

Abstract
Mutations in the APC or β-catenin genes are well established initiators of colorectal cancer (CRC), yet modifiers that facilitate the survival and progression of nascent tumor cells are not well defined. Using genetic and pharmacological approaches in mouse CRC and human CRC xenograft models, we show that incipient intestinal tumor cells activate CDC42, an APC-interacting small GTPase, as a crucial step in malignant progression. In the mouse, Cdc42 ablation attenuated the tumorigenicity of mutant intestinal cells carrying single APC or β-catenin mutations. Similarly, human CRC with relatively higher levels of CDC42 activity were particularly sensitive to CDC42 blockade. Mechanistic studies suggested that Cdc42 may be activated at different levels, including at the level of transcriptional activation of the stem-cell-enriched Rho family exchange factor Arhgef4. Our results suggest that early-stage mutant intestinal epithelial cells must recruit the pleiotropic functions of Cdc42 for malignant progression, suggesting its relevance as a biomarker and therapeutic target for selective CRC intervention.
INTRODUCTION
Colorectal cancer (CRC) remains the most prevalent digestive cancer, affecting nearly 150,000 people annually in the US and mortality being an outcome for a third of these patients (1). The view that APC mutation acts as a genetic initiator for most CRCs has been shaped by molecular pathological observations and studies of CRC animal models (2, 3). APC mutations are detected in 88% of non-hypermutated CRCs (4), and are found in the earliest microadenoma lesions containing only several dysplastic glands (5). Comparing to other somatic mutations with progressively higher mutation frequency in advanced adenocarcinomas, APC mutations have a similar rate in both microadenomas and advanced adenocarcinomas (2, 6). The earliest morphological changes in APCΔ716/+ mouse intestines (7) emerged as outpocketing epithelial pouches from the upper part of a crypt. These tumor-initiating cells then developed laterally into the neighboring villi forming the typical adenomatous polyps (7). Similar morphogenetic changes were described in mice carrying a dominant stable mutation in β-catenin (8), suggesting that some shared mechanisms might underlie the aberrant morphogenetic transformation in early-stage tumor cells.
APC directly binds to an APC-stimulated exchanging factor (Asef1, or Arhgef4 hereafter) through its conserved Armadillo repeat domain (9, 10). Both Arhgef4 and its homolog Spata13 (Asef2) are specific guanine nucleotide exchanging factors (GEFs) for cell division control 42 (Cdc42) (11, 12). Cdc42 is a small GTPase regulating various aspects of cell morphogenesis, division, and migration. APC directly interacts with Cdc42 in multiple in vitro interaction assays (13). The link between APC and Arhgef4 has been viewed as one of the most persuasive evidences for APC-mediated remodeling of cytoskeleton (14). However, with controversial mechanisms proposed for APC-mediated Cdc42 activation (12, 15, 16), the functional output of this regulatory cascade during intestinal tumorigenesis remains poorly understood.
Cdc42 was observed to be highly expressed in 60% of human CRCs with its level positively correlating with poorly differentiated CRCs (17). Arhgef4 was recently identified as one of the signature genes characteristic of Lgr5+ intestinal stem cells (18). Both Arhgef4 and Cdc42 have been proposed as tumor suppressors (12); however, in vivo evidence supporting this notion has been missing. We have recently showed that Cdc42 deletion impaired intestinal Lgr5+ stem cell homeostasis (19). Here, we provide genetic evidence that nascent mouse intestinal tumor cells carrying single mutations in either APC or β-catenin could activate Cdc42, possibly at different levels. Inhibition of Cdc42 by genetic ablation or small molecule inhibitor attenuated tumorigenicity of the fast-cycling microadenoma-constructing tumor cells, whose survivability depended on high levels of cell-autonomous Cdc42 activity. Human CRCs with higher Cdc42 levels are more sensitive to Cdc42 inhibition. Our results suggest that Cdc42 may be an immediate mediator of APC/β-catenin mutations in early-stage tumor cells, and may be used as a biomarker for selective targeting of some CRCs.
MATERIALS AND METHODS
Mice
Cdc42loxP (20), Catnb(ex3)fl (8), Lgr5EGFP-IRES-creERT2 (21), Rosa26REYFP (22), Villin-Cre (23), Villin-CreER (24), and ApcMin/+ mice (25) have been described previously. Mice were maintained at 129/BL6 mixed background. For comparing Cdc42 activities between 7-week ApcMin/+ and wild type mice, C57BL/6J mice of same genetic background were used as controls. All mouse experiments were exclusively performed on littermate animals, with 3~10 mice used for each genotype. Rutgers University Institutional Animal Care and Use Committee approved all mouse procedures.
Isolation of Lgr5+ intestinal stem cells from 3-month old Lgr5EGFP-IRES-creERT2 mice was performed with a 4-way MoFlo cell sorter (Beckman-Coulter) based on EGFP and EpCAM expression as described (26).
Human CRC Cell Culture
Human CRC cells, LIM1863, LIM1899, LIM2551, LIM2550, LIM1215, SW480, and Caco2 have been maintained in conditions described previously (27, 28). For LIM1863 cells that form tumor organoids in suspension, cells were grown in RPMI1640 (Corning, MT10-040-CM) with 10% FBS (Sigma, F2442), 1 μg/mL hydrocortisone (Sigma, H0888), 0.01μg/mL thioglycerol (Sigma, M1753), and 0.025 U/ml insulin (Sigma, I1882). For passaging, LIM1863 cells were mechanically pipetted up-and-down to break the organoids before seeding into fresh medium. All in vitro cell experiments were done in triplicates and repeated at different circumstances.
To determine the growth rate of different cell lines, cells (103/well) were seeded into a 96-well plate in triplicate. Cell growth was measured every 24 hrs using Cell Counting Kit-8 (CCK-8, Dojindo Laboratories, Japan). Briefly, WST-8 was reduced by dehydrogenases in cells to give an orange soluble formazan dye, which is directly proportional to the number of living cells. CCK-8 reagent (10 μL) was added into each well, incubated for 2 hrs, and subjected to absorbance measurement at 450nm. Cell culture media were gently replenished every 24 hrs to avoid losing viable cells. For CASIN treatment experiments, 3×103 cells/well were seeded in 100 μL culture media in the absence or presence of 10 μM CASIN. Cell growth was measured by CCK-8 every 24 hours. To avoid losing viable cells, suspended cells in culture media were also collected by centrifugation at 200 g and seeded back when culture media were replenished.
CASIN (C20H22N2O), or 2-((6-phenyl-2,3,4,9-tetrahydro-1H-carbazol-1-yl) amino) ethanol, was dissolved in DMSO and administrated to culture media at desired concentrations (19). Cells were treated with same volumes of DMSO in control groups. Lentiviral particle mediated Cdc42 knockdown has been described in details (19).
For cell cycle analysis by flow cytometry, cells were cultured until they reached a confluence of 70–80%, trypsinized, and harvested by centrifuge. After washing by PBS, 106 cells were re-suspended in 0.5 mL PBS, dispersed into single cells by pipetting. Cells were fixed by 70% ethanol at 4°C. Fixed cells were centrifuged, washed by PBS, incubated in 10 μg/mL Propidium Iodide (Sigma, P4170) with 0.1% (v/v) Triton X-100 and 100 μg/mL DNase-free RNaseA in PBS, at 37°C for 10 min, and subjected to flow cytometry analysis.
