

SUMMARY
Cancers arise through a process of somatic evolution that can result in substantial sub-clonal heterogeneity within tumors. The mechanisms responsible for the coexistence of distinct sub-clones and the biological consequences of this coexistence remain poorly understood. Here we used a mouse xenograft model to investigate the impact of sub-clonal heterogeneity on tumor phenotypes and the competitive expansion of individual clones. We found that tumor growth can be driven by a minor cell subpopulation, which enhances the proliferation of all cells within a tumor by overcoming environmental constraints and yet can be outcompeted by faster proliferating competitors, resulting in tumor collapse. We then developed a mathematical modeling framework to identify the rules underlying the generation of intratumor clonal heterogeneity. We found that non-cell autonomous driving, together with clonal interference, stabilizes sub-clonal heterogeneity, thereby enabling inter-clonal interactions that can lead to new phenotypic traits.
INTRODUCTION
Cancers result from genetic and epigenetic changes that fuel Darwinian somatic evolution1,2. Until recently, the evolution was assumed to proceed as a linear succession of clonal expansions triggered by acquisition of strong driver mutations that progressively increase cell fitness and lead to selective sweeps3. However, the recent explosion of data from tumor genome sequencing studies and single-cell based analyses has revealed substantial genetic heterogeneity within tumors, including sub-clonal differences in driver mutations4–8. This contradicts the linear succession model and challenges the assumption of tumor evolution being driven by mutations providing strong clone-specific selective advantages. Furthermore, clonal heterogeneity raises the possibility of biologically and clinically important interactions between distinct clones9,10.
Many oncogenic mutations confer a cell-autonomous fitness advantage by either providing independence from growth factors or abolishing apoptotic response. These mutations are thus expected to drive clonal expansions11. At the same time, tumor progression is frequently limited by microenvironmental constraints12–14 that cannot be overcome by a cell-autonomous increase in proliferation rates. Instead, progression depends on alterations of the microenvironment, mediated by non-cell-autonomously acting factors, such as metalloproteinases and cytokines. It is unclear whether these secreted factors preferentially benefit the “producer” clone (s) enabling their clonal dominance.
A model of clonal heterogeneity
Understanding clonal heterogeneity has been hindered by the lack of suitable experimental models. While patient tumor-derived xenograft studies using clonal tracing can be insightful, their utility is limited by the challenges in deciphering mechanisms that underlie biological differences between sub-clones. We aimed to bypass these challenges by experimentally defining sub-populations via overexpression of factors previously implicated in tumor progression. We decided to exploit a scenario of a tumor that is “stuck” in a microenvironmentally-constrained progression bottleneck, which is relevant for occult cancers, dormant micro-metastatic lesions, and perhaps early, clinically undetectable stages of tumor development. This scenario offers two key advantages. First, in contrast to a rapidly growing tumor, the constrained population size of non-growing tumors composed of rapidly cycling cells is expected to intensify competition for limited microenvironmental resources. This enhances the detection of differences in competitive fitness. Second, the indolent morphology and lack of net tumor growth should facilitate the detection of increase in tumor growth and metastasis.
In search of tumors satisfying these criteria, we analyzed a panel of breast cancer-derived cell lines for tumors formed by orthotopic transplantation into the mammary fat pads of immunodeficient Foxn1nu (nu) mice. Whereas most of the tested cell lines either failed to produce tumors or formed tumors that grew too rapidly (e.g., SUM149PT cells), MDA-MB-468 cell line formed indolent tumors which, upon reaching 2–5 mm in diameters, showed very slow growth rates (Fig. 1a and data not shown). Despite slow net growth, the tumor cells were actively proliferating: 80–90% of them were in cell cycle based on Ki-67 staining, and 20–30% were in S-phase based on BrdU incorporation (Fig. 1b). The slow net tumor growth indicated that cell proliferation was counterbalanced by cell death. Indeed, 1–3% of the cells were apoptotic. Tumors contained large necrotic areas implying significant necrotic cell death (Fig. 1b).
Figure 1. Experimental system.
