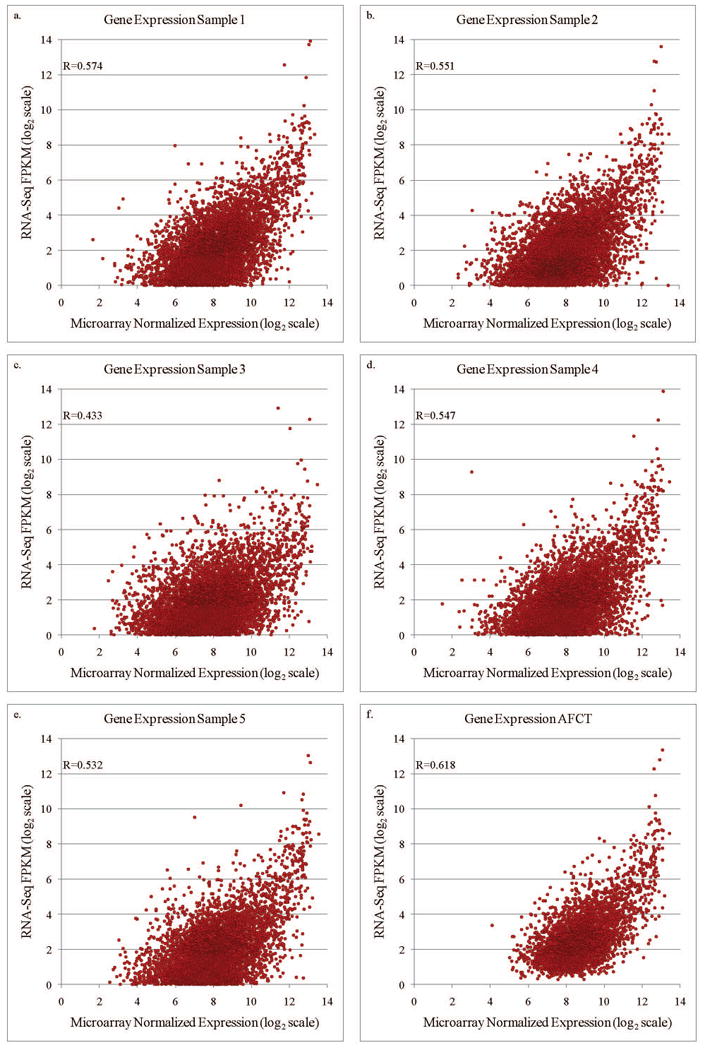
Figure 3.
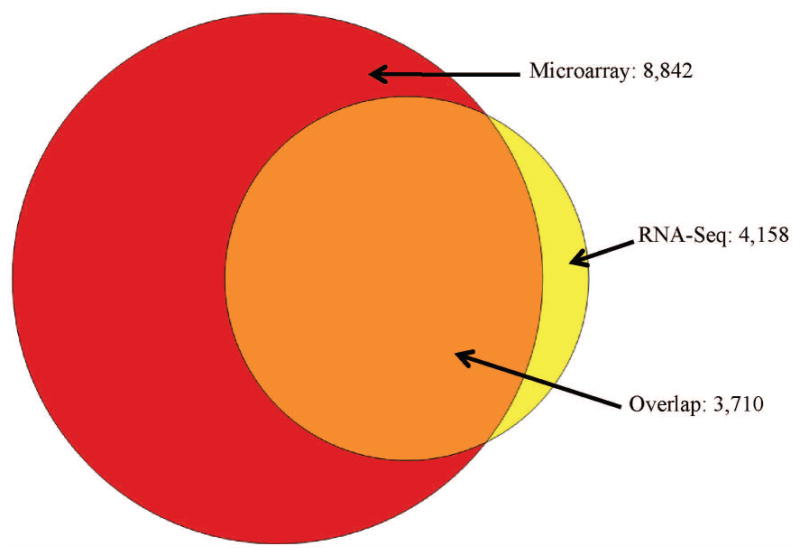
Table 2.
Comparison of the amniotic fluid core transcriptomes.
| Subseta | Microarray | RNA-Seq | % Overlap |
|---|---|---|---|
| Top 10% | 884 | 742 | 84% |
| Median 20% | 1,768 | 704 | 40% |
| Bottom 10% | 884 | 74 | 8% |
Subset based on ranking of the 8,842 genes in the microarray core transcriptome.