For live cell or cancer organoid imaging, small intestinal crypts or human CRC cells were cultured in glass bottom dishes (MatTek Corporation, Ashland, MA, Catalog # P35-1.5-10-C) and imaged using an inverted Zeiss Observer wide field microscope equipped with a CoolSnapHQ2 (Photometrics, Tucson, AZ). Axiovision 4.8 was used to acquire differential interference contrast images every 5 minutes using a 63 × water immersion objective for various durations depending on the experiments. The dish was maintained at 37°C in a 5% CO2 environment using temperature control 37-2 digital module and CTI Controller 3700 digital (Zeiss), respectively.
For CRC cell line xenograft assays, male athymic nude-Fox1 nu/nu mice (5-weeks old, Harlan Laboratories, Indianapolis, IN) were randomly allocated into 2 groups, 5 mice per group, and housed in plastic cages with filter tops and acclimated to the regular diet. CRC cells grown in regular culture media without antibiotics were divided into 2 groups: one treated overnight (~16 hrs) with 5 or 10 μM CASIN and the other with DMSO. Cells (1×106) were harvested in 100 μL of a 2:1 mixture of serum-free RPMI-1640 and Matrigel (BD, Franklin Lakes, NJ), and subcutaneously injected to the left and right flank of the mice (i.e., left for CASIN-treated cells, and right for control cells). For xenograft of CRC cells with stable Cdc42 knockdown, 2.5×106 cells of each group, i.e., control and Cdc42-knockdown cells, were subcutaneously injected to the left and right flank of the nude mice (n=5 for each cell line). Tumor volume, body weight, and food consumption were monitored once a week. Tumor size (length and width) was measured by a caliper and calculated based on the formula (tumor volume = length × width2 × 0.5). The tumors were dissected and weighed; one half of the tumor was fixed in 10% buffered formalin for histological and IHC analyses and the second half were snap-frozen for biochemical analysis.
Mouse Intestinal Crypt Isolation and Enteroid Culture
Procedures for crypt isolation and culture were essentially the same as reported (29). Proximal third of the intestines were dissected and flushed with 1× PBS, cut open and fragmented into smaller pieces (~2 cm sizes). Tissue fragments were then rinsed 3 times in PBS followed by 2 washes in cold chelating buffer (2 mM EDTA in PBS) at 4°C for 5 minutes and 40 minutes respectively. Intestinal fragments were then vigorously re-suspended in the cold chelation buffer and allowed to flow through 70 μm filter (BD Falcon, 352350) into pre-cooled PBS. A pellet of crypts was obtained by centrifuging this flow-through suspension at 200 g for 3mins at 4°C. The pellet was then washed twice and resuspended in cold PBS for counting. 100~200 crypts suspended in matrigel (BD Biosciences, 356231) were plated into each well of pre-warmed (at 37 °C) 24-well plates. After allowing the matrigel to solidify at 37°C for 10 minutes, 500 μl of ENR organoid culture medium was added to each well (30). Working ENR medium contains 2× N2 supplement (Life Tech Gibco. 17502-048), 0.5× B27 (Life Tech Gibco. 17504-044), 1 mM N-Acetyl Cysteine (Sigma, A9165), 0.05 μg/ml of EGF (Life Technologies, PMG8043), 0.1 μg/ml of Noggin (Peprotech, 250-38) and 50 μg of R-Spondin (R & D Systems, 3474-RS-050)] made in 50 ml of Basal Culture medium [Advanced DMEM/F12 (Life Technologies, 12634-010) with 1× Penicillin/Streptomycin (Life Technologies, 15140-122), 1× Glutamax (Life Technologies, 35050-061) and 10 mM HEPES Buffer (Life Technologies, 15630-080)]. Crypts were prepared from Cdc42ΔIEC mice and wild type littermates. One hundred crypts were seeded in 50 μL Matrigel in each well on a 48-well plate. Three hundred μL ENR medium was added to each well. Every following morning, surviving crypts in each well were counted under a Nikon TE2000 fluorescent microscope from 4~6 wells for each genotype. For co-culture experiments, wild type and EGFP+ knockout (Cdc42fl/fl;Vil-Cre;Rosa26REYFP) crypts, 100 total, were mixed at 3 different ratios: 25/75, 50,50, and 75/25, and surviving Cdc42 knockout crypts, identified by EYFP fluorescence, were monitored for 7~10 days with medium changed every day. Recombinant Mouse Wnt3a (R & D Systems, USA) dissolved in sterile PBS was added at final concentrations of 50 and 200 ng/mL, to crypt culture medium at the beginning of the culture, and the effects on Cdc42 knockout crypt survival were monitored continuously for 7 days. For tamoxifen induction of cultured Catnb(ex3)fl/fl; Villin-CreER and Catnb(ex3)fl/+; Villin-CreER organoids, 2.7 μM tamoxifen (Sigma T5648) dissolved in ethanol was administrated to culture medium. A total of 200 organoids of each genotype was used for RT-PCR or Cdc42 activation assays.
Propidium iodide staining of CASIN- and DMSO-treated wild type and β-catenin mutant organoids: organoids were cultured in above culture medium. 10 μM CASIN was added to the culture for 16 hrs and incubated with 500 nM propidium iodide (Sigma, P4170) in PBS solution at 37°C for 20 min. The organoids were washed with PBS and covered with new medium. Images were taken with Zeiss LSM510 confocal microscope.
Immunohistochemistry, Confocal Immunofluorescence, and RNA in Situ Hybridization
Procedures of immunohisochemistry and confocal immunofluorescent analyses have been described (31). Confocal fluorescent and differential interference contrast images were acquired using a Zeiss LSM 510 confocal microscope. TUNEL staining (32) and RNA in situ (33) were performed as previously described. Primary antibodies were described in Supplementary Information.
Western Blot Analysis
Western blot analyses for specific protein targets were performed on freshly prepared tissue or cell lysates following procedures previously described (19). To detect active Cdc42, GTP-bound Cdc42 was pulled down using GST-human PAK1-PBD supplied by Cdc42 activation kit (Cell Signaling, Catalog # 8819), and detected by Cdc42 antibody (Cell Signaling, #2466). Experiments were repeated on independent biological samples for multiple times.
Histopathology Scoring of Intestinal Polyps and Adenomas
For macroscopic assessment of intestinal adenomatous polyps in ApcMin/+ mice, animals were sacrificed at desired ages and the full-length intestines were flushed with PBS. The small intestines were subdivided into three equal segments corresponding to duodenum, jejunum, and ileum, opened longitudinally, and examined using a dissecting microscope. Indigo carmine (Sigma, 57000) (1%) was dropped directly on the mucosal surface immediately after PBS rinses to remove any overlying mucous. Adenomas were identified, counted, classified, and measured. Neoplastic lesions were categorized into three subclasses; aberrant crypts (<0.25 mm), small adenomas (0.25–1 mm), and large adenomas (>1 mm).
For assessment of microadenomas in β-catenin mutant mice, Intestinal tissues were dissected out from neonatal mice of 5~12 days of ages, fixed in 4% paraformaldehyde, and embedded in paraffin. H. & E. stained tissue sections were analyzed by 2 experienced independent observers. The numbers of aberrant crypt-like foci were counted for each villus. One hundred crypt-villus compartments were counted for each histology section for individual mice.