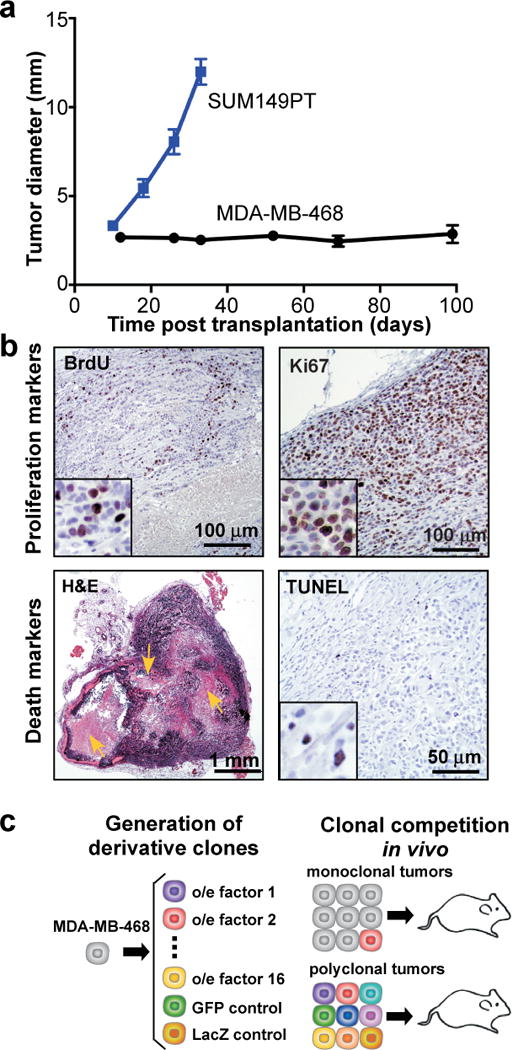
a, Growth of tumors upon fat pad transplantation of indicated cell lines, n=10/group, error bars indicate SEM. b, Representative images of indicated staining. Arrows indicate necrotic areas. c, Experimental scheme.
We used MDA-MB-468 cells to generate a panel of sub-lines (henceforth called “sub-clones”) defined by the lentiviral overexpression of a single secreted factor. Each factor had been previously implicated in tumor progression, along with reported high expression in patient’s breast carcinoma cells (Fig. 1c and Extended Data Table 1). Given the recently reported variability in clonal proliferation dynamics15, and to minimize the confounding influences of genetic/epigenetic heterogeneity within the cell line, we used pools of transduced cells rather than single cell-derived clones. This panel enabled us to compare phenotypic properties of tumors and clonal expansions under two circumstances: (i) each sub-clone competing against parental cells (monoclonal tumors), and (ii) sub-clones competing against all other sub-clones (polyclonal tumors) (Fig. 1c). We had 18 sub-clones in total. In order to maintain equal initial clonal proportions in all tumors, we employed the ratio of 1:18 between a sub-clone and parental competitors.
Non-cell autonomous tumor driving
We first investigated whether individual sub-clones, initially present as a minor sub-population competing against parental cells, could impact tumor properties. We focused on tumor growth and metastasis, features that are most relevant clinically and amenable to quantification. Whereas we observed variability between the groups in morphology, proliferation and vascularization (Extended data Fig. 1), only the chemokine (C-C motif) ligand 5 (CCL5) and interleukin 11 (IL11) overexpressing sub-clones were able to enhance tumor growth (Fig. 2a,b). None of the tumors were metastatic, as evaluated by in vivo bioluminescence imaging and examination of draining lymph nodes, peritoneal walls and bone marrow (data not shown).
Figure 2. Polyclonality affects tumor phenotypes.
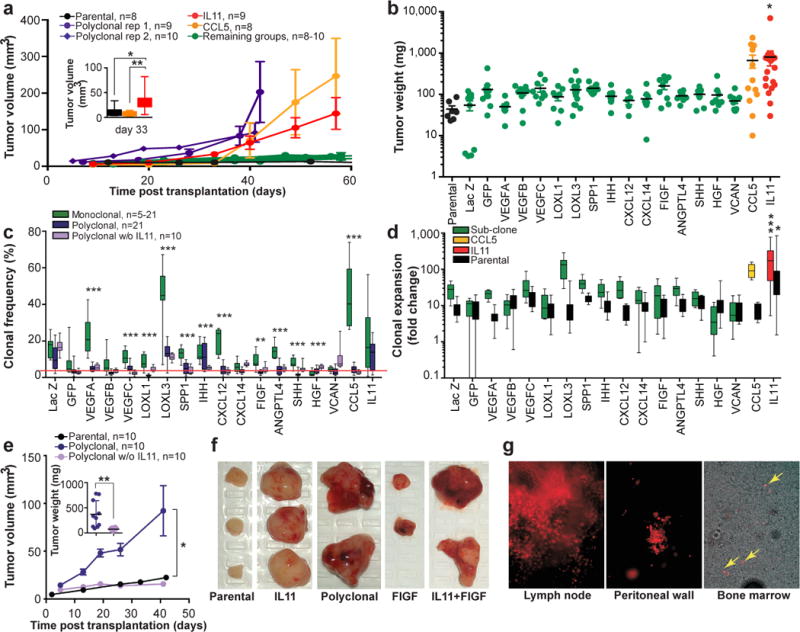
a, Tumor growth kinetics. b, Tumor weights. c, Sub-clones frequencies within tumors. Red line indicates initial frequency. d, Expansion (fold change over initial cell number) of sub-clones and parental cells from monoclonal tumors shown in c. e, Tumor growth kinetics and weights (inset). f, Representative images of tumors. g, Live fluorescent microscopy images of tumor cells (mCherry+) in tissues.*, ** and *** indicate p<0.05, p<0.01 and p<0.001, respectively of Student’s t test (a, c, e) or ANOVA multiple group comparison against parental (b) or LacZ (d) with Dunnet’s connection. Error bars indicate SEM.
