

Abstract

The scope of the [4 + 2] cycloaddition reactions of substituted 1,2,3-triazines, bearing noncomplementary substitution with electron-withdrawing groups at C4 and/or C6, is described. The studies define key electronic and steric effects of substituents impacting the reactivity, mode (C4/N1 vs C5/N2), and regioselectivity of the cycloaddition reactions of 1,2,3-triazines with amidines, enamines, and ynamines, providing access to highly functionalized heterocycles.
Inverse-electron-demand Diels–Alder reactions of electron-deficient heterocyclic azadienes often provide a powerful approach to the preparation of highly substituted heteroaromatic systems not easily accessed by conventional means.1 The most widely recognized of such heterocyclic azadienes are the 1,2,4,5-tetrazines,2 1,2,4-triazines,3 1,3,5-triazines,4 1,3,4-oxadiazoles,5 and 1,2-diazines.6 Subject to the impact of substituents that modulate their reactivity, alter their mode of reaction, and control their reaction regioselectivity, they have been shown to react with a variety of electron-rich or strained dienophiles to provide not only six-membered heterocyclic ring systems but also five-membered heteroaromatics,7 many of which represent core elements present in natural products.8−11 Recently, we disclosed studies on the cycloaddition reactions of 1,2,3-triazine (1)12 and the 5-substituted 1,2,3-triazines 2–413 bearing complementary substitution that enhance their reactivity, reinforce an intrinsic mode and regioselectivity of cycloaddition, and substantially expand the scope of such reactions (Figure 1).14,15 Herein, we report an examination of 4-substituted, 4,6-disubstituted, and 4,5,6-trisubstituted 1,2,3-triazines as additional participants in inverse-electron-demand Diels–Alder reactions, defining the steric and electronic effects of the noncomplementary placement of electron-withdrawing substituents on the scope of such cycloaddition reactions.
Figure 1.
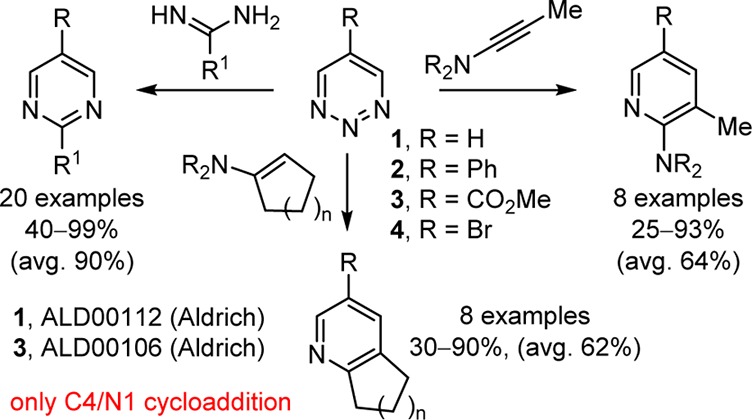
Selected Diels–Alder reactions of 1,2,3-triazines.
The 1,2,3-triazines 1–4 participate in inverse-electron-demand Diels–Alder reactions in which the C5 substituent was used to predictably enhance their reactivity (R = CO2Me > Ph > H) without impacting either the intrinsic mode (across C4/N1) or regioselectivity of the 1,2,3-triazine cycloaddition reaction. The rate of reactions for each dienophile class was also shown to follow a now predicable trend (amidine > ynamine > enamine) and the reactivity of 3, bearing a strong C5 electron-withdrawing group (CO2Me), extended to an even larger and remarkable range of additional dienophiles.13 The efficiency with which amidines were found to react with 1–4 to provide pyrimidines was unique and suggested that this reaction might exhibit an even broader scope.
