

Abstract
Synaptic dysfunction is thought to play an important role in the pathophysiology of neurodegenerative diseases, such as Alzheimer’s disease (AD) and Lewy body disease (LBD). To improve our understanding of synaptic alterations in health and disease, we investigated synaptosomes prepared from post-mortem human cerebral cortex, putamen, and two regions of the caudate nucleus, dorso-lateral (DL) and ventro-medial (VM), regions commonly affected in AD and LBD. We observed that the fraction of synaptosomal particles with reactivity for dopamine transporter (DAT) was significantly reduced in the putamen and VM caudate of patients with neuropathological diagnosis of LBD. As expected, these differences also were reflected in direct measurements of dopamine (DA) and its metabolite, 3,4-dihydroxyphenylacetic acid (DOPAC), in caudate and putamen of LBD patients. The fraction of synaptosomal particles positive for amyloid β (Aβ) was significantly increased in frontal cortical samples of patients with the neuropathological diagnosis of severe AD, and was positively correlated with disease progression. We also prepared synaptosomes from the striatum of mice with severe loss of DA neurons (Slc6a3-DTR mice) and wild-type littermate controls. We observed dramatically reduced levels of DAT-positive synaptosomes in Slc6a3-DTR mice following exposure to diphtheria toxin (DT). Striatal levels of DA and DOPAC in Slc6a3-DTR mice also were reduced significantly following DT exposure. We conclude that flow cytometric analysis of synaptosomes prepared from human or mouse brain provides an opportunity to study expression of pathology-associated proteins and also the specific loss of dopaminergic nerve terminals. Hence, we believe it is a valid method to detect pathological changes at the level of the synapse in LBD as well as AD.
Keywords: Synaptosome, dopamine, dementia, Lewy body disease, Alzheimer’s disease, amyloid beta, striatum, cortex
There remain significant gaps in knowledge regarding the precise neural substrates for cognitive dysfunction in LBD (1, 2). Neuropathological characteristics of LBD, which includes dementia with Lewy bodies (DLB) and Parkinson’s disease (PD), are well known and include loss of dopaminergic neurons, deposition of α-synuclein, innate immune activation, and oxidative damage among others (3). However, the mechanisms by which these pathologic processes yield motor and cognitive impairments are not well understood (4). There is growing evidence that the substrate of motor and cognitive impairment is the culmination of a complex interplay of neuronal dysfunction, reduced plasticity, and ultimately neuron death (5, 6). For example, recent evidence for LBD suggests that the initial neuronal change associated with LB formation may be diminished synaptic function and plasticity (7, 8). Since synaptic function is essential for cognition, studying the pathological changes and possible compensatory mechanisms in the surviving nerve terminals and their immediate connections is important for better understanding of the disease pathophysiology. Traditional neuropathological approaches, which typically work well for assessment of whole cells, do not work well for nerve terminals. This limitation has been addressed in part by application of ultrastructural or Golgi staining techniques (9). However, both have their own limitations, including the restriction of analysis to relatively small regions of brain, selective staining of neurons based on unclear mechanisms related to staining protocols, and technical challenges regarding molecular investigation (10). Thus, despite the likely key role in disease pathogenesis, molecular pathologic changes at the synaptic level remain largely unknown in humans.
Synaptosomes are resealed synaptic terminals obtained from fresh brain tissue through a series homogenization and centrifugation steps (11, 12). First isolated in 1958 by Hebb and Whittaker (13), freshly prepared synaptosomes maintain electrical potential and can be used for functional investigations. While the electrochemical integrity of human post-mortem synaptosomes is unreliable, these preparations have great potential to be used to test hypotheses about molecular alterations in human synapses (14). Recently, a group led by Gylys (15, 16) demonstrated the utility of combining antibody-based flow cytometry methodologies with synaptosome preparations to specifically interrogate synapse subtype and associations with amyloid β (Aβ) and tau species in AD.
The human striatum and frontal cortex are variably affected by LBD, micro-vascular brain injury (μVBI) and AD (17, 18). Selective degeneration of striatal dopaminergic terminals is characteristic of LBD, both PD and DLB, but is not known for AD or μVBI (1). Aβ accumulation in AD starts in the cerebral cortex and at intermediate pathologic stages of AD, Aβ accumulates in the striatum, although not typically accompanied by neurodegeneration or atrophy. In LBD, Lewy bodies (LBs), and Lewy neurites (LNs), which are inclusions containing phosphorylated species of α-synuclein, accumulate in cerebral cortex (19, 20). Micro-VBI, as assessed by neuropathologic consensus guidelines, is a hallmark of widespread small vessel-mediated injury to brain that often includes striatum and cortex (21). To better understand synaptic changes that occur in frontal cortex and striatum related to LBD, AD and μVBI, we have adapted the flow cytometry method of synaptosome analysis developed by Gylys and colleagues (22) to define molecular alterations in synaptosome particles prepared from brain regions by a rapid autopsy protocol for research subjects in the University of Washington Alzheimer’s Disease Research Center (ADRC) and the Adult Changes in Thought (ACT) study, a population-based study of brain aging in the Seattle metropolitan area (23).
MATERIALS AND METHODS
Human Subjects
This study was approved by the University of Washington Institutional Review Board. All cases were from the Neuropathology Core of the University of Washington. All study subjects were participants in the ADRC, Pacific Northwest Udall Center, ACT or Seattle longitudinal study. Brain tissue samples (1 g or less) were collected at autopsy in cases with a post-mortem interval (PMI) of < 8 hours. Tissue samples included middle frontal gyrus (MFG, Brodmann area 9), anterior portion of the head of the caudate nucleus, which was separated into dorso-lateral (DLC) and ventro-medial (VMC) parts, and anterior portion of the putamen (PT). In addition, for measurements of DAT, Aβ and α-synuclein phosphorylated at Ser129, we also collected some tissue samples from cerebellar cortex, a region that is relatively spared in AD and receives only sparse dopaminergic innervation. For this study, we selected 24 consecutive rapid autopsy cases, 14 men and 10 women, median age 87 years, who met study inclusion criteria (PMI ≤ 8 h, and had no gross lesions in the structures of interest). We used consensus neuropathological criteria to stratify cases (24, 25). Based on the neuropathological diagnosis, the subjects were divided into 4 groups: LBD (n = 5); AD (n = 11); μVBI with or without concomitant AD (μVBI±AD) (n = 3); control group (n = 5) (Table 1). For all cases, as shown in Supplementary Table S1, we determined Braak stage for neurofibrillary tangles (NFTs) (26), CERAD (Consortium to Establish a Registry for Alzheimer’s Disease) score for neuritic plaques (27), and Thal phase for Aβ deposits (28). In order to determine the effects of PMI on our assay, three temporal neocortical samples were obtained from adult patients undergoing surgery for medically intractable temporal lobe epilepsy. Deidentified temporal cortex samples (1 g or less) obtained beyond the primary area of pathology were received from the operating room, placed on wet ice, and immediately processed identically to the tissue samples obtained at autopsy. No other clinical or demographic information was provided with these samples in accordance with the University of Washington Institutional Review Board.
Table 1.
Patient data
| Group | Number of subjects | Age at time of death (years, median ± SD) | Post-mortem interval (hours, mean ± SEM) | Male to female ratio |
|---|---|---|---|---|
| LBD | 5 | 82 ± 4.0 | 5 ± 1.4 | 5:0 |
| AD | 11 | 90 ± 9.6 | 4 ± 1.0 | 9:2 |
| μVBI±AD | 3 | 93 ± 4.4 | 6 ± 1.6 | 3:0 |
| Control | 5 | 84 ± 4.9 | 5 ± 1.6 | 4:1 |
Experimental Animals
Slc6a3-DTR mice were generated by gene targeting. They have a cassette containing the human diphtheria toxin receptor (DTR) open reading frame inserted into the 5′-UTR region of exon 2, just upstream of the translation start codon of the mouse DAT gene (Slc6a3). All mice for these studies were on a C57Bl/6 genetic background. Only heterozygotes were used for breeding and produced litters of experimental (Slc6a3-DTR) and litter-mate control WT (wild-type) animals. At the age of 2–4 months, all animals received two intramuscular injections, separated by 48 h, each with 50 μg of diphtheria toxin (DT) per kg mouse. This technique has been demonstrated previously to result in the ablation of DTR-containing neurons, while leaving neuronal populations in wild-type mice intact (29). After 20–24 weeks, all mice were sacrificed and striatal tissue harvested for either synaptosome preparation or HPLC analysis of DA. Striatal mouse tissue contained both dorsal and ventral parts of the striatum. In addition, we also collected samples from the cerebellum of wild-type mice for synaptosome preparation. Due to its low level of DAT expression, we chose the cerebellum as an additional negative control for DAT antibody based labeling techniques. All animal procedures were conducted in accordance with the guidelines of the Institutional Animal Care and Use Committee at the University of Washington.