We then analyzed the population frequencies of individual sub-clones within the tumors using a genomic quantitative polymerase chain reaction (qPCR) approach, utilizing clone-specific and reference amplicons (Extended data Fig. 2). Surprisingly, we observed no strict correlation between the increase in sub-clonal frequencies and the growth rate of tumors (Fig. 2a–c). The LOXL3 sub-clone underwent the strongest (~10-fold) expansion in population frequency, yet failed to promote overall tumor growth. On the other hand, both CCL5 and IL11, each capable of driving outgrowth of tumors, exhibited ~8- and ~4-fold expansion, respectively. To address the link between clone-specific expansion and tumor growth more directly, we calculated rates of expansion in cell numbers over the initially transplanted cells using volume-based cellularity inferences (Fig. 2d, Extended Data Fig. 3a). Only IL11 was capable of non-cell autonomous driving. We saw enhanced expansion of both IL11-expressing and parental cells. Increased growth of CCL5-driven tumors was only attributable to cell-autonomous expansion of CCL5-expressing cells. This finding was consistent with the observed delay in tumor outgrowth driven by CCL5 compared to IL11-driven tumors (Fig. 2a, insert).
We did not observe a positive correlation between tumor weights and final percentages of IL11 expressing cells (Extended Data Fig. 4a). An increase in the initial frequency of the IL11 sub-clone did not further enhance tumor growth (Extended Data Fig. 4b). Parental cells expressed undetectable basal levels of IL11 (Extended Data Fig. 4c,d) and the non-cell autonomous driving of tumor growth was observed with four independent derivations of the IL11 overexpressing sub-clones using two distinct lentiviral backbones that provide different levels of expression (Extended Data Fig.4c–e). This strongly suggests that the phenomenon was IL11-specific and did not require additional stochastic events.
We then initiated tumors in which all the sub-clones, present at the initial 1:18 frequency, were set to compete against one another. These tumors grew faster than monoclonal tumors, suggesting additive growth-promoting interactions among the sub-clones (Fig. 2a). However, omitting the IL11 sub-clone (2:18 ratio of control LacZ sub-clone was used to maintain 1:18 ratio of the remaining sub-clones) blocked the increased growth of polyclonal tumors, reducing clonal expansions (Fig.2e and Extended data Fig. 5a). Therefore, non-cell autonomous stimulation by IL11 was both necessary and sufficient to drive tumor growth.
Sub-clonal cooperation in metastasis
In addition to accelerated growth rates, polyclonal tumors displayed regions of extensive hemorrhage and multiple cysts (Fig. 2f), indicative of increased blood and lymphatic vessel leakage. Consistently, a large fraction of polyclonal tumors were metastatic: 7/12 analyzed animals displayed lymph node metastases, 6/12 displayed metastatic nodes on the peritoneal wall, and 4/7 contained tumor cells in the bone marrow (Fig. 2g). Animals bearing polyclonal tumors accumulated peritoneal fluid and demonstrated signs of systemic toxicity, requiring euthanasia at earlier time points compared to other groups.