Quantitative real-time RT-PCR (qRT-PCR)
TaqMan qRT-PCR was carried out in triplicate for mouse Arhgef4, Cdc42, Spata13, and, APC, using probe sets mm00805525_m1, mm01194005_g1, mm01323215_m1, and mm00545872_m1, respectively (Applied Biosystems, Carlsbad, CA). All assays were designed to have primers/probes to span exon-exon junctions. Relative gene expression was calculated by the ratio of the target genes to β-actin (mm00607939_s1) on an ABI StepOnePlus Detection System. Primers for Sox9, Lgr5, and CD44 are listed in Supplementary Information. To examine mRNA expression from organoids, tamoxifen-treated wild type and Ex3fl/fl;Vil-CreER organoids were used for RNA extraction. To examine the possibility of stem cell specific transcriptional regulation of Arhgef4, we interrogated existing enhancer chromatin datasets (H3K4me2 ChIP-seq) for evidence of a stem cell specific enhancer at the Arhgef4 locus (datasets in GEO GSM1246037 and GSM851122). Data were normalized for promoter chromatin levels and then visualized using the Integrative Genomics Viewer.
Statistical Analysis
Oncomine analysis (34) was conducted in June, 2014 using the research edition (www.oncomine.org), and queried the following datasets (35–41). Kaplan-Meier curves were generated for survival analyses. Morphometric analyses of intestinal villi and crypts were performed using NIH Image J software. One-way Analysis of Variance (ANOVA) followed by Tukey’s post hoc test was used for the comparison of the differences between the control and experimental groups in nude mice tumorigenicity assays. Student’s t test was used to determine the difference between experimental and control groups. Statistical significance was indicated by p< 0.05 or p<0.01 in two-tailed comparison. Mean values are shown in all graphs with error bars representing the standard error of the mean (S.E.M.).
RESULTS
Cdc42 is essential for intestinal crypt maintenance
Previous studies showed that total loss of Cdc42 in intestinal epithelial cells (IECs) impaired crypt organization in Cdc42fl/fl;Villin-Cre (or Cdc42ΔIEC) mice (19), suggesting that crypt homeostasis depends on Cdc42. Western blot analyses using separated intestinal tissues showed the highest levels of Cdc42 and Cdc42-GTP (active form) in crypts, compared with villus and stromal compartments (Fig. 1A). Higher levels of Arhgef4, an APC-driven Cdc42-specific guanine nucleotide exchanging factor, were also detected in crypts encompassing both the Paneth and Lgr5+ stem cell domains (Fig. 1B). Adult Lgr5+ stem cells (EGFP+) isolated by fluorescence-activated cell sorting (FACS) of Lgr5EGFP-IRES-CreER mouse IECs showed significantly higher Arhgef4 levels than EGFP− IECs (Fig. 1C). This transcriptional enrichment of Arhgef4 in Lgr5+ stem cells was supported by the presence of a stem cell-specific enhancer region upstream of Arhgef4 promoter (see arrows, Fig. 1D). Crypts isolated from Cdc42ΔIEC mice (Fig. 1E) failed to establish intestinal organoids in culture, with 94% of these Cdc42-knockout (KO) crypts lost within 24 hrs (Fig. 1F) and less than 1% survived up to 72 hrs (Fig. 1G), collectively suggesting that Cdc42 activity is essential for crypt survival and maintenance.
Figure 1. Cdc42 is essential for organoid survival in vitro.

(A) Western blots showed increased total and active Cdc42 levels in separated adult mouse crypts comparing to villus and stromal compartments. Villin, Vimentin, and Lysozyme served as markers for individual tissue fractions.
(B) Confocal immunofluorescent analyses for Arhgef4 (red) and EGFP+ Lgr5+ cells (green) using Lgr5EGFP-IRES-creERT2 mouse intestinal sections. Arrows point to stem cells.
(C) Real-time RT-PCR showed enriched Arhgef4 in FACS-isolated adult Lgr5+ stem cells.
(D) A stem cell-specific enhancer (arrow), 80-Kb upstream of Arhgef4 locus, was found in H3K4me2 ChIP-seq dataset from Lgr5+ stem (green) but not villus cells (blue).
(E) Western blots showed deletion of Cdc42 from Cdc42ΔIEC intestines and crypts. Note that the trace amount of Cdc42 (top panel, 2nd lane) reflected non-epithelial Cdc42 in the total intestinal lysates.
(F) 70% control crypts formed polarized cysts, whereas 94% Cdc42ΔIEC crypts arrested after 24 hrs in culture.
(G) Addition of Wnt3a (50 and 200 ng/mL) could not rescue Cdc42 knockout (KO) crypts.
(H) Co-culture of wild type and Cdc42KO crypts, using various mixing ratios, could not rescue the knockout KO. Note Cdc42KO crypts were isolated from Cdc42fl/fl;Vil-Cre;Rosa26REYFP mice, therefore were labeled as EYFP+ to distinguish from wild type crypts. Scale bars, 2 μm.
Paneth cells provide niche factors and support Lgr5+ stem cell homeostasis (30), and were observed to be dislocated from Cdc42ΔIEC mouse crypts (19, 42), raising the possibility that impaired Paneth cell development might indirectly contribute to observed loss of Cdc42-KO crypts (Fig. 1F). However, neither exogenous Wnt3a protein (Fig. 1G) nor co-culture with wild type crypts rescued these Cdc42-KO crypts (KO crypts were labeled as EYFP+ by a Rosa26REYFP allele, Fig. 1H). One out of 100 Cdc42ΔIEC crypts (EYFP+) formed initial buds (Fig. 1I); however none of these buds propagated as wild type organoids for a prolonged duration of culture (not shown). These data suggested that Cdc42 deletion cell-autonomously affected stem cells.
Reduction of Cdc42 alleviated APCMin/+ mouse polyposis
Cdc42 is overexpressed in the large majority of CRCs (17). We used Oncomine analysis (34) and examined transcript changes in colon tumors compared to normal tissues from 8 primary colon tumor data sets containing expression data for SPATA13, a homolog of ARHGEF4. Of these, 7 showed a significant increase (p<0.001, Student’s t-test) in SPATA13 transcript levels in colon tumors (35–41). ARHGEF4 and CDC42 showed inconsistent patterns of expression. While transcript expression levels may hint at a functional role in tumor development, the activation status of Cdc42 small GTPase is arguably more relevant. Using APCMin/+ mice, we asked whether and when Cdc42 is activated during the intestinal tumorigenesis in this model. Western blots detected increased Cdc42-GTP levels in ApcMin/+ mouse small and large intestines around 6–7 weeks (Fig. 2A). At this stage, dysplastic crypts became evident histopathologically (Fig. 2C), consistent with the reported earliest detection of intestinal lesions in this mouse model (43). These distorted glands frequently possessed eosinophilic granular debris (see high magnifications, Fig. 2C), positively labeled by Paneth cell marker Lysozyme (Fig. S1A). Highly increased levels of both total Cdc42 and Cdc42-GTP levels were observed in older APCMin/+ mouse intestines bearing adenomas (Fig. 2A, right panel). The expressional domains of Arhgef4 extended into the adenomatous polyps in APCMin/+ mice (Fig. 2B), suggesting that this Cdc42 activator was upregulated during tumorigenesis. To determine whether the observed elevation of Cdc42 activity played a role in tumor progression, we bred ApcMin/+ mice to Cdc42fl/+;Vil-Cre mice to derive ApcMin/+ mice with IEC-specific deletion of one or both Cdc42 alleles. ApcMin/+ mice developed tumors preferentially in small intestines (25), matching the sites of Vil-Cre mediated Cdc42 deletion. Western blots showed the reductions of Cdc42 and Cdc42-GTP levels upon Cdc42 deletion from ApcMin/+ intestines (Fig. 2E). ApcMin/+;Cdc42fl/fl;Villin-Cre mice, with both Cdc42 alleles deleted, showed early postnatal growth retardation and diarrhea, phenotypes resulting from Cdc42-deficiency (19). Pronounced epithelial cystic formation and lack of Paneth cell were observed in the intestines of these mice; however, no polyps were detected (Fig. 2D, and Fig. S1B). We excluded ApcMin/+;Cdc42fl/fl;Villin-Cre mice from long-term tumorigenesis assays due to phenotypic complexity and reduced body conditions, and performed the analyses on ApcMin/+;Cdc42fl/+;Villin-Cre and their littermate controls.