Preparation of the crude synaptosome fraction
The synaptosome preparation protocol was adapted from Gylys et al. (22). Human tissue samples were obtained at autopsy or during surgery. Mouse tissue samples were obtained immediately after the animals were sacrificed. Samples were minced, slowly frozen in 0.32 M sucrose with 10% DMSO, and stored at −80 °C. To obtain the crude synaptosome fraction, tissue was thawed in a 37 °C water bath and homogenized in 10 mM Tris buffer (pH 7.4) with proteinase inhibitors (Roche, IN) and phosphatase inhibitors (Sigma-Aldrich, MO) using a glass/Teflon homogenizer (clearance 0.1–0.15 mm). The homogenate was centrifuged at 1,000 g at 4 °C for 10 min, the supernatant was removed and centrifuged again at 10,000 g at 4 °C for 20 min. Resulting pellets were resuspended in sucrose/Tris solution and stored at −80 °C.
Immunolabeling of the synaptosome fraction
For every incubation or wash, the synaptosome pellets were resuspended by gentle vortexing, and then collected by centrifugation (5,000 g for 4 min at 4 °C). The pellets were first thawed in a 37 °C degree water bath, washed twice with Krebs-Ringer phosphate buffer (118 mM NaCl, 5 mM KCl, 4 mM MgSO4, 1 mM CaCl2, 1 mM KH2PO4, 16 mM sodium phosphate buffer, and 10 mM glucose), fixed with 0.25% paraformaldehyde (PFA) for 1 hour at 4 °C, and washed with phosphate buffered saline (PBS; 137 mM NaCl, 2.7 mM KCl, 10 mM NaH2PO4, 1.8 mM KH2PO4). The pellets were then incubated with 0.2% Tween 20 in PBS at 37 °C for 15 min, washed first with PBS, and then with the blocking buffer (2% fetal calf serum in PBS). Afterwards, the pellets were incubated with the primary antibodies or isotype control antibodies (Table 2) in blocking buffer, washed twice with 0.2% Tween 20 in PBS and incubated with 1:200 dilution of respective AlexaFluor 488-conjugated secondary antibody (Jackson ImmunoResearch, PA) in blocking buffer. The pellets were then washed once with 0.2% Tween 20 in PBS, once with PBS and then resuspended in PBS for flow cytometry analysis.
Table 2.
Antibodies
| Antigen | Manufacturer, clonality, product number, isotype, host, dilution | Isotype control antibody, manufacturer, product number |
|---|---|---|
| Aβ (6E10) | Covance, monoclonal (clone 6E10), SIG-39320, mouse, IgG1, 1:500 | Mouse myeloma IgG1, Invitrogen, 02-6100 |
| Aβ (1-42 peptide) | Novus Biologicals whole antisera, rabbit, 1:1000 | Rabbit polyclonal IgG, Abcam, ab26478 |
| α-synuclein (phospho S129) | Abcam, monoclonal, ab51253 (EP1536Y), rabbit, 1:5,000 | Rabbit polyclonal IgG, Abcam, ab26478 |
| DAT | Abcam, monoclonal, ab5990, IgG, rat, 1:250 | Rat IgG, Abcam, ab37361 |
| SNAP-25 | Covance, monoclonal (clone SMI 81), SMI-81R, IgG1, mouse, 1:250 | Mouse myeloma IgG1, Invitrogen, 02-6100 |
| VGLUT1 | Millipore, monoclonal (clone 3C10.2), MAB5502, IgG1, mouse, 1:50 | Mouse myeloma IgG1, Invitrogen, 02-6100 |
Flow cytometry
The synaptosome suspension was analyzed using a BD FACSCalibur Flow cytometer (BD Biosciences, CA) equipped with 15 mW 488 nm (blue) and 633 nm (red) lasers. Polystyrene microspheres (Polysciences Inc, PA) of varying size (0.54 μm and 1.5 μm) were used as size standards (Fig. 1a). Size gate was set to include only particles of approximate size of 0.5 to 1.5 μm. At least ten thousand size-gated events per sample were collected for further analysis.
Figure 1.

Flow cytometry analysis of human cortical synaptosome preparation from a control sample. The gain settings on the flow cytometry machine were set to provide optimal visualization of the total population. The highly non-homogeneous nature of the preparation resulted in a wide spread of values along the ordinate, with a part of the positive gate extending into the 101 region of the axis (conventionally reserved for background signal). (a) Particle size in the synaptosome preparations compared to polystyrene size standards (shown as circles within the scatter plot; 1: 0.54 μm, 2: 1.5 μm). (b) Electron microscopy of crude synaptosome preparation. Arrows indicate post-synaptic density (black) and dense-core synaptic vesicles (white). (c) Immunolabeling of synaptosomal preparation with SNAP-25 antibody. (d) Isotype antibody control immunolabeling of synaptosomal preparation.
Each sample was stained with both the primary antibody selective to the protein of interest and the respective isotype control antibody, used as a negative control. The positive gate was set on the sample stained with the isotype control; we found that drawing the positive gate to include 5% ± 0.1% of the total size-gated particle population offered optimal discrimination between the positive and the negative sample and provided the best reproducibility in repeated experiments. Others have previously reported using a similar or more inclusive size gate (30). Experiments that involved DAT, Aβ, or α-synuclein antibody labeling also included a human or mouse cerebellum sample as additional negative control.
Analysis of synaptosomes by transmission electron microscopy
To confirm that our preparation indeed contained intact synaptosomes, we also processed a sample of human cortex according to the protocol described above up to, but not including, the wash with blocking buffer. Instead of washing the pellet with blocking buffer and consequent immunostaining, the pellet was fixed in 4% glutaraldehyde/2% PFA and processed for electron microscopy and ultrastructural analysis.
Catecholamine measurements
Human tissue samples were flash-frozen at the time of autopsy/dissection. All human samples were obtained from tissue sections that were adjacent to the sections taken for the synaptosome preparation, but not further subdivided into DL and VM part. In the mouse, tissue punches (1-mm diameter) were obtained from the striatum, flash-frozen immediately and stored at −80 °C. HPLC coupled with electrochemical detection (Coulochem III electrochemical detector, Thermo Fisher Scientific, MA) was used to measure DA and DOPAC content. All raw measurements of DA and DOPAC were normalized to tissue weight (human) or to total protein content (mouse).
Statistical Analysis
All experimental data are shown as mean ± standard error of the mean (SEM). Basic patient data, age of disease onset and post-mortem interval, are shown as median ± standard deviation (SD). Data were analyzed using one- or two-way analysis of variance (ANOVA), followed by Tukey’s post-test, or a two-tailed t-test. Correlation was assessed by calculation of Pearson’s correlation coefficient. Alpha was always set at p < 0.05.
RESULTS
General characteristics of our synaptosome fraction
Size comparison to polystyrene size standards showed that the majority of particles present in the synaptosome fraction were between 0.5–1.5 μm (Figure 1a), which is consistent with previously reported synaptosome preparations (31, 32). We also performed electron microscopy analysis of our preparation from human cortex that confirmed the presence of intact synaptosomes with both pre- or post-synaptic membrane- bound elements (presynaptic vesicles, synaptic densities) occasionally bound together at the synapse (Figure 1b). To validate further the synaptic origin of the preparation, we labeled synaptosome fractions with an antibody against the synaptosomal-associated protein 25 (SNAP-25), a component of the SNARE complex in presynaptic terminals. In human cortical samples, SNAP-25 immunoreactivity was present in 90.8 ± 3.4% of the size-gated synaptosome population (Figures 1c and 1d, Supplementary Figure S1), and in human striatal samples, 87.4 ± 4.2% of the particles were positive for SNAP-25 (Supplementary Figure S2). In the mouse striatum, SNAP-25 – positive particles accounted for the 92.3 ± 0.4% of the total size-gated population (Supplementary Figure S3).