In studies first examined in the total synthesis of (−)-pyrimidoblamic acid and P-3A, we successfully employed the reaction of amidines with two 1,2,3-triazines (7 and 9) bearing differentiated substitution at the 4,6- and 4,5,6-positions that represent noncomplementary substitution sites for promoting the [4 + 2] cycloaddition reactions.16 This success provided the incentive to more comprehensively investigate the reactivity of such 1,2,3-triazines with 5–9 (Figure 2). At the onset of these studies, it was not clear if 5–9 would behave analogously to 1–4 or if the addition of the electron-withdrawing substituents at the C4 and/or C6 positions would sterically slow the reactions or electronically redirect the mode of cycloaddition (C4/N1 vs C5/N2). Intuitively, one might expect the 4-substituted (5), 4,6-disubstituted (6–7), and 4,5,6-trisubstituted (8–9) 1,2,3-triazines to exhibit increased electrophilic character at the C5-position. The preparation of 7 and 9, enlisting an oxidative ring expansion (I2, KHCO3, 68 and 73% respectively) of the corresponding N-aminopyrazoles, was conducted as disclosed,16 and 1,2,3-triazines 5, 6, and 8 were prepared in an analogous fashion by N-amination of the starting pyrazoles followed by oxidative ring expansion using NaIO414 (5, 73%) or I215b (6, 46%; 8, 66%; Supporting Information).
Figure 2.

1,2,3-Triazines examined.
The initial dienophiles examined as the cycloaddition partners for the 1,2,3-triazines 5–9 were amidines. The reactions of 5 with the amidines at room temperature were monitored (TLC), and although product formation appeared complete after 15 min, the reaction mixtures were stirred for 1 h at 25 °C (0.3 M, CH3CN) to ensure complete conversion. Aromatic, benzylic, and aliphatic amidines were effective reaction partners for 5, providing the 2-substituted-4-carbomethoxypyrimidines in good yields (68–98%) without further optimization and without the appearance of competitive reaction products (Figure 3).
Figure 3.

Reaction of 1,2,3-triazine 5 with amidines.
The behavior of the 4,6-disubstituted 1,2,3-triazines 6 and 7 was especially interesting. With the exception of the phenylamidine 10a, the benzylic and aliphatic amidines provided the pyrimidine products in good to excellent yields (51–89%, CH3CN, 25 °C, 4 h), and the unsymmetrical 1,2,3-triazine 7, bearing the larger t-Bu ester, consistently provided higher yields than the symmetrical 1,2,3-triazine 6 (Figure 4). Unlike reactions of 1,2,3-triazine 5, which proceeded within 1 h, both 6 and 7 showed more gradual product formation over 4 h. Notably, substitution of the 1,2,3-triazine reacting C4 center did not preclude the cycloaddition reaction, and despite the noncomplementary electronic and steric bias, the C4/N1 (vs C5/N2) cycloaddition mode was not altered.
Figure 4.

Reaction of 1,2,3-triazines 6 and 7 with amidines.
Similarly, the fully substituted 1,2,3-triazines 8 and 9 were subjected to [4 + 2] cycloaddition with the amidines. Not only do they bear noncomplementary substitution at either of the reacting C4 centers but they also contain a nonactivating substituent at C5. Both 1,2,3-triazines reacted to provide the product pyrimidines in moderate to good yields (19–66%) without optimization (Figure 5). Expectedly, these 1,2,3-triazines exhibited a lower reactivity and required longer reaction times (CH3CN, 25 °C, 16 h) and, in one instance, an elevated temperature to complete product formation. Again, the unsymmetrical 1,2,3-triazine 9, bearing the larger t-Bu ester, consistently provided the pyrimidines in a more pronounced 2–3-fold higher yield, although the origin of this more effective behavior of 9 is not yet understood.
Figure 5.

Reaction of 1,2,3-triazines 8 and 9 with amidines.
Pairwise competition studies comparing the reactivity of the 1,2,3-triazines with amidine 10b revealed that they follow the trend of 3 > 6 > 5 > 8 and 1 in which the complementary C5 substitution with a single electron-withdrawing substituent provides the most reactive 1,2,3-triazine (3 > 6), that the noncomplementary 4,6-disubstitution with the two electron-withdrawing groups is more activating than C4 monosubstitution (6 > 5) despite the additional steric bias, that 8 was the least reactive of the substituted 1,2,3-triazines despite the diactivation substitution, and that not only 3 but also 5 and 6 were more reactive than 1,2,3-triazine (1) itself.