Figure 2. Cdc42 reduction alleviated APCMin/+ intestinal polyposis.

(A) Western blots showed increased levels of active Cdc42 in small and large intestines of 7-week APCMin/+ mouse intestines compared with age matched wild type mice of same genetic background (left panel, compare lanes 1–3 to lanes 4–6). Total and active Cdc42 were increased in older APCMin/+ mice (right panel). Jej: jejunum; Ile: ileum; Col: colon.
(B) Arhgef4 immunohistochemstry showed expansion of its expression domain into adenomatous tissues in APCMin/+ intestines.
(C) Dysplastic lesion in 40-day old APCMin/+ mice. Note that the distorted glands possess large eosinophilic granular debris, typically found in.Paneth cells.
(D) Total deletion of Cdc42 in Cdc42ΔIEC; APCMin/+ mice caused epithelial cystic formations. No dysplastic crypt was found. There is a complete loss of Paneth cell granule in crypts.
(E) Western blots for total, active Cdc42, and several Wnt targets. Note that Cdc42fl/+;Vil-Cre littermate mice, phenotypically wild type in terms of intestinal development and function (19), were used as reference.
(F) Cdc42 reduction decreased the tumor load in APCMin/+ mouse intestines. Macroscopic polyp detection by indigo carmine staining was followed by histology analyses and β-catenin immunohistochemistry on swiss-roll intestinal sections to identify adenoma. Note that Cdc42 inhibition reduced β-catenin nuclear but not epithelial junction staining. All comparison was made from same intestinal segments from littermate animals unless specified otherwise.
(G) Tumor numbers were counted and sizes were measured in duodenum, jejunum and ileum of mice with indicated genotypes. * indicates p<0.05 when compared to Cdc42L/+;Vil-Cre; and # indicates p<0.05 when compared to APCMin/+ mice. Scale bars, 5 μm.
At 4~5 months of age, ApcMin/+ mice developed in average 50 polyps per mouse revealed by indigo carmine staining (Fig. 2F). In contrast, the number and size of polyps were significantly decreased in ApcMin/+;Cdc42fl/+;Villin-Cre mice throughout the entire small intestinal tract (Fig. 2F, G). This alleviation of polyposis was exhibited at molecular levels, as Cdc42 reduction also reduced the levels of Tcf1, Tcf4, Sox9, C-Myc - a key mediator of Wnt signaling driven CRCs (44), and nuclear β-catenin (Fig. 2E, F), suggesting that high levels of Cdc42 might be required for APCMin/+ intestinal tumor progression.
Reduction of Cdc42 level attenuated intestinal tumorigenesis in β-catenin mutant mice
Cdc42 stabilizes β-catenin in skin cells through a GSK3β-mediated mechanism (20). We reasoned that if Cdc42-deletion triggered β-catenin degradation, it would explain the observed tumor suppression in APCMin/+ mice. (Fig. 2G). On the other hand, this tumor suppressive effect would not be observed in a β-catenin stable mutant genetic background. To explore whether Cdc42 inhibition indeed attenuated intestinal tumorigenesis by changing β-catenin stability, we examined the CRC mouse model carrying a dominant stable mutation, Catnbfl(ex3), in β-catenin (8). Cre-mediated deletion of β-catenin exon 3 in this mouse allele generated a truncated protein missing the GSK3β phosphorylation sites, leading to a dominant stable hence “activated” β-catenin (8). Western blots detected the truncated form of β-catenin in Catnb(ex3)/+;Vil-Cre mouse intestines carrying 1 allele of activated β-catenin (Fig. 3A). Remarkably, we detected a 14-fold increase in Cdc42-GTP level in these mutant intestines (Fig. 3B). Total Cdc42 levels were increased about 3-fold in the same tissues (Fig. 3B), suggesting that the mutant cells might have activated Cdc42 from different levels including transcriptional upregulation of Cdc42 itself. Approximately 50% of Catnb(ex3)/+;Vil-Cre pups died within the first week of their life, while none survived up to 12 days (red line, Fig. 3C). Numerous aberrant crypt-like foci (ACF), or microadenomas (8), were found throughout intestinal villus epithelia (see arrows, Fig. 3D). These lesions contained proliferative cells identified by both bromodeoxyuridine (BrdU) and pHH3 labeling (Fig. 3E, G).
Figure 3. Cdc42 reduction attenuates tumorigenesis in β-catenin stable mutant intestines.

(A) Western blots for β-catenin identified truncated protein in Catnblox(ex3)/+;Vil-Cre intestines.
(B) Western blots for showed 3- and 14-fold increases in the total and active Cdc42 levels in Catnblox(ex3)/+;Vil-Cre mouse intestines, comparing to littermate Cdc42fl/+;Vil-Cre mice.
(C) Cdc42 haploinsufficiency extended median survival time for 4 days (p<0.001).
(D) Cdc42 reduction reduced crypt-like foci (arrows) throughout the epithelia. Histological analyses were done on mouse small intestines from postnatal day 7 mice with indicated genotypes.
(E) Cdc42 reduction decreased tumor cell proliferation. Mice were pulse-labeled by BrdU for 1 hr. Note that residual villus proliferative cells remained to be detected (arrows).
(F) Numbers of crypt-like foci, BrdU+, and pHH3+ cells were reduced upon Cdc42 reduction.
(G) pHH3 staining (red) showed reduced mitotic tumor cells upon Cdc42 reduction. * indicates p<0.05 when compared to control; and # indicates p<0.05 when compared to Catnblox(ex3)/+;Vil-Cre mice. Scale bars, 5 μm.
We derived Catnb(ex3)/+;Cdc42fl/+;Villin-Cre mice, in which the Cdc42 gene dosage was reduced by half in the mutant IECs (grey bars, Fig. 3B). Western blots confirmed a reduction of Cdc42-GTP levels in these double heterozygous mice (Fig. 3B). The medium survival time of these double heterozygous mice was significantly prolonged from 9 to 13 days, compared with Catnb(ex3)/+;Villin-Cre littermates (p<0.001, Fig. 3C). 10% of animals were rescued up to 39 days of their life upon Cdc42 reduction (green line, Fig. 3C), accompanied by a significant gain of body weight (Fig. S2). This improved survival was also reflected by the significant reduction of villus lesions (Fig. 3D, F) and reduced tumor cell proliferation (Fig. 3E–G). The residual proliferative epithelial lesions in double heterozygous animals (Fig. 3E, F) indicated that reduction of Cdc42 by half might not sufficient to completely block tumorigenesis, because when Cdc42 was completely deleted in Catnb(ex3)/+;Cdc42fl/fl;Villin-Cre mice, no ACF, but severe epithelial cystic formation, was observed (Fig. S3). These data collectively suggested that genetic attenuation of Cdc42 also reduced intestinal tumorigenesis in a β-catenin stable mutant background.