To reduce the impact of the low alignment seen for sample 5, we also made reduced AFCTs for each platform that contained only the information from samples 1-4 (4,759 genes for RNA-Seq and 9,376 genes for microarray). Considering the 938 genes (top 10%) that were the most highly expressed in this four microarray sample set, 808 were also present in the four RNA-Seq sample set (86%). By contrast, considering the 938 genes that were the lowest expressed in the four samples, only 97 were also present in the four RNA-Seq sample set (10%). Thus, when excluding sample 5, the trend persists that highly expressed genes are more likely to be detected by both platforms.
Focusing again on the full AFCTs for each platform, and examining only the 3,710 genes that were present in both, we saw a correlation between the relative expression levels of R=0.62 (Figure 2f). Among the top 10% of these genes, as ranked by average microarray expression, there was 59% concordance in gene ID; among the median 20% of ranked genes there was 24% concordance in gene ID and in the bottom 10% of ranked genes there was 23% concordance in gene ID (Table 3).
Table 3.
Genes represented in the amniotic fluid core transcriptomes of both platforms.
| Subseta | Microarray | RNA-Seq | % Overlap |
|---|---|---|---|
| Top 10% | 371 | 217 | 59% |
| Median 20% | 742 | 176 | 24% |
| Bottom 10% | 371 | 85 | 23% |
Subset based on ranking of average microarray expression level across the five samples.
Functional Analyses
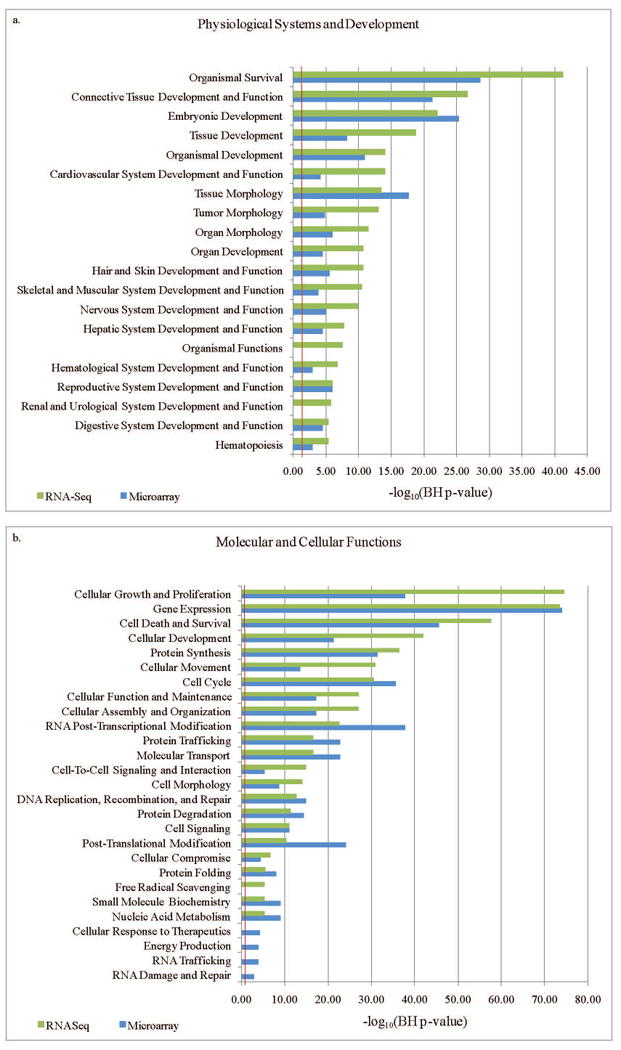
The 8,842 genes from the microarray AFCT and the 4,158 genes from the RNA-Seq AFCT were next imported to IPA to search for enriched pathways. Most of the top pathways in the “Physiological Systems Development and Function” category were shared between the two platforms, although RNA-Seq data yielded more significant p-values than the microarray data for many pathways (16 versus 2, respectively, Benjamini-Hochberg corrected p-value <0.05, Figure 4a). For the “Molecular and Cellular Functions” categories a similar number of pathways were most highly significant for microarray and RNA-Seq (15 versus 11, respectively, Figure 4b).