The reactions of three enamines with the 1,2,3-triazines 5, 6, and 8 were examined (Scheme 1). The reaction of methyl 1,2,3-triazine-4-carboxylate (5) proceeded with cycloaddition only across C6/N3 and with a regioselectivity where the nucleophilic carbon of the enamine adds to C6, providing exclusively the pyridine products in good yields (Scheme 1). Although nitrogen evolution was often observed upon mixture of the reactants at room temperature, indicating a rapid [4 + 2] cycloaddition followed by loss of N2, complete reaction required higher reaction temperatures (60 °C, CHCl3) in the presence of 4 Å molecular sieves (MS)3c to promote the slower aromatization step with the elimination of pyrrolidine.
Scheme 1.

The more substituted and symmetrical 1,2,3-triazines 6 and 8 behaved similarly with enamine addition across C4/N1 (C6/N3) to provide exclusively the pyridine products, albeit in reactions that progressed to completion at a slower rate. Examined in detail with the fully substituted 1,2,3-triazine 8, its reaction with enamines 16a and 16c at 25 °C led to an instantaneous color change, the observable evolution of N2, consumption of 8 within 15–30 min, and the generation of the isolated unaromatized cycloadducts 20 (77%) and 21 (57%) in excellent conversions (Scheme 2).
Scheme 2.

The extension of the reaction at higher temperatures (60 °C) for longer periods of time (12 h) even in the presence of 4 Å MS3c was insufficient to promote completion of the aromatization step. However, the addition of strong acid catalysts (HOAc, 12 h, 54%; MeSO3H, 2 h, 67%)3c or m-CPBA (N-oxide formation, 0 °C, 1 h, 78%) to the reaction mixtures, following the consumption of 8 upon addition of 16a (40 min, 25 °C), led to effective aromatization to provide the pyridine product. Notably and despite the noncomplementary substitution, the mode of cycloaddition with enamines (C4/N1 vs C5/N2) was not altered, and they exhibited an enhanced reactivity relative to 1,2,3-triazine (1) but diminished relative to 3 bearing the complementary C5 substitution.
The [4 + 2] cycloaddition reactions of 5, 6, and 8 with ynamines proved especially interesting (Scheme 3). The reactions proceeded rapidly at room temperature (22 °C, CHCl3, 1 h) to provide predominately (5 and 8, >8:1) or exclusively (6) the pyridazine products 23, 25, and 26 (64–85%) in excellent yields and derived from cycloaddition across C5/N2 with the nucleophilic carbon of the ynamine attaching to C5.
Scheme 3.

This preferential C5/N2 mode of cycloaddition not only represents a switch in the mode of cycloaddition relative to their reactions with amidines and enamines but also that of the ynamine cycloadditions observed with 1 and the C5 activated 1,2,3-triazines 2–4, where only C4/N1 cycloaddition is observed. In the case of 5, where C6 is unsubstituted and sterically accessible, and 8, where C5 is substituted and sterically less accessible, a small amount of C4/N1 (C6/N3) cycloaddition is observed, resulting in the minor pyridine products 24 and 27 (0–12%). Unlike their behavior toward amidines and enamines, the C4 and C4/C6 substitution with electron-withdrawing groups redirects the mode of cycloaddition (C5/N2 vs C4/N1) as well as enhances the reactivity of the 1,2,3-triazines relative to 1 toward ynamines by virtue of this now complementary substitution. Just as interesting, the initial ynamine-derived C5/N2 cycloadduct derived from 5 exclusively undergoes a retro-Diels–Alder loss of HCN (vs MeO2CCN) providing a single pyridazine product.
A competition study comparing the reactivity of 5, 6, and 8 toward ynamine 22a (1.2 equiv, 22 °C, CHCl3, 30 min) defined a clear trend (6 > 5 > 8) where the now complementary C4 and C6 disubstitution (CO2Et) electronically enhances the reactivity of 6 relative to 5 bearing a single electron-withdrawing group (CO2Me), and the C5 methyl substitution of 8 sterically slows the cycloaddition relative to not only 6 but also 5, despite the C4/C6 disubstitution activation of 8 (Figure 6).