DAT-positive synaptosomes and catecholamine levels in human striatum
We next investigated the expression of DAT in striatal synaptosomes to assess the integrity of striatal dopaminergic synapses. Post-mortem synaptosome preparations of the putamen and two regions of caudate (DL and VM) from the human autopsy cohort were labeled with an antibody against DAT, and the percentage of DAT-positive terminals was assessed using flow cytometry. We observed significant effects of disease status on the percentage of DAT-positive particles (Figure 2a, Supplementary Figure S4) in the putamen (ANOVA F3, 20 = 8.38, p < 0.01) and in the VM caudate (ANOVA F3, 20 = 4.01, p < 0.05), but not in the DL caudate. In the putamen, the percentage of DAT-positive particles in the LBD group (5%) was significantly lower (p < 0.05) than in all other groups (each 25%) and in the VM caudate, the level of DAT-positive particles in the LBD group (11%) was only significantly lower than in the Control group (27%). This level was only slightly higher than in the cerebellum (4 ± 0.3%, data not shown in the figure), a structure that receives only minor dopaminergic input (33). When we examined each of the LBD cases individually (Figure 2b), we observed that for 4 of the 5 patients, the levels of DAT-positive particles in the VM caudate were either similar or slightly higher than in the DL caudate or putamen, which is consistent with the pathophysiology of PD, a subtype of LBD. However, in one of the patients, the level of DAT-positive particles in DL caudate was notably higher than in the VM portion of the caudate. In addition, we observed significant positive correlations (p < 0.01) between the levels of DAT-positive particles in the DL and VM caudate nucleus (R2 = 0.76, Figure 2c) and also between the levels in the putamen and the DL caudate (R2 = 0.46), VM caudate (R2 = 0.49) and combined (DL + VM) caudate nucleus (R2 = 0.55, Figure 2d).
Figure 2.

Flow cytometry analysis of synaptosome preparation of human striatum from control (n = 5), LBD (n = 5), AD (n = 11) and μVBI±AD (n = 3) groups labeled with an antibody against DAT. (a) Percentage of DAT-positive particles in striatal synaptosomes of all groups. (b) DAT-positive particles in striatal synaptosomes of individual patients with LBD compared to average number of DAT-positive particles in control and combined AD + μVBI±AD groups. (c) Percentage of DAT-positive particles in the DLC plotted against percentage of DAT-positive particles in the VMC. (d) Percentage of DAT-positive particles in the averaged DLC and VM caudate nucleus synaptosomes plotted against percentage of DAT-positive particles in putamen synaptosomes. All data are presented as mean ± SEM. * p < 0.05. Significant pairwise comparisons of groups are indicated by brackets.
Dopamine content in flash-frozen whole-tissue preparations, as measured by HPLC, was significantly affected by disease status in the putamen (ANOVA F3, 20 = 5.14, p < 0.01; Figure 3a), but not in the caudate (not separated into DL and VM parts; Figure 3b). In the putamen, levels of DA in the LBD group were significantly lower (p < 0.01) than in the Control group. Similarly, tissue content of the DA metabolite DOPAC was also significantly affected by disease status in the putamen (ANOVA F3, 20 = 4.53, p < 0.01; Figure 3c), but not in the caudate (Figure 3d). Again, DOPAC levels in the putamen of the LBD group were significantly lower (p < 0.01) than in the Control group.
Figure 3.
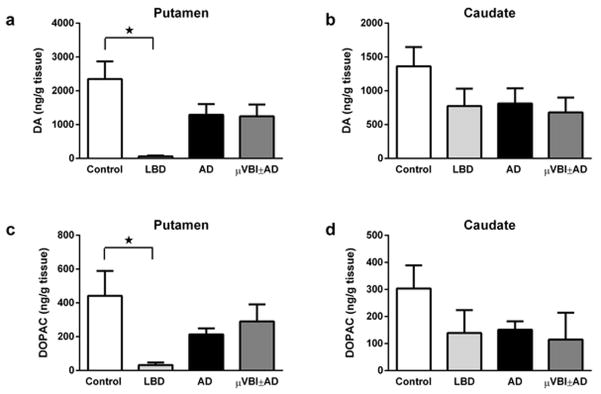
Catecholamine levels in post-mortem human striatum from control (n = 5), LBD (n = 5), AD (n = 11) and μVBI±AD (n = 3) groups, measured by HPLC and normalized to tissue weight. (a) DA in Putamen. (b) DA in the caudate nucleus. (c) DOPAC in Putamen. D. DOPAC in the caudate nucleus. All data is presented as mean ± SEM. * p < 0.05. Significant pairwise comparisons of groups are indicated by brackets.
DAT-positive synaptosomes and catecholamine levels in mouse striatum
The majority of dopaminergic neurons are characterized by expression of DAT. To determine the specificity of our antibody-based staining technique and to demonstrate the feasibility of the technique in the main species of laboratory animals, we used mice in which the human DT receptor is expressed in DAT neurons (Figure 4a).
Figure 4.

Mouse model of loss of striatal DA projections. (a) Schematic representation illustrating the Slc6a3-hDTR model, in which expression of the human DTR in all DAT neurons allows ablation of DA neurons. (b) Percentage of DAT-positive particles in striatal synaptosomes from DT-treated WT (n = 6) and Slc6a3-DTR mice (n = 5). Tissue content of (c) DA and (d) DOPAC in the striatum of DT-treated WT (n = 6) and Slc6a3-DTR mice (n = 5) measured by HPLC and normalized to protein content. All data are presented as mean ± SEM. ** p < 0.01. Significant pairwise comparisons of groups are indicated by brackets.
Synaptosome preparations of the striatum from DT-treated Slc6a3-DTR (n = 5) and wild-type mice (n = 6) were labeled with an antibody against DAT. Compared to DT-treated wild-type mice, DT-treated Slc6a3-DTR mice had a significantly (p < 0.01) decreased percentage of DAT-positive particles in striatal synaptosomes (Figure 4b, Supplementary Figure S5). Interestingly, the percentage of DAT-positive particles in mouse striatal synaptosomes (35%) was comparable to our human data for caudate and putamen, and the percentage found in our mutant mouse model for severe loss of DA (3%) also was similar to that found in the LBD group. Most important, this level was only slightly higher than in synaptosomes derived from wild-type mouse cerebellum (0.7 ± 0.4%, data not shown in the figure), a structure that does not receive significant dopaminergic input (34).
The striatal levels of DA (Figure 4c), and its major metabolite, DOPAC (Figure 4d), as measured by HPLC, were significantly reduced in the striatum of DT-treated Slc6a3-DTR mice (each p < 0.01).
Aβ accumulation in human cortical synaptosomes
We examined synaptosome preparations from post-mortem human cortical samples to determine if we could detect synaptosomal accumulation of Aβ. Different types of Aβ-containing aggregates are known to accumulate in the human brain with advanced age, but we expected to see higher levels of Aβ-positive particles in subjects with neuropathological changes of advanced AD (35). The synaptosomal preparation was labeled with the 6E10 antibody, an antibody which is reactive to amino acid residues 1-16 of the Aβ protein, and is commonly used in neuropathologic evaluation, as well as an antibody specific to 1-42 Aβ peptide (Aβ-42), which is considered to be a more toxic form of Aβ and is the predominant form present in senile plaques (36). The 24 subjects from the autopsy cohort were divided into 3 groups based on different approaches to pathologic staging of AD (see Supplementary Table S1): the Braak stage system for NFT distribution (26), CERAD score for neuritic plaque density (27), or Thal phase for Aβ plaque distribution (28). In the samples stained with the 6E10 antibody, we confirmed significant effects of Braak stage (ANOVA F2, 21 = 4.61, p < 0.05; Figure 5a), CERAD score (ANOVA F2, 21 = 3. 45, p < 0.05; Figure 5b) and Thal phase (ANOVA F2, 21 = 4.83, p < 0.01; Figure 5c) on the percentage of Aβ-positive particles. Samples from patients with the highest neuropathological AD burden, as assessed by either Braak stage (Braak stage V–VI), CERAD score (3, or “Frequent”), or Thal phase (Thal phase 4–5), had significantly more Aβ-positive particles than samples with the lowest neuropathologic burden of AD (each p < 0.05). Levels of synaptosomal Aβ in the cerebral cortex of the low neuropathologic AD groups were still higher (each p < 0.05) than in the cerebellum (5.3 ± 0.3%, data not shown in the figure), which is one of the least-affected structures in AD (37). When the subjects were again analyzed as the four original cohorts (AD, μVBI±AD, LBD, and control), subjects diagnosed with AD or μVBI±AD had the highest values of synaptosomal Aβ (27.1 ± 3.1 and 28.3 ± 5% respectively), followed by the LBD cohort (23.8%). The control group had the lowest values (15.8%), but the differences did not reach statistical significance.