Figure 4.
Alternative Splicing
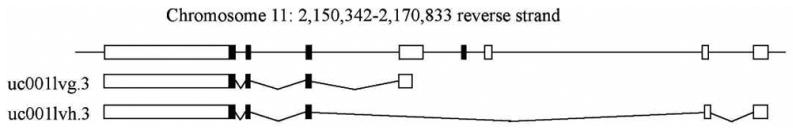
Using RNA-Seq, we detected several examples of known alternative splicing in genes such as ANXA2, and SPRR3. Additional examples include genes for which alternative splicing had previously been examined specifically in the human fetus or amniocytes, such as H19, IGF2 and RBPJ.19,20,21 These gene transcripts were observed in all samples (Table S1), but specific samples have been highlighted for brevity (Table 4, Figure 5). In several of these examples, one isoform was observed at a much greater level than the alternative transcripts.
Table 4.
A selection of known alternative splice variants present in two samples of RNA-Seq data.
| Sample | Gene symbol | UCSC variant | FPKM |
|---|---|---|---|
| 1 | ANXA2 | uc002agk.3 | 1.42 |
| 1 | ANXA2 | uc002agl.3 | 190.59 |
| 1 | ANXA2 | uc002agn.3 | <1 |
| 1 | H19 | uc001lva.4 | 1,940.00 |
| 1 | H19 | uc021qby.1 | 13,115.60 |
| 1 | H19 | uc021qbz.1 | 199.08 |
| 1 | IGF2 | uc001lvg.3 | 3,621.01 |
| 1 | IGF2 | uc001lvh.3 | 1.00 |
| 1 | RBPJ | uc003gsa.2 | 2.20 |
| 1 | RBPJ | uc003gsb.2 | <1 |
| 1 | SPRR3 | uc001fax.4 | 15.78 |
| 1 | SPRR3 | uc001faz.4 | 791.66 |
| 5 | ANXA2 | uc002agk.3 | 53.35 |
| 5 | ANXA2 | uc002agl.3 | 78.19 |
| 5 | ANXA2 | uc002agn.3 | 10.37 |
| 5 | H19 | uc001lva.4 | 916.92 |
| 5 | H19 | uc021qby.1 | 7,375.20 |
| 5 | H19 | uc021qbz.1 | 81.87 |
| 5 | IGF2 | uc001lvg.3 | 1,843.93 |
| 5 | IGF2 | uc001lvh.3 | <1 |
| 5 | RBPJ | uc003gsa.2 | 1.99 |
| 5 | RBPJ | uc003gsb.2 | 2.94 |
| 5 | SPRR3 | uc001fax.4 | 11.22 |
| 5 | SPRR3 | uc001faz.4 | 330.50 |
FPKM, fragments per kilobase of transcript per million mapped fragments.
Figure 5.
Discussion
In this pilot study we used two commonly selected platforms for gene expression analysis to survey gene expression in second trimester fetuses. A strength of this approach is that RNA was extracted from a single biological sample and analyzed on two different platforms, allowing us to compare the efficacy of each technical approach while minimizing biological variation. The results from each analysis share many commonalities, and validate previously published work on the second trimester fetal transcriptome,4 demonstrating the success of each approach.
Gene Expression
The extent of overlap between the two platforms was greatest for the most highly expressed genes (Table 2); the genes with the lowest microarray-detected expression values were less likely to be detected by RNA-Seq (83% versus 8%). In the design of our experiment, we purposefully limited the depth of sequencing to equate the cost of the two types of experimental approaches. Therefore, only the more highly expressed genes were assessed by RNA-Seq (Table 2). Differences in the underlying technology of the two techniques make RNA-Seq particularly sensitive to missing low expressed genes in the case of insufficient sequencing depth: microarray will yield a signal for all transcripts present in the sample that hybridize to the array, whereas the sampling method of RNA-Seq results in a set of reads comprised primarily of the most highly expressed genes.