Figure 6.

Ynamine competition study.
Although not comprehensively explored and unlike methyl 1,2,3-triazine-5-carboxylate (3),13 which bears a complementary activating C5 electron-withdrawing substituent, additional electron-rich alkenes (ketene acetals, enol ethers) and alkynes (ethoxyacetylene) failed to effectively react with 5–9.
A systematic study of the cycloaddition reactions of 1,2,3-triazines substituted with noncomplementary C4 and/or C6 electron-withdrawing groups was conducted. Such substituents were found to enhance the reactivity of the 1,2,3-triazine (5–9 > 1) despite the noncomplementary electronic and steric effects. Remarkably, the mode of cycloaddition for amidines and enamines relative to 1 was not altered (C4/N1), whereas that of ynamines was redirected to C5/N2. Significantly, the studies provide additional heterocyclic azadienes, complementary to the 1,2,4- and 1,3,5-triazines, that participate in effective cycloaddition reactions, extending the range of accessible heterocyclic ring systems.
Acknowledgments
We gratefully acknowledge the financial support of the National Institutes of Health (CA042056). A.S.D. was an American Cancer Society postdoctoral fellow (2012–2014, PF-12-122-01-CDD).
Supporting Information Available
Full experimental details. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Boger D. L. Tetrahedron 1983, 39, 2869. [Google Scholar]; b Boger D. L. Chem. Rev. 1986, 86, 781. [Google Scholar]; c Boger D. L.; Weinreb S. M.. Hetero Diels–Alder Methodology in Organic Synthesis; Academic: San Diego, 1987. [Google Scholar]
- a Carboni R. A.; Lindsey R. V. J. Am. Chem. Soc. 1959, 81, 4342. [Google Scholar]; b Sauer J.; Wiest H. Angew. Chem., Int. Ed. Engl. 1962, 1, 269. [Google Scholar]; c Boger D. L.; Panek J. S. Tetrahedron Lett. 1983, 24, 4511. [Google Scholar]; d Boger D. L.; Panek J. S.; Coleman R. S.; Sauer J.; Huber F. X. J. Org. Chem. 1985, 50, 5377. [Google Scholar]; e Boger D. L.; Sakya S. M. J. Org. Chem. 1988, 53, 1415. [Google Scholar]; f Sakya S. M.; Groskopf K. K.; Boger D. L. Tetrahedron Lett. 1997, 38, 3805. [Google Scholar]; g Boger D. L.; Schaum R. P.; Garbaccio R. M. J. Org. Chem. 1998, 63, 6329. [DOI] [PubMed] [Google Scholar]; h Soenen D. R.; Zimpleman J. M.; Boger D. L. J. Org. Chem. 2003, 68, 3593. [DOI] [PubMed] [Google Scholar]; i Hamasaki A.; Ducray R.; Boger D. L. J. Org. Chem. 2006, 71, 185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Steigel A.; Sauer J. Tetrahedron Lett. 1970, 3357. [Google Scholar]; b Neunhoffer H.; Fruhauf H.-W. Leibigs Ann. Chem. 1972, 758, 120–125. [Google Scholar]; c Boger D. L.; Panek J. S. J. Org. Chem. 1981, 46, 2179. [Google Scholar]; d Boger D. L.; Panek J. S.; Meier M. M. J. Org. Chem. 1982, 47, 895. [Google Scholar]; e Boger D. L.; Panek J. S. J. Org. Chem. 1982, 47, 3763. [Google Scholar]; f Boger D. L.; Panek J. S. Tetrahedron Lett. 1984, 25, 3175. [Google Scholar]