Figure 5.
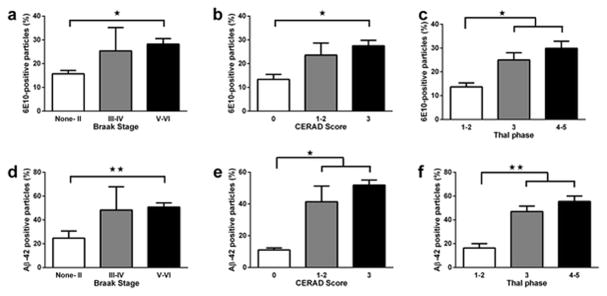
Human cortical synaptosome preparation labeled with the 6E10 (a–c) and the Aβ-42 (d–f) antibody against amyloid β. (a and d) Subjects were divided into 3 groups according to the Braak and Braak staging of AD: “None-II” (n = 14), “III–IV”, (n = 3),“V–VI” (n = 7). (b and e) Subjects were divided into 3 groups according to the CERAD score: “0” (n = 5), “1–2” (n = 5), and “3” (n = 14). (c and f) Subjects were divided into 3 groups according to the Thal phase: “1–2” (n = 6), “3” (n = 8) and “4–5” (n = 10). All data are presented as mean ± SEM. * p < 0.05, ** p < 0.01. Significant pairwise comparisons of groups are indicated by brackets.
When the samples were labeled with the antibody to Aβ42, the results appeared very similar to the results of staining with 6E10. We observed significant effects of Braak stage (ANOVA F2, 21 = 5.61, p < 0.05; Figure 5d), as well as CERAD score (ANOVA F2, 21 = 13.47, p < 0.001; Figure 5e), and Thal phase (ANOVA F2, 21 = 18.12, p < 0.0001; Figure 5f) on the percentage of Aβ-42 - positive particles. Again, samples from patients with the highest AD burden had significantly more Aβ-42 - positive particles than samples with the lowest neuropathologic burden of AD (each p < 0.05) or cerebellum samples (7.4 ± 1.0%, data not shown in the figure). When the subjects were again analyzed as the four original cohorts (AD, μVBI±AD, LBD, and control), subjects diagnosed with AD or μVBI±AD had the highest values of synaptosomal Aβ (50.2 ± 5.4 and 55.0 ± 5.6% respectively), followed by the LBD cohort (41.8 ± 9.1%). The control group had the lowest values (20.6 ± 6.5%). The difference between the AD and Control group was statistically significant (p < 0.05), data not shown.
Accumulation of phosphorylated α-synuclein in human cortical synaptosomes
The pathological hallmark of LBD is the presence of cortical LBs and LNs, structures that contain, among other proteins, a phosphorylated form of α-synuclein (38). To see if the phosphorylated α-synuclein was also present in the cortical synaptosomes from patients with LBD, we labeled cortical samples immunologically with the antibody against α-synuclein phosphorylated at Ser129. We only observed minimal signal in the majority of our samples: 4.8 ± 0.7% in the control group, 5.0 ± 1.1% in the LBD group, 4.3 ± 0.4% and 4.0 ± 1.0% in the AD and μVBI±AD group respectively (Supplementary Figure S6). Even though the highest value observed (9%) did indeed come from a patient with LBD, the differences between groups were not statistically significant. Furthermore, signals observed in all samples were not significantly higher than in cerebellum (4.32 ± 0.23%, n=3, data not shown). We did not observe any correlation between cortical levels of α-synuclein phosphorylated at Ser129 and the percentage of DAT-positive terminals in the striatum.
VGLUT1-positive synaptosomes in post-mortem samples of human cortex and striatum
To assess potential disease-associated changes in glutamatergic terminals within the cerebral cortex (39, 40), we examined cortical synaptosome preparations with an antibody against the vesicular glutamate transporter 1 (VGLUT1). We did not detect any significant changes in cortical VGLUT1 expression between our analyzed groups (Figure 6a). Because cerebral cortical glutamatergic input to the striatum may play a role in the pathophysiology of LBD (41), we also examined VGLUT1 expression in putamen, DL caudate and VM caudate. Again, we did not detect any significant changes in striatal VGLUT1 expression between our analyzed groups (Figure 6a, Supplementary Figure S7). Interestingly, when we excluded LBD cases and analyzed all other groups together, we found a significant negative correlation between the levels of DAT-positive particles and VGLUT1-positive particles in the putamen (R2 = 0.25; Figure 6b), but not in the DL or VM caudate.
Figure 6.
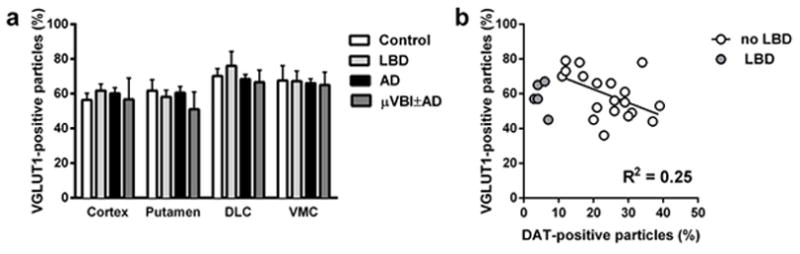
Flow cytometry analysis of the synaptosome preparation of human cortex and striatum labeled with the antibody against vesicular glutamate transporter 1 (VGLUT1). (a) Percentage of VGLUT1-positive particles in the post-mortem samples of cortex, putamen, and caudate nucleus. Data is presented as mean ± SEM. (b) Correlation between the number of synaptosome particles in human putamen positive for VGLUT1 and DAT. “No LBD” group (n = 19) includes all cases without the diagnosis Lewy body disease. Correlation coefficient was calculated for the “No LBD” group only.
VGLUT1 in surgical specimens
Finally, we wanted to determine if our results obtained in the post-mortem tissue samples are comparable to fresh tissue samples. To address that question, we prepared synaptosomes from samples of human cortex resected during surgery for intractable temporal lobe epilepsy, and labeled them with the antibody against VGLUT1. The levels of VGLUT1-positive particles in the surgically resected samples were not significantly different from the samples obtained at an autopsy (66 ± 10.4%, n = 3 in the surgical samples, 59.3 ± 2.3%, n = 24 in the post-mortem samples, p > 0.05).
DISCUSSION
To evaluate specific alterations in synaptic function in neurodegenerative disease, we generated synaptosome preparations from brain samples obtained from 24 consecutive autopsies of research subjects without significant neurodegenerative neuropathology or diagnosed pathologically as having LBD, AD and μVBI. Because we did not specifically select cases on the basis of the neuropathological diagnosis, but assigned them to appropriate groups after the diagnosis was made, the number of cases varied between disease groups. We also used a mouse model for severe DA denervation to assess the integrity of striatal dopaminergic synapses. Using these synaptosome preparations, we applied flow cytometry techniques to quantify expression of proteins that are directly associated with LBD (DAT) and AD (Aβ) and also expression of VGLUT1, a protein expressed in excitatory synapses.
We found that in synaptosome preparations from patients with a neuropathologic diagnosis of LBD, but not AD or μVBI, the percentage of synaptosomal particles with DAT expression was significantly decreased in putamen and the ventro-medial (VM) portion of the caudate nucleus, both areas are reported to show dopaminergic degeneration in LBD. Furthermore, we confirmed our method for quantification of synaptic DAT-expression in Slc6a3-hDTR mice, an animal model for severe loss of dopaminergic neurons. In these mutant mice, injection of diphtheria toxin causes severe ablation of DAT-expressing dopaminergic neurons, which results in a drastic loss of both striatal DA content and striatal DAT expression in synaptosomes. This further confirms the specificity of our synaptosome DAT relative quantification and also demonstrates that our method can be applied to both post-mortem clinical and fresh-frozen experimental samples obtained from standard laboratory animals. Interestingly, one patient with LBD (case LBD-1), who appeared notably different from the rest of the LBD group by having high levels of DAT-positive synaptosome particles in the caudate nucleus, also had the shortest disease duration. This subject was diagnosed with PD only 2 years prior to death, while the average disease duration in the rest of the group was 8.5 ± 1.6 years. The pronounced difference in the levels of DAT-positive particles in the two regions of the striatum (dramatically decreased in the putamen, but close to the normal levels in the caudate) in this subject coincides with the typical progression pattern of PD, with the putamen being affected earlier than caudate nucleus (42).