FIGF was the only other sub-clone displaying elevated vascular leakage in monoclonal tumors, albeit with incomplete penetrance. Hence we asked whether the combination of IL11 and FIGF could recapitulate the metastatic phenotypes of polyclonal tumors. Indeed, FIGF/IL11 tumors displayed an increase in tumor volume and extensive hemorrhage (Fig.2f, Extended Data Fig. 5b), with 4/4 animals presenting both lymph node and peritoneal wall metastases. Therefore, our data suggest that biological interactions between distinct sub-populations can lead to the emergence of new tumor phenotypes.
Contexts of polyclonal tumors strongly inhibited the expansion of individual sub-clones in comparison to monoclonal tumors (Fig. 2c). This phenomenon is known as clonal interference. When multiple clones with higher than average fitness emerge in a population at the same time, they interfere with each other. This slows down the rate of clonal evolution16. The reduced expansion of individual sub-clones in IL11-driven polyclonal tumors could also be the result of a growing population. Therefore, to distinguish between the effects of clonal interference and expanding tumor volume, we determined clonal expansions in slower growing polyclonal tumors without IL11 (Fig. 2c). We found that while the removal of IL11 significantly affected clonal composition of the tumors (p<0.0001 for the interaction factor in a 2-way ANOVA), expansion of most of the sub-clones remained inhibited. This suggests that clonal interference is a major determinant of the differences in the competitive dynamics in polyclonal tumors.
Mathematical model of tumor clonality
To investigate the rules of tumor growth and to predict clonal dynamics on a longer timescale, we developed a mathematical framework incorporating clonal interference and heterogeneity. First, we investigated the growth behavior of monoclonal tumors, finding that tumors exhibited an exponential growth pattern (Extended Data Fig. 3b). We then estimated the clone-specific exponential growth rates for each monoclonal growth experiment. With these rates we predicted tumor sizes in polyclonal tumors adding a dynamic interaction term (Fig. 4a, Extended Data Fig. 3c,d
Figure 4. Impact of IL11 on clonal dynamics.
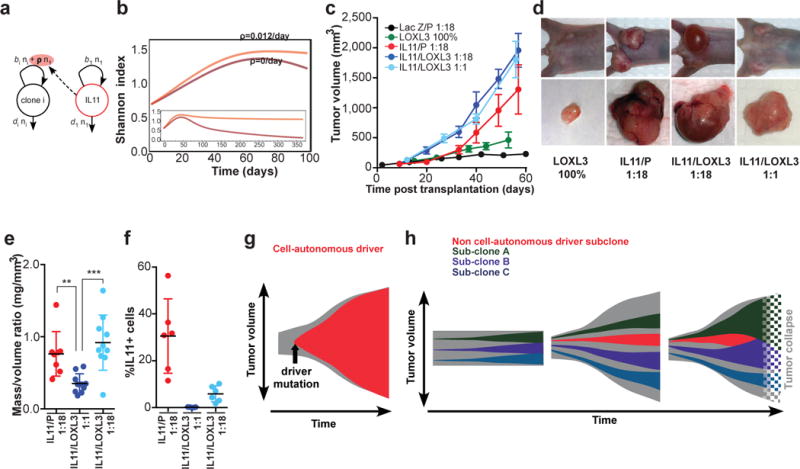
a, Outline of the linear model that best explains polyclonal dynamics (see SI): b, Prediction of diversity over time without (dark) or with (light) non-cell autonomous driver. c, Rumor growth kinetics n=10/group, d, Representative images e, Weight/volume ratios of tumors in c–e excluding cyst fluid, each dot represents individual tumor. ** and *** indicate p<0.01 and p<0.001, respectively; error bars indicate SEM, f, Final population frequencies of IL11+ cells in the indicated tumors. g, Models of cell-autonomous and, h, non-cell autonomous driving of tumor growth.
In order to account for interactions between a driver clone and other clones, we investigated a hierarchy of nested, increasingly complex mathematical descriptions of clonal dynamics for their ability to predict data from individual polyclonal growth experiments. The null hypothesis of no clonal interactions was easily rejected. The best agreement between model predictions and experimental observations in polyclonal tumors was achieved by including a constant positive growth effect of the IL11 clone on all other clones. Higher-order interactions involving multiple drivers did not improve the predictive power of the model. The best-fitting model can then be used to predict heterogeneity in polyclonal tumors over longer time scales. In the absence of IL11, clonal heterogeneity is predicted to eventually vanish, since clones with the highest proliferation rates outcompete less fit clones. In contrast, non-cell autonomous stimulation of cell growth supports clonal diversity over clinically relevant time scales (Fig. 4b).