- a Neunhoeffer H.; Bachmann M. Chem. Ber. 1975, 108, 3877. [Google Scholar]; b Boger D. L.; Schumacher J.; Panek J. S.; Mullican M. D.; Patel M. J. Org. Chem. 1982, 47, 2673. [Google Scholar]; c Boger D. L.; Dang Q. Tetrahedron 1988, 44, 3379. [Google Scholar]; d Boger D. L.; Dang Q. J. Org. Chem. 1992, 57, 1631. [Google Scholar]; e Boger D. L.; Kochanny M. J. J. Org. Chem. 1994, 59, 4950. [Google Scholar]
- a Wilkie G. D.; Elliott G. I.; Blagg B. S. J.; Wolkenberg S. E.; Soenen D. R.; Miller M. M.; Pollack S.; Boger D. L. J. Am. Chem. Soc. 2002, 124, 11292. [DOI] [PubMed] [Google Scholar]; b Elliott G. I.; Fuchs J. R.; Blagg B. S. J.; Ishikawa H.; Yuan Z.-Q.; Boger D. L. J. Am. Chem. Soc. 2006, 128, 10589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Neunhoeffer H.; Werner G. Liebigs Ann. Chem. 1973, 437–1955. [Google Scholar]; b Boger D. L.; Coleman R. S. J. Org. Chem. 1984, 49, 2240. [Google Scholar]; c Kessler S. N.; Wegner H. A. Org. Lett. 2010, 12, 4062. [DOI] [PubMed] [Google Scholar]; d Sumaria C. S.; Tuerkmen Y. E.; Rawal V. H. Org. Lett. 2014, 16, 3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Review:; a Joshi U.; Pipelier M.; Naud S.; Dubreuil D. Curr. Org. Chem. 2005, 9, 261. [Google Scholar]; b Boger D. L.; Coleman R. S.; Panek J. S.; Yohannes D. J. Org. Chem. 1984, 49, 4405. [Google Scholar]; c Boger D. L.; Baldino C. M. J. Org. Chem. 1991, 56, 6942. [Google Scholar]; d Yeung B. K. S.; Boger D. L. J. Org. Chem. 2003, 68, 5249. [DOI] [PubMed] [Google Scholar]; e Joshi U.; Josse S.; Pipeleir M.; Chevallier F.; Pradere J. P.; Hazard R.; Legoupy S.; Huet F.; Dubreuil D. Tetrahedron Lett. 2004, 45, 1031. [Google Scholar]
- a Boger D. L.; Panek J. S. J. Am. Chem. Soc. 1985, 107, 5745. [Google Scholar]; b Boger D. L.; Panek J. S.; Duff S. R.; Yasuda M. J. Org. Chem. 1985, 50, 5790. [Google Scholar]; c Boger D. L.; Duff S. R.; Panek J. S.; Yasuda M. J. Org. Chem. 1985, 50, 5782. [Google Scholar]; d Boger D. L.; Coleman R. S. J. Am. Chem. Soc. 1987, 109, 2717. [Google Scholar]; e Boger D. L.; Coleman R. S. J. Am. Chem. Soc. 1987, 110, 4796. [Google Scholar]; f Boger D. L.; Zhang M. J. Am. Chem. Soc. 1991, 113, 4230. [Google Scholar]; g Boger D. L.; Menezes R. F.; Honda T. Angew. Chem., Int. Ed. Engl. 1993, 32, 273. [Google Scholar]; h Boger D. L.; Honda T.; Menezes R. F.; Colletti S. L.; Dang Q.; Yang W. J. Am. Chem. Soc. 1994, 116, 82. [Google Scholar]; i Boger D. L.; Honda T. J. Am. Chem. Soc. 1994, 116, 5647. [Google Scholar]; j Boger D. L.; Colletti S. L.; Honda T.; Menezes R. F. J. Am. Chem. Soc. 1994, 116, 5607. [Google Scholar]; k Boger D. L.; Honda T.; Dang Q. J. Am. Chem. Soc. 1994, 116, 5619. [Google Scholar]; l Boger D. L.; Hong J.; Hikota M.; Ishida M. J. Am. Chem. Soc. 1999, 121, 2471. [Google Scholar]; m Boger D. L.; Wolkenberg S. E. J. Org. Chem. 2000, 65, 9120. [DOI] [PubMed] [Google Scholar]