The data presented in Table 3 were limited to genes present in the core transcriptomes of both platforms. They indicate that the lower a gene's expression, the more likely it was that the two platforms disagreed on the expression level. This finding was mirrored by the strongly positive, but incomplete, correlations seen in Figure 2. This finding is intriguing given that prior studies have found RNA-Seq to be more affected by technical variation than microarray.12,13 The varied RNA-Seq percent alignment shown in Table 1 contrasts with the relatively uniform microarray hybridization percentages, indicating that in our study, the microarray approach was less affected than the RNA-Seq analysis by variations in sample quality. Inclusion in the AFCT requires consistency among all five samples, thus increased sensitivity of the RNA-Seq platform to inter-sample biological or technical variation may contribute to the decreased size of its AFCT.
Functional Analyses
Many of the top pathways in the “Physiological Systems Development and Function” include organogenesis, tissue development, skin development, and nervous system development, all known to be active in the second-trimester fetus. The lower p-values seen with the RNA-Seq data in IPA likely reflect the smaller size of this AFCT, as IPA p-values are determined in part by the number of input molecules. Given that only the most abundant transcripts were detected by this RNA-Seq experiment, the sample set uploaded to IPA was in essence pre-selected for the genes most actively expressed during fetal development, resulting in lower p-values for the associated pathways (Figure 4).
Alternative Splicing
Using RNA-Seq data to create a comprehensive census of known and novel splice isoforms requires a longer read length and greater depth of sequencing than we used here, but we were able to detect isoforms of many genes already known to be alternatively spliced. The AFCT generated by RNA-Seq included reports of alternative splicing for many genes of interest, including H19, IGF2, and RBPJ (Figure 5, Table 4, Table S1). H19 and IGF2 are well-studied imprinted genes, with proper expression necessary for the regulation of fetal and placental growth. The abundance of different isoforms of IGF2 and H19 appears to be tissue-specific and has been previously studied in the human fetus.19,20 RBPJ is part of the NOTCH signaling pathway and plays an important role in stem-cell maintenance. The alternative splicing of RBPJ in amniocytes, which have many stem cell-like qualities, has previously been identified.21 The existence of prior studies on alternative splicing of these three genes in fetal cells further supports our finding of alternative transcripts in the second trimester AFS cff RNA. This ability to detect fine-scale alternative splicing is one advantage of an RNA-Seq based approach over the microarray platform used here.
Technical Challenges
Direct comparisons between our findings and prior studies are made here with the knowledge that previous studies used different thresholds for assessing gene expression, with accepted metrics including raw read count, FPKM, and statistical significance for differential gene expression.8,9,10,11,12 Each study defined its own threshold for minimum transcript detection. Our choice of an FPKM of 1 as a minimum was based on prior work,13,22 which found that this was a sensitive threshold. Several previous studies have focused on the ability to detect differential gene expression (DGE), and have found that RNA-Seq performs as well or better than the microarray for DGE analysis at highly expressed levels,10,12 while for lowly expressed genes, technical variation among RNA-Seq samples was problematic.12,13 Unlike prior studies, the work presented here focuses on the related but distinct question of presence or absence of gene expression. In this pilot analysis, we found that the underperformance of RNA-Seq was due to two main issues: the technical challenges posed by AFS cff RNA quality and quantity, and the insufficient depth of sequencing at an equivalent cost to microarrays.
Working with dilute, degraded cell-free fetal RNA presents technical challenges, which we have overcome in our microarray workflow, but which somewhat hindered the RNA-Seq based approach. Compared to samples prepared with cellular RNA, a low concentration of sequenceable molecules was seen within the completed libraries (personal communication, Hudson Alpha). The difficulties in library preparation contributed to the fact that the RNA-Seq AFCT is smaller than the microarray AFCT. Technical issues affected the sequencing depth attained for each sample as well as the percentage of aligned reads (Table 1), thus reducing the size of the RNA-Seq AFCT. The variability in percent alignment between samples (Table 1) most likely resulted from issues in Illumina sequence enrichment created during the library construction process. The library preparation method included a bead-based cleanup step, which excluded primer-dimers. Bioanalysis of the finished library did not reveal characteristic primer-dimer peaks at 120 bp, so the over-enrichment in Illumina sequence was unexpected. The absence of a 120 bp peak is likely because the over-enrichment was not in the form of simple primer-dimers, but rather, the 50 bp reads contained some transcript sequence, flanked by some Illumina sequence. The over-enrichment may have been partly caused by the degraded nature of the input RNA. AFS cff RNA typically has a low RNA integrity number and varies in quality. These variations are accentuated by the amplification steps in the library creation as well as the cluster generation that precedes sequencing, resulting in a wide variation of final data quality. Enrichment for adaptor sequences can also result from insufficient input of double-stranded cDNA, either due to low cDNA input or to the input being a mixture of single- and double-stranded cDNA. To mitigate the effects of Illumina sequence enrichment, data analysis was undertaken using the STAR aligner.17 This alignment software distinguishes itself through its ability to handle short reads and find the seed-length of maximum mappability without necessitating prior trimming to remove error-prone bases.