The proportions of particles containing VGLUT1 were not significantly different among LBD, AD, μVBI, and control groups, suggesting that, unlike striatal dopaminergic particles in LBD, VGLUT1-containing cerebral cortical or striatal synapses were not preferentially lost in LBD, AD and μVBI. This finding is consistent with an earlier study, which compared the levels of VGLUT1-positive terminal in the cortex of patients with AD and control cases (30). Interestingly, we observed that in the putamen of patients without the clinical diagnosis of LBD, levels of the DAT-positive synaptosomal particles were negatively correlated with the levels of VGLUT1-positive particles, suggesting a reciprocal relationship between glutamatergic input and nigral dopaminergic input to the striatum in the absence of LBD. Similar results have been previously reported in animal models of experimental Parkinsonism; in moderately to severely affected DA-depleted mice and rats, loss of dopaminergic neurons results in significantly increased glutamatergic activity in the striatum (43, 44). Reports from patients with LBD have been mixed, possibly as a result of the effect of therapeutic interventions which may differentially influence the pathophysiology of the disease (45). Interpretation of findings in this group is further complicated because similar treatment can differentially affect individuals; for instance, increased activity of glutamatergic projections to striatum has been observed with patients with L-dopa induced dyskinesias, but not in other patients receiving L-dopa (46). Our current finding suggests that other than in the presence of severe DA denervation that occurs in LBD, there is an inverse relationship between dopaminergic and glutamatergic innervation of the human putamen of older adults.
We also measured levels of Aβ-containing particles in the frontal cortex samples from patients with varying levels of neuropathologic burden of AD. We used two different antibodies to Aβ: 6E10, which is commonly used in neuropathologic evaluation, but is known to exhibit some cross-reactivity to the amyloid precursor protein, and an Aβ-42 antibody, which is expected to react specifically with the more toxic from of Aβ, 1-42 Aβ peptide, but not 1-40 peptide or amyloid precursor protein. The absolute values obtained with the Aβ-42 antibody were, on average, higher than obtained with the 6E10 antibody, which could be explained either by different binding properties of the two antibodies, or more likely, by a suboptimal concentration of isotype control used in the staining with Aβ-42 antibody. Because this antibody came as whole antiserum, we could not calculate perfect matching concentration of the isotype control. Other than this difference in the absolute values, results obtained with the two antibodies were very similar. Severe AD as assessed by different staging systems had significantly higher levels of synaptosomal particles positive for Aβ protein compared to the subjects with no or low neuropathologic evidence of AD. Our finding confirms the seminal reports of Gylys and colleagues who observed increased number of Aβ-positive synaptosomes in the cerebral cortex of patients with severe AD (16, 35). The proportion of Aβ-containing particles was numerically higher in our samples from patients with LBD than in control subjects, but not significantly different. This finding is expected because the extent of Aβ deposition in cerebral cortex is lower on average in LBD than in AD (47, 48).
We also measured levels of phosphorylated α-synuclein in cortical synaptosomes. Even though we did observe a small increase in the number of particles positive for phosphorylated α-synuclein in the LBD group, compared to the rest of the cohort, the difference was not statistically significant. One possible explanation may be low abundance of phosphorylated α-synuclein in our brain samples. Phosphorylated α-synuclein is concentrated in the LBs and LNs, but LBs are only present in neuronal perikarya, and we are not aware of any reports of LNs being present in a synaptosome preparation. On the other hand, even though pathologic species of α-synuclein may be a critical factor in causing neurodegeneration in LBD, it is not clear how much of the phosphorylated protein is actually present in the presynaptic terminal. Evidence from in vitro studies suggests that formation of LN-like inclusions in neuronal processes is accompanied by a decrease in the amount of α-synuclein in presynaptic terminals (49). Furthermore, in patients with LBD, phosphorylated α-synuclein is predominantly localized in LBs and LNs, rather than in synaptic terminals (38), which are the major components of synaptosomal preparations.
We also showed, by preparing and immunostaining surgically obtained human tissue samples and comparing them with clinical post-mortem samples that a postmortem interval of up to 8 hours did not affect immunoreactivity with our method, exemplified by staining of VGLUT1 expression.
Taken together, we demonstrated that the use of flow cytometry to analyze crude synaptosomal fraction prepared from post-mortem human brain, a method pioneered by Gylys and coworkers (22), can also be successfully applied to study diseases of the dopaminergic system in human and mouse models. In the future, we hope to extend our knowledge of LBD pathophysiology by using flow cytometry analysis of synaptosomes to study changes in other neurotransmitter systems, which are known to be affected in LBD, such as projections by cholinergic neurons in the pedunculopontine tegmental nucleus or by glutamatergic neurons in the centre-median/parafascicular complex of the thalamus and the subthalamic nucleus (50–53).
Supplementary Material
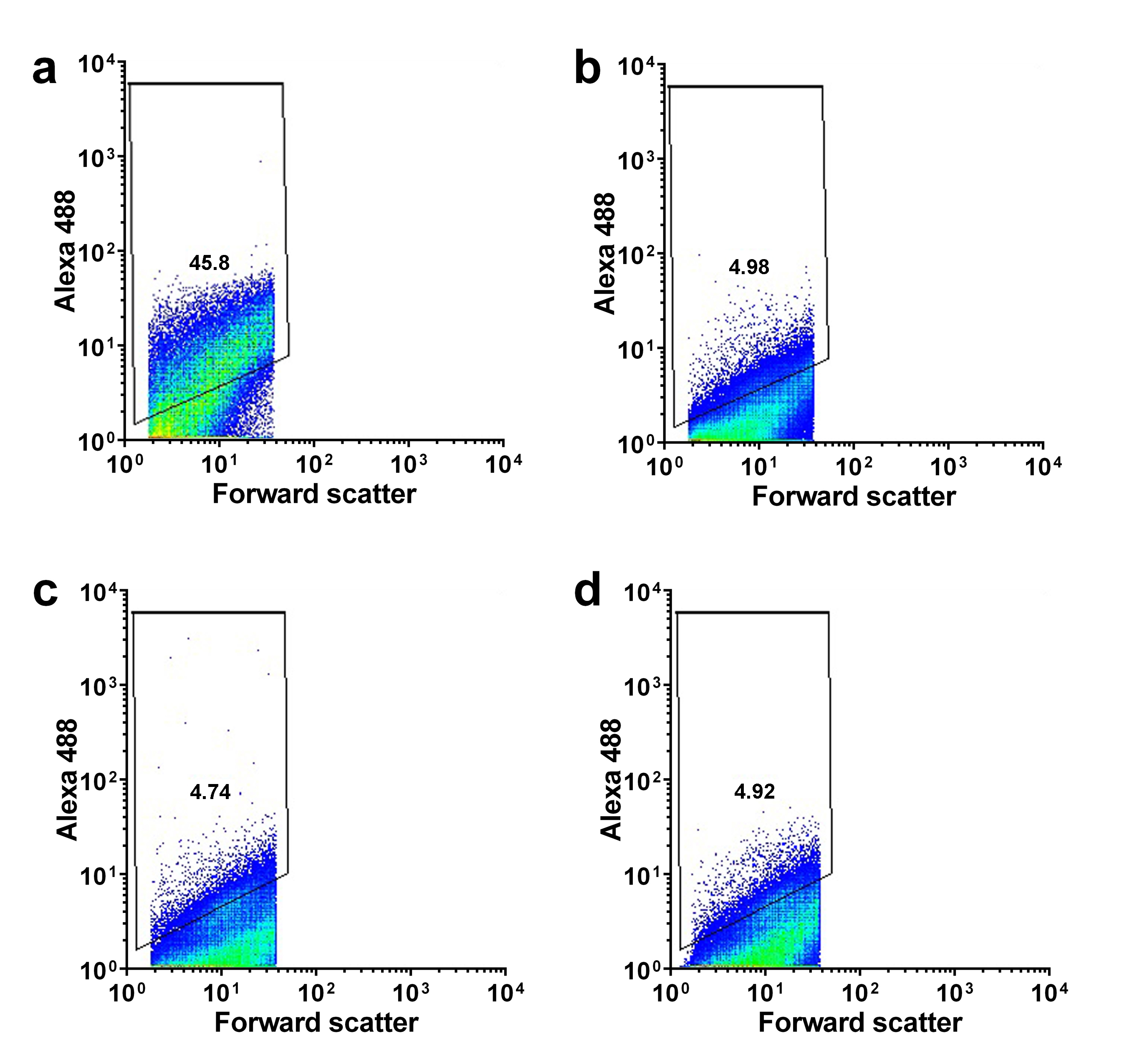
Representative density plots of human cortical synaptosome preparations labeled with SNAP-25 and isotype control (IgG) antibodies. SNAP-25 antibody staining of synaptosome preparations obtained from (a) control group, (b) LBD group, (c) AD group and (d) μVBI±AD group. Isotype control staining of synaptosome preparations obtained from (e) control group, (f) LBD group, (g) AD group and (h) μVBI±AD group. For each sample pair (a–e, b–f, c–g, d–h), the respective isotype control-labeled sample (e, f, g, h) was used to set the positive gate for the corresponding sample labeled with SNAP-25 antibody (a, b, c, d). Due to the varying levels of non-specific binding of the antibodies between individual cases, the position of the positive gates for each sample pair is slightly different.