Chemotherapy boosts clonal competition
Since anti-cancer therapy exerts selective pressures that can affect evolutionary dynamics, we investigated the impact of treatment with doxorubicin, a commonly used chemotherapeutic agent in breast cancer. Two rounds of doxorubicin administration substantially inhibited tumor growth and cell proliferation in polyclonal tumors (Extended Data Fig. 6a,b). Instead of the expected changes in the expansion of specific sub-clones differing in drug sensitivity, we found that the amplitude of clonal expansion and contractions was increased compared to untreated tumors, reducing clonal diversity (Extended Data Fig. 6c,d). Therefore, even in the absence of selection for resistant subpopulations, doxorubicin treatment nonspecifically amplified the effects of differences in competitive fitness. This was most likely a result of increased competition due to treatment-induced stabilization of the population size.
Mechanisms of IL11-driven tumor growth
Elevated tumor growth implies an increase in net cell proliferation rates, either by stimulating proliferation or by inhibiting death. IL11-driven tumors displayed a subtle, but significant, increase in proliferation rates compared to parental tumors (Fig. 3a). Apoptosis rates were similar (Extended Data Fig.1b). This increase in cellular proliferation could result either from a direct autocrine/paracrine stimulation of cell growth or from indirect effects mediated by the microenvironment. IL11 signals through a unique and specific receptor, IL11Rα, that forms a signaling complex with the GP130 co-receptor shared with other IL6 cytokine family members17. IL11 promotes growth of gastric carcinoma via direct stimulation of epithelial cells18,19. Similar stimulation of tumor growth via non-cell autonomous signaling between tumor cells, involving two related cytokines, IL6 and LIF, was reported in glioblastomas20. We therefore asked whether modulation of IL11Rα expression in carcinoma cells affects the ability of IL11 to induce tumor growth. Neither overexpression, nor shRNA-mediated down-regulation of IL11Rα affected IL11-driven tumor growth (Fig. 3b and Extended Data Fig. 7). Furthermore, IL11 significantly promoted growth of 2/4 additional breast cancer cell lines despite low or undetectable levels of IL11Rα (Fig.3c,d).
Figure 3. IL11 drives tumor cell proliferation via microenvironmental changes.
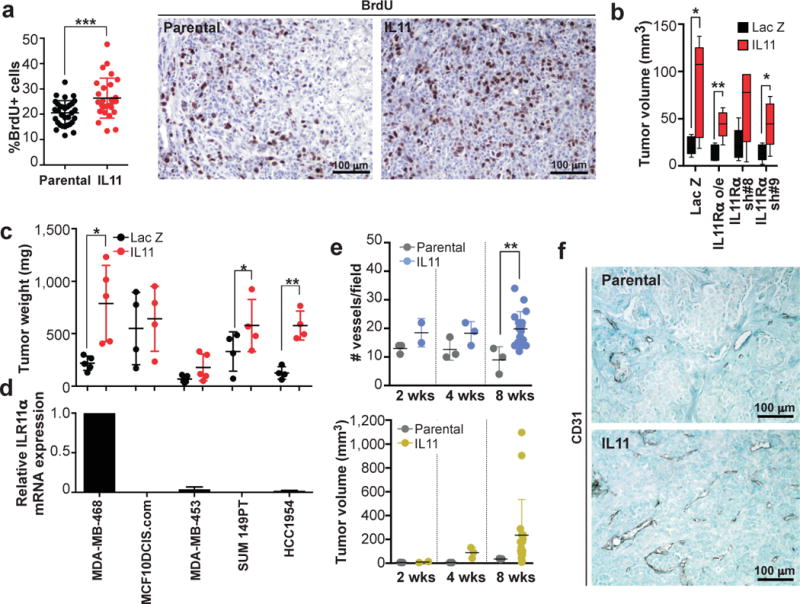
a, Quantitation and representative images of anti-BrdU immunohistochemical staining in control and IL11-driven tumors. b, Tumor volumes 31 days post transplantation of parental MDA-MB-468 cells, cells over-expressing or down-regulated IL11Rα n=5/group c, Tumor weights of contra-lateral parental and IL11 expressing tumors formed by indicated cell lines. d, Levels of expression of IL11Rα mRNA in indicated cell lines, normalized to MDA-MB-468. e, Quantitation of average number of CD31+ vessels/field and tumor volumes. f, Representative images of anti-CD31 immunohistochemical staining. *, ** and *** indicate p<0.05, p<0.01 and p<0.001, respectively of unpaired (a, b, e) or paired (c) student’s t-test. Error bars indicate SEM.