Future Studies
Increasing the sequencing depth in future studies will increase the number of observed genes, but the technical issues leading to decreased read alignment must also be overcome to maximize the RNA-Seq AFCT. Alternative approaches to library preparation can increase the sequence yield and alignment success for RNA-Seq. Head et al.23 have shown that the NuGEN RNA-Seq kit can generate single-stranded DNA with hairpin structures that interfere with downstream Illumina library preparation. In their studies, they found that treatment of the NuGEN product with a single stranded nuclease, such as Promega's S1 nuclease, significantly enhanced downstream library yield. Following the use of the NuGEN RNA-Seq kit, treatment with a single-stranded nuclease is thus recommended for future experiments involving cff RNA from AFS.
Summary
Here we compared the benefits and limitations of two different approaches to profiling the cell-free amniotic fluid fetal transcriptome. Gene expression levels reported by the two approaches were strongly and positively correlated. While the microarray data afforded a broader and more consistent view of gene expression for AF cff RNA, the MPS data provided a better focus on alternative splicing and specific biological pathways relevant to the developing fetus. We also determined that the low-concentration and degraded nature of the cell-free RNA in AF significantly affects library preparation and suggest ways of overcoming these technical challenges.
Supplementary Material
What's Already Known about This Topic.
Cell-free fetal (cff) RNA in amniotic fluid supernatant (AFS), is a pure source of fetal transcripts from multiple organs, including fetal brain.
Prior studies in adult tissues comparing expression microarray and massively parallel sequencing found that the RNA-Seq yielded comparable or greater information than the expression microarray.
What Does This Study Add
In this pilot study, we found that while the expression microarray data gave a broader view of gene expression, particularly in low-concentration or degraded samples, the RNA-Seq data provided a better focus on alternative splicing and specific biological pathways relevant to the developing fetus.
Degraded cell-free fetal RNA in amniotic fluid presents a technical challenge for RNA-Seq library preparation.
Acknowledgments
The authors wish to thank Joshua Ainsley, PhD, Lakshmanan Iyer, PhD and Albert Tai, PhD of Tufts University for helpful bioinformatics discussions, Donna Slonim, PhD of Tufts University for critical reading, and Janet Cowan, PhD of Tufts Medical Center Cytogenetics Laboratory for providing the amniotic fluid supernatant samples.
Funding: This work was supported by Eunice Kennedy Shriver National Institute of Child Health and Human Development (R01 HD 042053-10 to Dr. Bianchi).
Disclosures: DWB reported grants from the National Institute of Child Health and Human Development during the course of the study. Grants and personal fees are received by DWB from Illumina (formerly Verinata Health), outside the submitted work. In addition, DWB has a patent pending on the use of cell-free RNA in amniotic fluid to develop novel fetal therapies.