{kind=link}
Representative density plots of human striatal synaptosome preparations labeled with SNAP-25 and isotype control (IgG) antibodies. SNAP-25 antibody staining of synaptosome preparations obtained from (a) control group, (b) LBD group, (c) AD group and (d) μVBI±AD group. Isotype control staining of synaptosome preparations obtained from (e) control group, (f) LBD group, (g) AD group and (h) μVBI±AD group. For each sample pair (a–e, b–f, c–g, d–h), the respective isotype control-labeled sample (e, f, g, h) was used to set the positive gate for the corresponding sample labeled with SNAP-25 antibody (a, b, c, d). Due to the varying levels of non-specific binding of the antibodies between individual cases, the position of the positive gates for each sample pair is slightly different.
{kind=link}
Representative density plots of mouse striatal synaptosome preparations labeled with SNAP-25 and isotype control (IgG) antibodies. SNAP-25 antibody staining of synaptosome preparations obtained from (a) DT-treated WT and (b) DT-treated Slc6a3-DTR mice. Isotype control staining of synaptosome preparations obtained from (c) DT-treated WT and (d) DT-treated Slc6a3-DTR mice. For each sample pair (a–c, b–d), the respective isotype control-labeled sample (b, d) was used to set the positive gate for the corresponding sample labeled with SNAP-25 antibody (a, b). Due to the varying levels of non-specific binding of the antibodies between individual mice, the position of the positive gates for each sample pair is slightly different.
{kind=link}
Representative plots of human striatal synaptosome preparations labeled with DAT and isotype control (IgG) antibodies. DAT-antibody staining of synaptosome preparations obtained from (a) control group, (b) LBD group, (c) AD group and (d) μVBI±AD group. Isotype control staining of synaptosome preparations obtained from (e) control group, (f) LBD group, (g) AD group and (h) μVBI±AD group. For each sample pair (a–e, b–f, c–g, d–h), the respective isotype control-labeled sample (e, f, g, h) was used to set the positive gate for the corresponding sample labeled with DAT antibody (a, b, c, d). Due to the varying levels of non-specific binding of the antibodies between individual cases, the position of the positive gates for each sample pair is slightly different.
{kind=link}
Representative plots of mouse striatal synaptosome preparations labeled with DAT and isotype control (IgG) antibodies. Samples from a DT-treated WT mouse labeled with an antibody against DAT (a) and isotype control antibody (b). Samples from a DT-treated Slc6a3-DTR mouse, labeled with the DAT antibody (c) and an isotype control antibody (d). For each sample pair (a–c, b–d), the respective isotype control-labeled sample (b, d) was used to set the positive gate for the corresponding sample labeled with DAT antibody (a, b). Due to the varying levels of non-specific binding of the antibodies between individual mice, the position of the positive gates for each sample pair is slightly different.
{kind=link}
Flow cytometry analysis of the synaptosome preparation of human cortex from control (n = 5), LBD (n = 5), AD (n = 11) and μVBI±AD (n = 3) groups labeled with the antibody against α-synuclein phosphorylated at Ser129. Percentage of α-synuclein-positive particles in the post-mortem samples is shown. Data is presented as mean ± SEM.
{kind=link}
Representative density plots of human cortical synaptosome preparations labeled with VGLUT1 and isotype control (IgG) antibodies. VGLUT1 antibody staining of synaptosome preparations obtained from (a) control group, (b) LBD group, (c) AD group, (d) μVBI±AD group and (e) from a human cortical sample resected during surgery. IgG staining of synaptosome preparations obtained from (f) control group, (g) LBD group, (h) AD group, (i) μVBI±AD group and (j) from a human cortical sample resected during surgery. For each sample pair (a–f, b–g, c–h, d–i, e–j), the respective isotype control-labeled sample (f, g, h, i, j) was used to set the positive gate for the corresponding sample labeled with VGLUT1 antibody (a, b, c, d, e). Due to the varying levels of non-specific binding of the antibodies between individual cases, the position of the positive gates for each sample pair is slightly different.
{kind=link}
Acknowledgments
This work was supported by grants from the NIH (U01 AG06781, P50 NS062684 and P50 AG05136) and the Nancy and Buster Alvord Endowment.
We thank Dr. Richard Palmiter, University of Washington, for generating the Slc6a3-DTR mice. We thank Dr. Karen Gylys and Dr. Gregory Cole for their helpful comments during the manuscript preparation. We thank Dr. Kathiravetpillai Arumuganathan, Director of Flow Cytometry Core Lab (Benaroya Research Institute at Virginia Mason), for assistance and support with cytometry. We also thank Angela Wilson, Nikolas Jorstad, Samantha Rice, Kim Howard, Hanna Kokko, Jeffrey Gibbs and Chiyen Miller for help with tissue collection and processing, electron microscopy and animal maintenance. We thank Aimee Schantz and Carol Arnold for administrative support. This work was supported by grants from the NIH (U01 AG06781, P50 NS062684 and P50 AG05136) and the Nancy and Buster Alvord Endowment.
Abbreviations
- AD
Alzheimer’s disease
- LB
Lewy body
- LBD
Lewy body disease
- μVBI
micro-vascular brain injury
- DL
dorso-lateral
- VM
ventro-medial
- Aβ
amyloid β
- DAT
dopamine transporter
- DA
dopamine
- DOPAC
3,4-dihydroxyphenylacetic acid
- hDTR
human diphtheria toxin receptor
Footnotes
DISCLOSURE/CONFLICT OF INTEREST
The authors declare no conflict of interest.