- a Boger D. L.; Patel M. J. Org. Chem. 1988, 53, 1405. [Google Scholar]; b Boger D. L.; Baldino C. M. J. Am. Chem. Soc. 1993, 115, 11418. [Google Scholar]; c Boger D. L.; Boyce C. W.; Labroli M. A.; Sehon C. A.; Jin Q. J. Am. Chem. Soc. 1999, 121, 54. [Google Scholar]; d Boger D. L.; Soenen D. R.; Boyce C. W.; Hedrick M. P.; Jin Q. J. Org. Chem. 2000, 65, 2479. [DOI] [PubMed] [Google Scholar]; e Boger D. L.; Hong J. J. Am. Chem. Soc. 2001, 123, 8515. [DOI] [PubMed] [Google Scholar]; f Hamasaki A.; Zimpleman J. M.; Hwang I.; Boger D. L. J. Am. Chem. Soc. 2005, 127, 10767. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Oakdale J. S.; Boger D. L. Org. Lett. 2010, 12, 1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Boger D. L.; Wolkenberg S. E. J. Org. Chem. 2002, 67, 7361. [DOI] [PubMed] [Google Scholar]; b Yuan Q.; Ishikawa H.; Boger D. L. Org. Lett. 2005, 7, 741. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Ishikawa H.; Elliott G. I.; Velcicky J.; Choi Y.; Boger D. L. J. Am. Chem. Soc. 2006, 128, 10596. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Ishikawa H.; Colby D. A.; Seto S.; Va P.; Tam A.; Kakei H.; Rayl T. J.; Hwang I.; Boger D. L. J. Am. Chem. Soc. 2009, 131, 4904. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Ishikawa H.; Colby D. A.; Boger D. L. J. Am. Chem. Soc. 2008, 130, 420. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Campbell E. L.; Zuhl A. M.; Liu C. M.; Boger D. L. J. Am. Chem. Soc. 2010, 132, 3009. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Kato D.; Sasaki Y.; Boger D. L. J. Am. Chem. Soc. 2010, 132, 3685. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Sasaki Y.; Kato D.; Boger D. L. J. Am. Chem. Soc. 2010, 132, 13533. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Lajiness J. P.; Jiang W.; Boger D. L. Org. Lett. 2012, 14, 2078. [DOI] [PMC free article] [PubMed] [Google Scholar]; j Xie J.; Wolfe A. L.; Boger D. L. Org. Lett. 2013, 15, 868. [DOI] [PMC free article] [PubMed] [Google Scholar]; k Lee K.; Boger D. L. J. Am. Chem. Soc. 2014, 136, 3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Boger D. L.; Cassidy K. C.; Nakahara S. J. Am. Chem. Soc. 1993, 115, 10733. [Google Scholar]; b Boger D. L.; Hüter O.; Mbiya K.; Zhang M. J. Am. Chem. Soc. 1995, 117, 11839. [Google Scholar]; c Boger D. L.; Hong J. J. Am. Chem. Soc. 1998, 120, 1218. [Google Scholar]; d Boger D. L.; Ichikawa S.; Jiang H. J. Am. Chem. Soc. 2000, 122, 12169. [Google Scholar]; e Blagg B. S. J.; Boger D. L. Tetrahedron 2002, 58, 6343. [Google Scholar]; f Schnermann M. J.; Boger D. L. J. Am. Chem. Soc. 2005, 127, 15704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson E. D.; Boger D. L. Org. Lett. 2011, 13, 2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson E. D.; Boger D. L. J. Am. Chem. Soc. 2011, 133, 12285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Sugita T.; Koyama J.; Tagahara K.; Suzuta Y. Heterocycles 1985, 23, 2789. [Google Scholar]; b Okatani T.; Koyama J.; Tagahara K.; Suzuta Y. Heterocycles 1987, 26, 595. [Google Scholar]
- Ohsawa A.; Kaihoh T.; Itoh T.; Okada M.; Kawabata C.; Igeta H. Chem. Pharm. Bull. 1988, 36, 3838. [Google Scholar]
- Duerfeldt A. D.; Boger D. L. J. Am. Chem. Soc. 2014, 136, 2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.