Cdc42 inhibition impaired survival of nascent microadenoma cells
The morphogenesis of crypt-like foci in early-stage APC-mutant (7) and β-catenin-mutant intestines (8) represented a typical pathological feature of intestinal tumorigenesis. As Cdc42 regulates actin and microtubule organization, cell migration, and morphogenesis through an array of well-documented effectors (45), we asked whether Cdc42 might be directly involved in promoting such microadenoma morphology. Indeed, reduction of Cdc42 levels did significantly reduce crypt-like foci in the villi (Fig. 3D, F). Of note, tumor cells within these foci strongly expressed Paneth cell marker – Lysozyme (Fig. 4A), as well as Wnt-specific transporter (Gpr177) and Wnt ligand (Wnt3) (Fig. 4A). Double staining demonstrated that tumor cells within these microadenoma foci were proliferative (see arrows pointing to BrdU+ nuclei, Fig. 4A, B). Upon genetic Cdc42 reduction, the number of these proliferative microadenoma was reduced (Fig. 4B, Fig. S4), accompanied by a significant increase of cell death within the same microadenoma foci (Fig. 4D). Western blots for cleaved Caspase-3 further confirmed this increased apoptotic activity triggered by Cdc42 inhibition (Fig. 4C). Therefore, compared to cells outside of the foci, the fast-cycling tumor cells within these microadenoma foci appeared to be more sensitive or vulnerable to Cdc42 inhibition. Loss of these microadenoma cells was further supported by drastic reductions of Tcf4, CD44 and Sox9 levels following Cdc42 reduction (Fig. 4E).
Figure 4. Fast-cycling microadenoma cells are sensitive to Cdc42 inhibition.

(A) Cells within the villus microadnoma foci in Catnblox(ex3)/+;Vil-Cre mouse intestines expressed Lysozyme, Gpr177, and Wnt3. Co-staining of Gpr177 and BrdU showed proliferative tumor cells (arrow) within the microadenoma foci.
(B) Cdc42 inhibition reduced microadnoma foci (arrows) identified by co-staining Gpr177 (pink) and BrdU (brown nuclei). Note that residual tumor cells were still detected at villi (arrowheads).
(C) Western blots for cleaved Caspase-3 showed increased apoptosis upon Cdc42 reduction.
(D) TUNEL analyses showed increased apoptotic cells (arrows) after Cdc42 reduction. Note that cell deaths were detected specifically in the ACF lesions (dotted boxes). * indicated p<0.05 comparing to wild type (Wt), and # indicated p<0.05 comparing to β-catenin mutant intestines.
(E) Loss of microadenoma cells after Cdc42 inhibition was supported by reduced levels of Tcf4, CD44 and Sox9. Note that the full length β-catenin proteins produced from the remaining wild type allele in Catnblox(ex3)/+;Vil-Cre tumor cells might be hyper-phosphorylated by GSK3β at a level higher than in control cells. Scale bars, 2 μm.
Incipient intestinal tumor cells activated Cdc42 after acquiring mutation
To investigate whether Cdc42 activation is an early molecular event driving tumor progression from incipient intestinal tumor cells, we inducibly introduced dominant active β-catenin mutations into Catnb(ex3)fl/fl;Villin-CreER intestinal organoids in culture via tamoxifen administration and examined Cdc42 activation statuses. Induced Catnb(ex3)fl/fl;Villin-CreER organoids developed cyst-like morphology, markedly different from the wild type organoids (Fig. 5A). Real time RT-PCR detected significantly increased levels of Arhgef4, along with expected Wnt target genes CD44 and Sox9 (Fig. 5C), suggesting that β-catenin mutation activated Arhgef4 at the transcriptional level. Despite a lack of transcriptional induction of Cdc42 expression (Fig. 5C), the total and activated Cdc42 protein levels were increased in Catnb(ex3)fl/fl;Villin-CreER tumor organoids (Fig. 5D). Of note, the weak but clearly detectable Cdc42-GTP induction reflected a quantitative Cdc42 activation from 200 organoids; similar induction was not observed in Catnb(ex3)fl/+;Villin-CreER organoids, suggesting that the Cdc42-GTP levels of the latter were below detection limit. Acute administration of C20H22N2O, a Cdc42 Activity Specific Inhibitor (CASIN) (19, 46, 47) abrogated the growth of both wild type and Catnb(ex3)fl/fl;Villin-CreER tumor organoids (Fig. 5A), by inducing cell death revealed by propidium iodide staining (Fig. 5B) suggesting that these tumor organoids could not tolerate the total inhibition of Cdc42 activity.
Figure 5. Cdc42 is activated following mutation acquisition.

(A) β-catenin stable mutation was induced by tamoxifen (2.7 μM) treatment of cultured Ex3fl/fl;Vil-CreER organoids. Note that mutant organoids, but not the wild type ones, changed morphology indicative of β-catenin activation. Induced Ex3fl/fl;Vil-CreER organoids failed to tolerate Cdc42 inhibitor CASIN.
(B) DMSO- or CASIN-treated Ex3fl/fl;Vil-CreER organoids were stained by propidium iodide to identify dead cells (red). Limited or no staining was found in over 95% DMSO-treated organoids (left panels). CASIN-treated organoids were almost all strongly stained positive (right panels).
(C) Real-time RT-PCR showed increased Arhgef4 levels in β-catenin-activated organoids compared to tamoxifen-treated wild type organoids. Cdc42 expression was not changed.
(D) Western blots showed that total and active Cdc42 levels were increased in organoids with induced β-catenin mutation. Data represent Cdc42 activities from seeded 200 organoids of each genotype.
Cdc42 inhibition reduced the tumorigenicity of Cdc42High human CRC cells
Intestinal tumor stem cells (TSCs) might require higher Cdc42 activities to sustain their divisions. To determine whether the active Cdc42 level could serve as a useful indicator of effective tumor suppression by inhibiting this molecule, we examined a panel of well-characterized human CRC cell lines carrying distinct APC or/and β-catenin mutations (28). These cells were chosen due to their molecular, morphological, and behavioral representations of clinical CRC features. Microsatellite stable (MSS) LIM1863 cells contain a truncated APC at amino acid 1,564 and wild type β-catenin (28); LIM2550 cells, microsatellite instability (MSI) CRC, are hypermutated at APC, K-RAS and PI3K loci; and LIM1215 cells (also MSI) are metastatic and carrying a deletion in β-catenin exon 3 (28). Western blots showed that LIM1863, Caco2, LIM1899, and LIM2551 cells contained higher Cdc42 levels than other cell types (Fig. 6A). Given that the fast-cycling Lgr5 stem cells expressed higher Arhgef4 than villus cells (Fig. 1C), we speculated that cell cycle status might influence Cdc42 activities. We performed cell cycle analysis using flow cytometry on individual above CRC cell lines to examine whether a particular phase within a cell cycle would contribute to the observed differential Cdc42 activities. When compared cell line to cell line, the relative Cdc42 levels were not precisely correlated with their percentage of distribution at any certain cell cycle phase (Fig. S5A). Interestingly, LIM1863 showing the highest Cdc42 activity grew in liquid medium as floating organoids (Fig. 6B), previously described as crypt-like structures (48). Caco2 (19, 27), and LIM1899 cells were capable of forming 3-dimensional epithelial cyst-like tumor organoids in Matrigel, whereas other cell lines tested under same conditions showed irregular or invasive growth behavior in 3-dimension (data not shown).
Figure 6. Human CRCs with higher Cdc42 activities were sensitive to Cdc42 inhibition.

(A) Western blots showed different total and active Cdc42 levels in a panel of human CRC cells.
(B) LIM1863 cells grew as suspending organoids in liquid medium.
(C) LIM1863 treated by 10 μM CASIN for 16 hrs showed reduced tumorigenicity in xenograft model. The left and right body flanks of the same nude mice were injected with equivalent numbers of CASIN-treated (T) versus control (C) cells. Mice were imaged at day 70 after cell implantation. Tumors were dissected and photographed from corresponding mice.