References
- 1.Burke RE, O’Malley K. Axon degeneration in Parkinson’s disease. Exp Neurol. 2013;246:72–83. doi: 10.1016/j.expneurol.2012.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jellinger KA. Significance of brain lesions in Parkinson disease dementia and Lewy body dementia. Front Neurol Neurosci. 2009;24:114–125. doi: 10.1159/000197890. [DOI] [PubMed] [Google Scholar]
- 3.Collins LM, Toulouse A, Connor TJ, Nolan YM. Contributions of central and systemic inflammation to the pathophysiology of Parkinson’s disease. Neuropharmacology. 2012;62(7):2154–2168. doi: 10.1016/j.neuropharm.2012.01.028. [DOI] [PubMed] [Google Scholar]
- 4.Irwin DJ, White MT, Toledo JB, et al. Neuropathologic substrates of Parkinson disease dementia. Ann Neurol. 2012;72(4):587–598. doi: 10.1002/ana.23659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kurz A, Perneczky R. Neurobiology of cognitive disorders. Curr Opin Psychiatry. 2009;22(6):546–551. doi: 10.1097/YCO.0b013e328330588b. [DOI] [PubMed] [Google Scholar]
- 6.Braak H, Rub U, Del Tredici K. Cognitive decline correlates with neuropathological stage in Parkinson’s disease. J Neurol Sci. 2006;248(1–2):255–258. doi: 10.1016/j.jns.2006.05.011. [DOI] [PubMed] [Google Scholar]
- 7.Bellucci A, Zaltieri M, Navarria L, Grigoletto J, Missale C, Spano P. From alpha-synuclein to synaptic dysfunctions: new insights into the pathophysiology of Parkinson’s disease. Brain Res. 2012;1476:183–202. doi: 10.1016/j.brainres.2012.04.014. [DOI] [PubMed] [Google Scholar]
- 8.Scott DA, Tabarean I, Tang Y, Cartier A, Masliah E, Roy S. A pathologic cascade leading to synaptic dysfunction in alpha-synuclein-induced neurodegeneration. J Neurosci. 2010;30(24):8083–8095. doi: 10.1523/JNEUROSCI.1091-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Milatovic D, Montine TJ, Zaja-Milatovic S, Madison JL, Bowman AB, Aschner M. Morphometric analysis in neurodegenerative disorders. Curr Protoc Toxicol. 2010;Chapter 12(Unit 12):16. doi: 10.1002/0471140856.tx1216s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meinertzhagen IA, Takemura SY, Lu Z, et al. From form to function: the ways to know a neuron. J Neurogenet. 2009;23(1–2):68–77. doi: 10.1080/01677060802610604. [DOI] [PubMed] [Google Scholar]
- 11.Booth RF, Clark JB. A rapid method for the preparation of relatively pure metabolically competent synaptosomes from rat brain. Biochem J. 1978;176(2):365–370. doi: 10.1042/bj1760365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gray EG, Whittaker VP. The isolation of nerve endings from brain: an electron-microscopic study of cell fragments derived by homogenization and centrifugation. J Anat. 1962;96:79–88. [PMC free article] [PubMed] [Google Scholar]
- 13.Hebb CO, Whittaker VP. Intracellular distributions of acetylcholine and choline acetylase. J Physiol. 1958;142(1):187–196. doi: 10.1113/jphysiol.1958.sp006008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arold S, Sullivan P, Bilousova T, et al. Apolipoprotein E level and cholesterol are associated with reduced synaptic amyloid beta in Alzheimer’s disease and apoE TR mouse cortex. Acta Neuropathol. 2012;123(1):39–52. doi: 10.1007/s00401-011-0892-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Henkins KM, Sokolow S, Miller CA, et al. Extensive p-tau pathology and SDS-stable p-tau oligomers in Alzheimer’s cortical synapses. Brain Pathol. 2012;22(6):826–833. doi: 10.1111/j.1750-3639.2012.00598.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tai LM, Bilousova T, Jungbauer L, et al. Levels of soluble apolipoprotein E/amyloid-beta (Abeta) complex are reduced and oligomeric Abeta increased with APOE4 and Alzheimer disease in a transgenic mouse model and human samples. J Biol Chem. 2013;288(8):5914–5926. doi: 10.1074/jbc.M112.442103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cholerton B, Larson EB, Baker LD, et al. Neuropathologic correlates of cognition in a population-based sample. J Alzheimers Dis. 2013;36(4):699–709. doi: 10.3233/JAD-130281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cholerton BA, Zabetian CP, Quinn JF, et al. Pacific Northwest Udall Center of excellence clinical consortium: study design and baseline cohort characteristics. J Parkinsons Dis. 2013;3(2):205–214. doi: 10.3233/JPD-130189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24(2):197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 20.Jellinger KA. Formation and development of Lewy pathology: a critical update. J Neurol. 2009;256 (Suppl 3):270–279. doi: 10.1007/s00415-009-5243-y. [DOI] [PubMed] [Google Scholar]
- 21.White L, Petrovitch H, Hardman J, et al. Cerebrovascular pathology and dementia in autopsied Honolulu-Asia Aging Study participants. Ann N Y Acad Sci. 2002;977:9–23. doi: 10.1111/j.1749-6632.2002.tb04794.x. [DOI] [PubMed] [Google Scholar]
- 22.Gylys KH, Fein JA, Cole GM. Quantitative characterization of crude synaptosomal fraction (P-2) components by flow cytometry. J Neurosci Res. 2000;61(2):186–192. doi: 10.1002/1097-4547(20000715)61:2<186::AID-JNR9>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 23.Montine KS, Montine TJ. Anatomic and clinical pathology of cognitive impairment and dementia. J Alzheimers Dis. 2013;33 (Suppl 1):S181–184. doi: 10.3233/JAD-2012-129032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hyman BT, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement. 2012;8(1):1–13. doi: 10.1016/j.jalz.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Montine TJ, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol. 2012;123(1):1–11. doi: 10.1007/s00401-011-0910-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82(4):239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 27.Mirra SS, Heyman A, McKeel D, et al. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology. 1991;41(4):479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 28.Thal DR, Rub U, Orantes M, Braak H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58(12):1791–1800. doi: 10.1212/wnl.58.12.1791. [DOI] [PubMed] [Google Scholar]
- 29.Luquet S, Perez FA, Hnasko TS, Palmiter RD. NPY/AgRP neurons are essential for feeding in adult mice but can be ablated in neonates. Science. 2005;310(5748):683–685. doi: 10.1126/science.1115524. [DOI] [PubMed] [Google Scholar]
- 30.Sokolow S, Luu SH, Nandy K, et al. Preferential accumulation of amyloid-beta in presynaptic glutamatergic terminals (VGluT1 and VGluT2) in Alzheimer’s disease cortex. Neurobiol Dis. 2012;45(1):381–387. doi: 10.1016/j.nbd.2011.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dunkley PR, Jarvie PE, Heath JW, Kidd GJ, Rostas JA. A rapid method for isolation of synaptosomes on Percoll gradients. Brain Res. 1986;372(1):115–129. doi: 10.1016/0006-8993(86)91464-2. [DOI] [PubMed] [Google Scholar]
- 32.Nagy A, Delgado-Escueta AV. Rapid preparation of synaptosomes from mammalian brain using nontoxic isoosmotic gradient material (Percoll) J Neurochem. 1984;43(4):1114–1123. doi: 10.1111/j.1471-4159.1984.tb12851.x. [DOI] [PubMed] [Google Scholar]
- 33.Melchitzky DS, Lewis DA. Tyrosine hydroxylase- and dopamine transporter-immunoreactive axons in the primate cerebellum. Evidence for a lobular- and laminar-specific dopamine innervation. Neuropsychopharmacology. 2000;22(5):466–472. doi: 10.1016/S0893-133X(99)00139-6. [DOI] [PubMed] [Google Scholar]
- 34.Bjorklund A, Dunnett SB. Dopamine neuron systems in the brain: an update. Trends Neurosci. 2007;30(5):194–202. doi: 10.1016/j.tins.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 35.Gylys KH, Fein JA, Yang F, Wiley DJ, Miller CA, Cole GM. Synaptic changes in Alzheimer’s disease: increased amyloid-beta and gliosis in surviving terminals is accompanied by decreased PSD-95 fluorescence. Am J Pathol. 2004;165(5):1809–1817. doi: 10.1016/s0002-9440(10)63436-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Iwatsubo T, Odaka A, Suzuki N, Mizusawa H, Nukina N, Ihara Y. Visualization of A beta 42(43) and A beta 40 in senile plaques with end-specific A beta monoclonals: evidence that an initially deposited species is A beta 42(43) Neuron. 1994;13(1):45–53. doi: 10.1016/0896-6273(94)90458-8. [DOI] [PubMed] [Google Scholar]
- 37.Kreisl WC, Lyoo CH, McGwier M, et al. In vivo radioligand binding to translocator protein correlates with severity of Alzheimer’s disease. Brain. 2013;136(Pt 7):2228–2238. doi: 10.1093/brain/awt145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fujiwara H, Hasegawa M, Dohmae N, et al. alpha-synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol. 2002;4(2):160–164. doi: 10.1038/ncb748. [DOI] [PubMed] [Google Scholar]