References
- 1.Larrabee PB, Johnson KL, Lai C, et al. Global gene expression analysis in the living human fetus using amniotic fluid: a feasibility study. JAMA. 2005;293:836–42. doi: 10.1001/jama.293.7.836. [DOI] [PubMed] [Google Scholar]
- 2.Massingham LJ, Johnson KL, Bianchi DW, et al. Proof of concept study to assess fetal gene expression in amniotic fluid by nanoarray PCR. J Mol Diagn. 2011;13:565–70. doi: 10.1016/j.jmoldx.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hui L, Wick HC, Edlow AG, et al. Global gene expression analysis of term amniotic fluid cell-free fetal RNA. Obstet Gynecol. 2013;121:1248–54. doi: 10.1097/AOG.0b013e318293d70b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hui L, Slonim DK, Wick HC, et al. The amniotic fluid transcriptome: a source of novel information about human fetal development. Obstet Gynecol. 2012;119:111–8. doi: 10.1097/AOG.0b013e31823d4150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Slonim DK, Koide K, Johnson KL, et al. Functional genomic analysis of amniotic fluid cell-free mRNA suggests that oxidative stress is significant in Down syndrome fetuses. Proc Natl Acad Sci USA. 2009;106:9425–9. doi: 10.1073/pnas.0903909106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koide K, Slonim DK, Johnson KL, et al. Transcriptomic analysis of cell-free fetal RNA suggests a specific molecular phenotype in trisomy 18. Hum Genet. 2011;129:295–305. doi: 10.1007/s00439-010-0923-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hui L, Wick HC, Moise KJ, Jr, et al. Global gene expression analysis of amniotic fluid cell-free RNA from recipient twins with twin-twin transfusion syndrome. Prenat Diagn. 2013;33:873–83. doi: 10.1002/pd.4150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marioni JC, Mason CE, Mane SM, et al. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008;18:1509–17. doi: 10.1101/gr.079558.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lahiri P, Lee LJ, Frey BJ, et al. Transcriptional profiling of endocrine cerebro-osteodysplasia using microarray and next-generation sequencing. PLoS One. 2011;6:e25400. doi: 10.1371/journal.pone.0025400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sirbu A, Kerr G, Crane M, et al. RNA-Seq vs dual- and single-channel microarray data: sensitivity analysis for differential expression and clustering. PLoS One. 2012;7:e50986. doi: 10.1371/journal.pone.0050986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Su Z, Li Z, Chen T, et al. Comparing next-generation sequencing and microarray technologies in a toxicological study of the effects of aristolochic acid on rat kidneys. Chem Res Toxicol. 2011;24:1486–93. doi: 10.1021/tx200103b. [DOI] [PubMed] [Google Scholar]
- 12.Liu S, Lin L, Jiang P, et al. A comparison of RNA-Seq and high-density exon array for detecting differential gene expression between closely related species. Nucleic Acids Res. 2011;39:578–88. doi: 10.1093/nar/gkq817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Raghavachari N, Barb J, Yang Y, et al. A systematic comparison and evaluation of high density exon arrays and RNA-seq technology used to unravel the peripheral blood transcriptome of sickle cell disease. BMC Medical Genomics. 2012;5:28. doi: 10.1186/1755-8794-5-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hui L, Bianchi DW. Cell-free fetal nucleic acids in amniotic fluid. Hum Reprod Update. 2011;17:362–71. doi: 10.1093/humupd/dmq049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dietz JA, Johnson KL, Massingham LJ, et al. Comparison of extraction techniques for amniotic fluid supernatant demonstrates improved yield of cell-free fetal RNA. Prenat Diagn. 2011;31:598–9. doi: 10.1002/pd.2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–10. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dobin A, Davis CA, Schlesinger F, et al. STAR: Ultrafast universal RNA-Seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trapnell C, Williams BA, Pertea G, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–5. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matouk I, Ayesh B, Schneider T, et al. Oncofetal splice-pattern of the human H19 gene. Biochem Biophys Res Commun. 2004;318:916–9. doi: 10.1016/j.bbrc.2004.04.117. [DOI] [PubMed] [Google Scholar]
- 20.Monk D, Sanches R, Arnaud P, et al. Imprinting of the IGF2 P0 transcript and novel alternatively spliced INS-IGF2 isoforms show differences between mouse and human. Hum Mol Genet. 2006;15:1259–69. doi: 10.1093/hmg/ddl041. [DOI] [PubMed] [Google Scholar]
- 21.Jezierski A, Ly D, Smith B, et al. Novel RBPJ transcripts identified in human amniotic fluid cells. Stem Cell Rev. 2010;6:677–84. doi: 10.1007/s12015-010-9162-1. [DOI] [PubMed] [Google Scholar]
- 22.Mortazavi A, Williams BA, McCue K, et al. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–8. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 23.Head SR, Komori HK, Hart GT, et al. Method for improved Illumina sequencing library preparation using NuGEN Ovation RNA-Seq System. Biotechniques. 2011;50:177–80. doi: 10.2144/000113613. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.