(D) Tumor volumes from DMSO and CASIN-treated LIM1863-CA, LIM2550-CA and LIM1215-CA cells were graphed for individual tumors. * indicates p<0.05. Note that LIM2550 and LIM1215 tolerated the transient CASIN treatment.
(E–F) CASIN caused an immediate growth arrest in some CRCs (E) but a delayed inhibition in others (F). Cell lines with relatively high Cdc42 activities were shown in red, otherwise in green.
(G–H) Stable knockdown of Cdc42 by lentiviral shRNA particles caused growth inhibition in vitro.
(I) LIM2550 and LIM1899 cells with stable Cdc42 knockdown showed significantly reduced tumorigenicity in xenograft assays, compared to control knockdown cells (p<0.05 for both tumor volume and tumor weight). Tumors were harvested at day 30 after injection for analyses.
Live cell imaging of tumor organoids showed that CASIN treatment abolished the proliferative activities normally observed at the peripheral of LIM1863 tumor organoids, causing a drastic accumulation of membrane protrusions at tissue surfaces (Fig. S5B). The growth of all LIM1863 organoids was arrested within 48 hours by CASIN in culture (Fig. S5B). Significantly reduced tumor formation was detected in nude mice injected with LIM1863 treated with 10 μM CASIN (~16 hr) (Fig. 6C–E). In contrast, LIM1215 and LIM2550 cells (with low Cdc42) exposed to 16 hr CASIN treatment formed tumors after subcutaneous injection (Fig. 6D). As LIM1899 and LIM2550 cells tolerated the short period of CASIN treatment (no significant growth arrest at 24 hr, Fig. 6F), but not a sustained CASIN treatment especially at their exponential growth phases (p<0.01 at 48 hr, Fig. 6F), we speculated that these Cdc42-low tumor cells might be sensitive to a sustained Cdc42 deprivation. Thus, we performed stable Cdc42 knockdown (KD) in LIM1899, LIM2550, Caco2, and LIM1215 using Cdc42-specific lentiviral shRNA particles, and successfully established LIM1899, LIM2550, and Caco2 cell clones with stable Cdc42-KD (Fig. 6G); no stable clone was established for LIM1215 under same condition. Significant inhibition of tumor cell growth and tumorigenicity were observed in these Cdc42-KD CRC cells (Fig. 6H and I). These data suggested that Cdc42 inhibition was effective to a variety of CRCs, in particular to those with high Cdc42 activities.
DISCUSSION
Over 90% of CRCs have APC and/or β-catenin mutations (4), and the majority overexpresses Cdc42 (17). The proposed molecular regulation of Cdc42 by full-length (12) versus truncated APC (10) were controversial (9, 12), thus the role of this regulatory cascade during CRC pathogenesis was not clear. Both Cdc42 and Arhgef4 have been proposed to be tumor suppressors (12). Through genetic and pharmacological analyses of mouse and human CRC models in vitro and in vivo, we have obtained several lines of evidence to support that some of CRC cells activate Cdc42 for immediate cell survival and microadenoma morphogenesis.
Aberrant morphogenesis of crypt-like foci represents one of the hallmarks of CRC pathogenesis (49). The molecular bases of such morphogenetic transformations, especially the very initial erratic movement of incipient tumor cells (7), have not been fully elucidated. We provided in vivo evidences that Cdc42 was strongly elevated and activated in both APCMin/+ and β-catenin mutant mouse intestines. Of note, Cdc42 was immediately activated in IEC organoids following the inducible introduction of β-catenin mutations. In this APCwild-type genetic background, Cdc42 activation was likely activated by an increased amount of Arhgef4, whose transcriptional levels was significantly elevated upon β-catenin mutation acquisition. This transcriptional activation of Arhgef4 was consistent with recently reported enrichment of this factor in Lgr5+ stem cell population, and was further affirmed by our detection of a stem cell-specific enhancer upstream of Arhgef4.
What advantages do tumor cells gain from Cdc42 activation? First, our results suggested that higher levels of Cdc42 activities might promote survivability of fast-cycling microadnoma-constructing tumor cells (Fig. 7). Intestinal crypts and stem cells haploid insufficient of Cdc42 (Cdc42fl/+;Vil-Cre) behave normally, whereas intestinal tumor cells with β-catenin mutation underwent increased apoptosis following haploid insufficiency in Cdc42, suggesting that TSCs might demand higher Cdc42 for survival comparing to wild type stem cells. This notion was also supported by the facts that Cdc42 deletion attenuated APCMin/+ tumorigenesis and human CRCs with higher Cdc42 activities were less tolerant to Cdc42 inhibitor. Prosurvival factors downstream of Cdc42, such as PAK1 (50), have been well documented and our Cdc42 activation assays demonstrated consistently elevated Cdc42-PAK1 binding in both mouse and human CRCs throughout the entire study. Cdc42 activation may also indirectly influence GSK3β activity through its direct target atypical PKC (20), ultimately promoting β-catenin-mediated cell survival and proliferation. A micro RNA, miR-185, that reduced Cdc42 expression also effectively induced cell cycle arrest and apoptosis in cultured CRCs (51). Thus, Cdc42 activation may cell-autonomously favor fast-cycling TSCs to survive and progress.
Figure 7. Intestinal tumor cells activate Cdc42 for survival and progression.
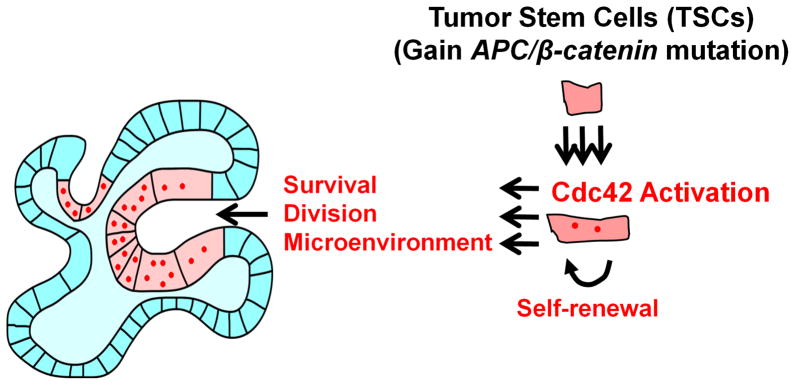
A model depicting that intestinal tumor cells, once acquiring APC or β-catenin mutation, activate Cdc42 to facilitate tumor cell survivability and microadenoma formation.
Second, we provided evidence that genetic inhibition of Cdc42 reduced the number of crypt-like foci and tumors in several mouse CRC models, whereas a Cdc42 inhibitor abrogated both mouse and human tumor organoids. Cdc42 activation in incipient tumor cells may immediately alter cell morphology through its well-established effectors such as IQGAP (52), reorganizing actin and microtubule cytoskeletons. This would directly favor tumor cell migration and aberrant tumor cell behaviors such as chromosomal mis-segregation and aneuploidy, all of which could drive CRC progression.
Third, we showed that Cdc42 deletion also affected cells (e.g., Paneth cells) within the normal or tumor stem cell compartments. Total loss of Cdc42 perturbed the development of Paneth cells, (19), the primary Wnt3 secretors in intestinal crypts. Loss of Wnt3 arrested the growth of intestinal organoids in vitro (53), a phenotype similar to Cdc42ΔIEC crypts. However, unlike Wnt3-deficient crypts, Cdc42ΔIEC crypts failed to be rescued by Wnt3a protein or co-culture of wild type crypts, indicating that Cdc42 was cell-autonomously required by stem cells for survival. The impact of Cdc42 on Paneth-like tumor cells was most likely indirect. On the other hand, reduction of Cdc42 clearly abrogated the formation of microadnoma foci that contained tumor cells with Wnt-producing capacity, suggesting that Cdc42 activation may also indirectly favor TSC growth through a non-cell-autonomous mechanism by assembling a tumorigenic microenvironment (Fig. 7).