- 39.Chen KH, Reese EA, Kim HW, Rapoport SI, Rao JS. Disturbed neurotransmitter transporter expression in Alzheimer’s disease brain. J Alzheimers Dis. 2011;26(4):755–766. doi: 10.3233/JAD-2011-110002. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 40.Price DL, Rockenstein E, Ubhi K, et al. Alterations in mGluR5 expression and signaling in Lewy body disease and in transgenic models of alpha-synucleinopathy--implications for excitotoxicity. PLoS One. 2010;5(11):e14020. doi: 10.1371/journal.pone.0014020. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 41.Blandini F. An update on the potential role of excitotoxicity in the pathogenesis of Parkinson’s disease. Funct Neurol. 2010;25(2):65–71. [PubMed] [Google Scholar]
- 42.Brucke T, Djamshidian S, Bencsits G, Pirker W, Asenbaum S, Podreka I. SPECT and PET imaging of the dopaminergic system in Parkinson’s disease. J Neurol. 2000;247(Suppl 4):IV/2–7. doi: 10.1007/pl00007769. [DOI] [PubMed] [Google Scholar]
- 43.Chassain C, Bielicki G, Keller C, Renou JP, Durif F. Metabolic changes detected in vivo by 1H MRS in the MPTP-intoxicated mouse. NMR Biomed. 2010;23(6):547–553. doi: 10.1002/nbm.1504. [DOI] [PubMed] [Google Scholar]
- 44.Gao HC, Zhu H, Song CY, et al. Metabolic changes detected by ex vivo high resolution 1H NMR spectroscopy in the striatum of 6-OHDA-induced Parkinson’s rat. Mol Neurobiol. 2013;47(1):123–130. doi: 10.1007/s12035-012-8336-z. [DOI] [PubMed] [Google Scholar]
- 45.Brichta L, Greengard P, Flajolet M. Advances in the pharmacological treatment of Parkinson’s disease: targeting neurotransmitter systems. Trends Neurosci. 2013;36(9):543–554. doi: 10.1016/j.tins.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 46.Ahmed I, Bose SK, Pavese N, et al. Glutamate NMDA receptor dysregulation in Parkinson’s disease with dyskinesias. Brain. 2011;134(Pt 4):979–986. doi: 10.1093/brain/awr028. [DOI] [PubMed] [Google Scholar]
- 47.Donaghy P, Thomas AJ, O’Brien JT. Amyloid PET Imaging in Lewy Body Disorders. Am J Geriatr Psychiatry. 2013 doi: 10.1016/j.jagp.2013.03.001. [DOI] [PubMed] [Google Scholar]
- 48.Shi M, Huber BR, Zhang J. Biomarkers for cognitive impairment in Parkinson disease. Brain Pathol. 2010;20(3):660–671. doi: 10.1111/j.1750-3639.2009.00370.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Volpicelli-Daley LA, Luk KC, Patel TP, et al. Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron. 2011;72(1):57–71. doi: 10.1016/j.neuron.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bohnen NI, Albin RL. Cholinergic denervation occurs early in Parkinson disease. Neurology. 2009;73(4):256–257. doi: 10.1212/WNL.0b013e3181b0bd3d. [DOI] [PubMed] [Google Scholar]
- 51.Brooks D, Halliday GM. Intralaminar nuclei of the thalamus in Lewy body diseases. Brain Res Bull. 2009;78(2–3):97–104. doi: 10.1016/j.brainresbull.2008.08.014. [DOI] [PubMed] [Google Scholar]
- 52.Halliday GM. Thalamic changes in Parkinson’s disease. Parkinsonism Relat Disord. 2009;15 (Suppl 3):S152–155. doi: 10.1016/S1353-8020(09)70804-1. [DOI] [PubMed] [Google Scholar]
- 53.Hirsch EC, Graybiel AM, Duyckaerts C, Javoy-Agid F. Neuronal loss in the pedunculopontine tegmental nucleus in Parkinson disease and in progressive supranuclear palsy. Proc Natl Acad Sci U S A. 1987;84(16):5976–5980. doi: 10.1073/pnas.84.16.5976. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Representative density plots of human cortical synaptosome preparations labeled with SNAP-25 and isotype control (IgG) antibodies. SNAP-25 antibody staining of synaptosome preparations obtained from (a) control group, (b) LBD group, (c) AD group and (d) μVBI±AD group. Isotype control staining of synaptosome preparations obtained from (e) control group, (f) LBD group, (g) AD group and (h) μVBI±AD group. For each sample pair (a–e, b–f, c–g, d–h), the respective isotype control-labeled sample (e, f, g, h) was used to set the positive gate for the corresponding sample labeled with SNAP-25 antibody (a, b, c, d). Due to the varying levels of non-specific binding of the antibodies between individual cases, the position of the positive gates for each sample pair is slightly different.
Representative density plots of human striatal synaptosome preparations labeled with SNAP-25 and isotype control (IgG) antibodies. SNAP-25 antibody staining of synaptosome preparations obtained from (a) control group, (b) LBD group, (c) AD group and (d) μVBI±AD group. Isotype control staining of synaptosome preparations obtained from (e) control group, (f) LBD group, (g) AD group and (h) μVBI±AD group. For each sample pair (a–e, b–f, c–g, d–h), the respective isotype control-labeled sample (e, f, g, h) was used to set the positive gate for the corresponding sample labeled with SNAP-25 antibody (a, b, c, d). Due to the varying levels of non-specific binding of the antibodies between individual cases, the position of the positive gates for each sample pair is slightly different.
Representative density plots of mouse striatal synaptosome preparations labeled with SNAP-25 and isotype control (IgG) antibodies. SNAP-25 antibody staining of synaptosome preparations obtained from (a) DT-treated WT and (b) DT-treated Slc6a3-DTR mice. Isotype control staining of synaptosome preparations obtained from (c) DT-treated WT and (d) DT-treated Slc6a3-DTR mice. For each sample pair (a–c, b–d), the respective isotype control-labeled sample (b, d) was used to set the positive gate for the corresponding sample labeled with SNAP-25 antibody (a, b). Due to the varying levels of non-specific binding of the antibodies between individual mice, the position of the positive gates for each sample pair is slightly different.
Representative plots of human striatal synaptosome preparations labeled with DAT and isotype control (IgG) antibodies. DAT-antibody staining of synaptosome preparations obtained from (a) control group, (b) LBD group, (c) AD group and (d) μVBI±AD group. Isotype control staining of synaptosome preparations obtained from (e) control group, (f) LBD group, (g) AD group and (h) μVBI±AD group. For each sample pair (a–e, b–f, c–g, d–h), the respective isotype control-labeled sample (e, f, g, h) was used to set the positive gate for the corresponding sample labeled with DAT antibody (a, b, c, d). Due to the varying levels of non-specific binding of the antibodies between individual cases, the position of the positive gates for each sample pair is slightly different.
Representative plots of mouse striatal synaptosome preparations labeled with DAT and isotype control (IgG) antibodies. Samples from a DT-treated WT mouse labeled with an antibody against DAT (a) and isotype control antibody (b). Samples from a DT-treated Slc6a3-DTR mouse, labeled with the DAT antibody (c) and an isotype control antibody (d). For each sample pair (a–c, b–d), the respective isotype control-labeled sample (b, d) was used to set the positive gate for the corresponding sample labeled with DAT antibody (a, b). Due to the varying levels of non-specific binding of the antibodies between individual mice, the position of the positive gates for each sample pair is slightly different.
Flow cytometry analysis of the synaptosome preparation of human cortex from control (n = 5), LBD (n = 5), AD (n = 11) and μVBI±AD (n = 3) groups labeled with the antibody against α-synuclein phosphorylated at Ser129. Percentage of α-synuclein-positive particles in the post-mortem samples is shown. Data is presented as mean ± SEM.
Representative density plots of human cortical synaptosome preparations labeled with VGLUT1 and isotype control (IgG) antibodies. VGLUT1 antibody staining of synaptosome preparations obtained from (a) control group, (b) LBD group, (c) AD group, (d) μVBI±AD group and (e) from a human cortical sample resected during surgery. IgG staining of synaptosome preparations obtained from (f) control group, (g) LBD group, (h) AD group, (i) μVBI±AD group and (j) from a human cortical sample resected during surgery. For each sample pair (a–f, b–g, c–h, d–i, e–j), the respective isotype control-labeled sample (f, g, h, i, j) was used to set the positive gate for the corresponding sample labeled with VGLUT1 antibody (a, b, c, d, e). Due to the varying levels of non-specific binding of the antibodies between individual cases, the position of the positive gates for each sample pair is slightly different.