A large fraction of APC mutations in CRCs, including the Min mutation, result in premature stop codon and truncated proteins that retain the conserved N-terminus armadillo domain responsible for Arhgef4-, Cdc42- and IQGAP-binding (54). Structural and cell culture analyses led to conflicting interpretations of APC-mediated Cdc42 activation (9–12, 15, 16). Our data suggested that Cdc42 might be activated in CRCs from multiple levels, including mechanisms depending on Arhgef4 and ones influencing the level of Cdc42 itself. Together with the observation that deletion of Arhgef4 reduced APCMin/+ mouse tumorigenesis (55), our data supported the tumor-promoting effect of this Arhgef4-Cdc42 cascade.
Cdc42 inhibition suppressed the in vivo tumorigenicity of several CRC cell lines, especially those with higher Cdc42 levels such as LIM1863. The growth of LIM1863 cells as floating organoids and Caco2 cells as epithelial cysts probably reflected some benign features of these cell types. In contrast, cells with low Cdc42 activities, for example, the hypermutated LIM2550 cells and metastatic LIM1215 cells were relative resistant to Cdc42 inhibition, and might represent advanced CRC types. Our xenograft assays suggested that the growth of even these cell lines was inhibited by stable Cdc42 depletion. The relatively high tolerance by these cells could attribute to their widespread genome anomalies. K-RAS activating mutation in LIM2550 cells may elicit a parallel survival pathway. Our mouse CRC studies suggested that Cdc42 activation might be an early molecular event, blockage of which could attenuate CRC progression. Determination of Cdc42 level of a given tumor cell type may identify its potential sensitivity to Cdc42 inhibition. Cdc42-inactivating mutations are rare in CRCs (4), hinting its indispensible role in initial tumor cell survival. Cdc42 might be considered as a useful marker and a potential target for selective intervention of CRCs.
Supplementary Material
Acknowledgments
This work is supported by NIH grants DK085194, CA178599, DK093809, DK102934 to N.G.; NIH grants DK088868, DK099251 to M.P.V.; Charles and Johanna Busch Memorial Award (659160) to N.G.; Rutgers University Faculty Research Grant (281708) to M.P.V. and N.G.; NIH (CA120915S2) to C.S.Y.; NIH (CA106308 and DE015654) to W.H.; New Jersey Commission on Cancer Research (NJCCR) Postdoctoral Fellowship (DFHS13PPC016) to S.Y; NJCCR Doctoral Fellowship (DFHS13PPC057) to A.H.
Footnotes
Authors have no conflict of interest to disclose.
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–49. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 2.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–67. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 3.Fearon ER. Molecular genetics of colorectal cancer. Annu Rev Pathol. 2011;6:479–507. doi: 10.1146/annurev-pathol-011110-130235. [DOI] [PubMed] [Google Scholar]
- 4.Network CGA. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–7. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87:159–70. doi: 10.1016/s0092-8674(00)81333-1. [DOI] [PubMed] [Google Scholar]
- 6.Polakis P. The many ways of Wnt in cancer. Curr Opin Genet Dev. 2007;17:45–51. doi: 10.1016/j.gde.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 7.Oshima H, Oshima M, Kobayashi M, Tsutsumi M, Taketo MM. Morphological and molecular processes of polyp formation in Apc(delta716) knockout mice. Cancer Res. 1997;57:1644–9. [PubMed] [Google Scholar]
- 8.Harada N, Tamai Y, Ishikawa T, et al. Intestinal polyposis in mice with a dominant stable mutation of the beta-catenin gene. Embo J. 1999;18:5931–42. doi: 10.1093/emboj/18.21.5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kawasaki Y, Senda T, Ishidate T, et al. Asef, a link between the tumor suppressor APC and G-protein signaling. Science. 2000;289:1194–7. doi: 10.1126/science.289.5482.1194. [DOI] [PubMed] [Google Scholar]
- 10.Kawasaki Y, Sato R, Akiyama T. Mutated APC and Asef are involved in the migration of colorectal tumour cells. Nat Cell Biol. 2003;5:211–5. doi: 10.1038/ncb937. [DOI] [PubMed] [Google Scholar]
- 11.Hamann MJ, Lubking CM, Luchini DN, Billadeau DD. Asef2 functions as a Cdc42 exchange factor and is stimulated by the release of an autoinhibitory module from a concealed C-terminal activation element. Mol Cell Biol. 2007;27:1380–93. doi: 10.1128/MCB.01608-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mitin N, Betts L, Yohe ME, Der CJ, Sondek J, Rossman KL. Release of autoinhibition of ASEF by APC leads to CDC42 activation and tumor suppression. Nat Struct Mol Biol. 2007;14:814–23. doi: 10.1038/nsmb1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sudhaharan T, Goh WI, Sem KP, Lim KB, Bu W, Ahmed S. Rho GTPase Cdc42 is a direct interacting partner of Adenomatous Polyposis Coli protein and can alter its cellular localization. PLoS ONE. 2011;6:e16603. doi: 10.1371/journal.pone.0016603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bienz M, Clevers H. Linking colorectal cancer to Wnt signaling. Cell. 2000;103:311–20. doi: 10.1016/s0092-8674(00)00122-7. [DOI] [PubMed] [Google Scholar]
- 15.Zhang Z, Chen L, Gao L, et al. Structural basis for the recognition of Asef by adenomatous polyposis coli. Cell Res. 2011;22:372–86. doi: 10.1038/cr.2011.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murayama K, Shirouzu M, Kawasaki Y, et al. Crystal structure of the rac activator, Asef, reveals its autoinhibitory mechanism. J Biol Chem. 2007;282:4238–42. doi: 10.1074/jbc.C600234200. [DOI] [PubMed] [Google Scholar]
- 17.Gomez Del Pulgar T, Valdes-Mora F, Bandres E, et al. Cdc42 is highly expressed in colorectal adenocarcinoma and downregulates ID4 through an epigenetic mechanism. Int J Oncol. 2008;33:185–93. [PubMed] [Google Scholar]
- 18.Munoz J, Stange DE, Schepers AG, et al. The Lgr5 intestinal stem cell signature: robust expression of proposed quiescent ‘+4’ cell markers. Embo J. 2012;31:3079–91. doi: 10.1038/emboj.2012.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sakamori R, Das S, Yu S, et al. Cdc42 and Rab8a are critical for intestinal stem cell division, survival, and differentiation in mice. J Clin Invest. 2012;122:1052–65. doi: 10.1172/JCI60282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu X, Quondamatteo F, Lefever T, et al. Cdc42 controls progenitor cell differentiation and beta-catenin turnover in skin. Genes Dev. 2006;20:571–85. doi: 10.1101/gad.361406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barker N, van Es JH, Kuipers J, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449:1003–7. doi: 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- 22.Srinivas S, Watanabe T, Lin CS, et al. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol. 2001;1:4. doi: 10.1186/1471-213X-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Madison BB, Dunbar L, Qiao XT, Braunstein K, Braunstein E, Gumucio DL. Cis elements of the villin gene control expression in restricted domains of the vertical (crypt) and horizontal (duodenum, cecum) axes of the intestine. J Biol Chem. 2002;277:33275–83. doi: 10.1074/jbc.M204935200. [DOI] [PubMed] [Google Scholar]
- 24.el Marjou F, Janssen KP, Chang BH, et al. Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis. 2004;39:186–93. doi: 10.1002/gene.20042. [DOI] [PubMed] [Google Scholar]
- 25.Moser AR, Pitot HC, Dove WF. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science. 1990;247:322–4. doi: 10.1126/science.2296722. [DOI] [PubMed] [Google Scholar]
- 26.Yu DH, Ware C, Waterland RA, et al. Developmentally programmed 3′ CpG island methylation confers tissue- and cell-type-specific transcriptional activation. Mol Cell Biol. 2013;33:1845–58. doi: 10.1128/MCB.01124-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gao N, Kaestner KH. Cdx2 regulates endo-lysosomal function and epithelial cell polarity. Genes Dev. 2010;24:1295–305. doi: 10.1101/gad.1921510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang HH, Walker F, Kiflemariam S, et al. Selective inhibition of proliferation in colorectal carcinoma cell lines expressing mutant APC or activated B-Raf. Int J Cancer. 2009;125:297–307. doi: 10.1002/ijc.24289. [DOI] [PubMed] [Google Scholar]
- 29.Sato T, Vries RG, Snippert HJ, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459:262–5. doi: 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- 30.Sato T, van Es JH, Snippert HJ, et al. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature. 2011;469:415–8. doi: 10.1038/nature09637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gao N, White P, Kaestner KH. Establishment of intestinal identity and epithelial-mesenchymal signaling by Cdx2. Dev Cell. 2009;16:588–99. doi: 10.1016/j.devcel.2009.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gao N, Davuluri G, Gong W, et al. The nuclear pore complex protein Elys is required for genome stability in mouse intestinal epithelial progenitor cells. Gastroenterology. 2011;140:1547–55. e10. doi: 10.1053/j.gastro.2011.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu HM, Jin Y, Fu J, Hsu W. Expression of Gpr177, a Wnt trafficking regulator, in mouse embryogenesis. Dev Dyn. 2010;239:2102–9. doi: 10.1002/dvdy.22336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rhodes DR, Kalyana-Sundaram S, Mahavisno V, et al. Oncomine 3. 0: genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia. 2007;9:166–80. doi: 10.1593/neo.07112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sabates-Bellver J, Van der Flier LG, de Palo M, et al. Transcriptome profile of human colorectal adenomas. Mol Cancer Res. 2007;5:1263–75. doi: 10.1158/1541-7786.MCR-07-0267. [DOI] [PubMed] [Google Scholar]
- 36.Skrzypczak M, Goryca K, Rubel T, et al. Modeling oncogenic signaling in colon tumors by multidirectional analyses of microarray data directed for maximization of analytical reliability. PLoS One. 2010;5 doi: 10.1371/journal.pone.0013091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–7. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gaspar C, Cardoso J, Franken P, et al. Cross-species comparison of human and mouse intestinal polyps reveals conserved mechanisms in adenomatous polyposis coli (APC)-driven tumorigenesis. Am J Pathol. 2008;172:1363–80. doi: 10.2353/ajpath.2008.070851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hong Y, Downey T, Eu KW, Koh PK, Cheah PY. A ‘metastasis-prone’ signature for early-stage mismatch-repair proficient sporadic colorectal cancer patients and its implications for possible therapeutics. Clinical & experimental metastasis. 2010;27:83–90. doi: 10.1007/s10585-010-9305-4. [DOI] [PubMed] [Google Scholar]
- 40.Gaedcke J, Grade M, Jung K, et al. Mutated KRAS results in overexpression of DUSP4, a MAP-kinase phosphatase, and SMYD3, a histone methyltransferase, in rectal carcinomas. Genes Chromosomes Cancer. 2010;49:1024–34. doi: 10.1002/gcc.20811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kaiser S, Park YK, Franklin JL, et al. Transcriptional recapitulation and subversion of embryonic colon development by mouse colon tumor models and human colon cancer. Genome Biol. 2007;8:R131. doi: 10.1186/gb-2007-8-7-r131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Melendez J, Liu M, Sampson L, et al. Cdc42 coordinates proliferation, polarity, migration, and differentiation of small intestinal epithelial cells in mice. Gastroenterology. 2013;145:808–19. doi: 10.1053/j.gastro.2013.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roberts RB, Min L, Washington MK, et al. Importance of epidermal growth factor receptor signaling in establishment of adenomas and maintenance of carcinomas during intestinal tumorigenesis. Proc Natl Acad Sci U S A. 2002;99:1521–6. doi: 10.1073/pnas.032678499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sansom OJ, Meniel VS, Muncan V, et al. Myc deletion rescues Apc deficiency in the small intestine. Nature. 2007;446:676–9. doi: 10.1038/nature05674. [DOI] [PubMed] [Google Scholar]
- 45.Park HO, Bi E. Central roles of small GTPases in the development of cell polarity in yeast and beyond. Microbiol Mol Biol Rev. 2007;71:48–96. doi: 10.1128/MMBR.00028-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Florian MC, Dorr K, Niebel A, et al. Cdc42 activity regulates hematopoietic stem cell aging and rejuvenation. Cell Stem Cell. 2012;10:520–30. doi: 10.1016/j.stem.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peterson JR, Lebensohn AM, Pelish HE, Kirschner MW. Biochemical suppression of small-molecule inhibitors: a strategy to identify inhibitor targets and signaling pathway components. Chem Biol. 2006;13:443–52. doi: 10.1016/j.chembiol.2006.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Whitehead RH, Jones JK, Gabriel A, Lukies RE. A new colon carcinoma cell line (LIM1863) that grows as organoids with spontaneous differentiation into crypt-like structures in vitro. Cancer Res. 1987;47:2683–9. [PubMed] [Google Scholar]
- 49.Boivin GP, Washington K, Yang K, et al. Pathology of mouse models of intestinal cancer: consensus report and recommendations. Gastroenterology. 2003;124:762–77. doi: 10.1053/gast.2003.50094. [DOI] [PubMed] [Google Scholar]
- 50.Zhu G, Wang Y, Huang B, et al. A Rac1/PAK1 cascade controls beta-catenin activation in colon cancer cells. Oncogene. 2012;31:1001–12. doi: 10.1038/onc.2011.294. [DOI] [PubMed] [Google Scholar]
- 51.Liu M, Lang N, Chen X, et al. miR-185 targets RhoA and Cdc42 expression and inhibits the proliferation potential of human colorectal cells. Cancer Lett. 2011;301:151–60. doi: 10.1016/j.canlet.2010.11.009. [DOI] [PubMed] [Google Scholar]
- 52.Watanabe T, Wang S, Noritake J, et al. Interaction with IQGAP1 links APC to Rac1, Cdc42, and actin filaments during cell polarization and migration. Dev Cell. 2004;7:871–83. doi: 10.1016/j.devcel.2004.10.017. [DOI] [PubMed] [Google Scholar]
- 53.Farin HF, Van Es JH, Clevers H. Redundant Sources of Wnt Regulate Intestinal Stem Cells and Promote Formation of Paneth Cells. Gastroenterology. 2012;143:1518–29. doi: 10.1053/j.gastro.2012.08.031. [DOI] [PubMed] [Google Scholar]
- 54.McCartney BM, Nathke IS. Cell regulation by the Apc protein Apc as master regulator of epithelia. Curr Opin Cell Biol. 2008;20:186–93. doi: 10.1016/j.ceb.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 55.Kawasaki Y, Tsuji S, Muroya K, et al. The adenomatous polyposis coli-associated exchange factors Asef and Asef2 are required for adenoma formation in Apc(Min/+)mice. EMBO Rep. 2009;10:1355–62. doi: 10.1038/embor.2009.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.