Independence of tumor growth from direct stimulation of tumor cells by IL11 prompted us to investigate changes in the tumor microenvironment. IL11-driven tumors displayed higher intratumoral vascular density compared to parental ones (Fig. 3e,f), more dispersed patterns of collagen organization and had more stromal fibroblasts (Extended Data Fig.8). Both increased vascularization and reorganization of the extracellular matrix have been implicated in the promotion of tumor growth21,22, suggesting that the tumor-promoting effects of IL11 may be attributable to microenvironmental changes.
The lack of strict correlation between clonal expansion and tumor growth prompted examination of the competition between IL11 and LOXL3 sub-clones. Latter showed the strongest expansion in population frequency without being able to drive tumor growth (Fig. 2d). IL11 accelerated the growth of tumors with LOXL3 competitors beyond the growth rates seen with IL11/P controls (Fig. 4c), consistent with the ability of faster proliferating LOXL3 cells to obtain additional benefit from IL11. However, upon harvest, 1:18 IL11/LOXL3 tumors contained very little solid tissue. Most of the volume was filled with interstitial fluid, likely a remnant of necrotic liquefaction, while 1:18 IL11/P and 1:1 IL11/LOXL3 tumors remained solid (Fig.4d,e).
Analysis of clonal composition revealed that LOXL3 had outcompeted the IL11 sub-clone below the detectability threshold in 1:18 IL11/LOXL3 tumors. In contrast, 1:1 IL11/LOXL3 tumors contained reduced, but significant proportions of IL11 cells (Fig.4f). Loss of IL11 cells most likely reflects differences in proliferation rates, rather then apoptotic elimination of slower dividing cells seen elsewhere23. We did not observe elevated rates of apoptosis in IL11 cells bordering LOXL3 in 1:1 IL11/LOXL3 tumors, and occasional IL11+ cells could still be detected in 1:18 IL11/LOXL3 tumors (Extended Data Fig 9). Additionally, the resulting clonal frequencies were consistent with predictions of our mathematical model (SI). Most likely, elimination of IL11 sub-clone restored microenvironmental barriers, thereby prohibiting the maintenance of a large tumor. These findings provide experimental support for the idea that a clone responsible for driving tumor outgrowth can be outcompeted by a clone with faster proliferation, leading to tumor collapse24,25.
DISCUSSION
Widespread tumor heterogeneity challenges the common assumption that tumor growth and malignant phenotypes are driven by dominant clones that have highest cell-autonomous fitness advantages (Fig. 4g). Previous studies in Drosophila and mouse models demonstrated that tumor growth can be supported by a small population of cells via direct non-cell autonomous stimulation20,26,27. Furthermore, the cross-talk between subpopulations of tumor cells was implied in metastasis28. Our results suggest that tumors can be driven by a sub-population of cells that des not have higher fitness, but instead stimulates growth of all tumor cells non-cell autonomously by inducing tumor-promoting microenvironmental changes (Fig.4h, center). Conversely, non-cell autonomous clonal expansion does not necessarily translate into increased tumor growth rates (Fig.4h, left). The non-cell autonomous driver sub-clone can be outcompeted by a sub-clone with higher proliferative output, thus collapsing the tumor (Fig.4h, right). Notably, in our experiments IL11-expressing cells were initially intermingled with the competitors. Under the scenario of stochastic activation of expression, benefits of secretion of non-cell autonomously acting factors might be skewed to the producer clone due to spatial considerations. Therefore, while extensive intermingling of evolutionarily diverged sub-populations has been reported for primary tumors29, it will be important to evaluate the impact of topology in future studies.
Our results provide direct experimental evidence that clonal interference limits clonal expansions in tumors. Our modeling predicts that non-cell autonomous driving of tumor growth can maintain clonal diversity over clinically relevant timeframes. In turn, clonal diversity can lead to clinically important phenotypic properties as suggested by the emergence of metastatic dissemination due to interaction between IL11- and FIGF-expressing subpopulations. Non-cell autonomous driving of tumor growth and inter-clonal interactions suggest that experimental analysis and clinical diagnostics focusing only on the most abundant sub-population of tumor cells might be misleading.
METHODS
Cell lines
Breast cancer cell lines were obtained from the following sources: MDA-MB-468, MDA-MB-453, and HCC1954 from ATCC; MCF10DCIS.com from Dr. F. Miller (Karmanos Cancer Institute, Detroit, MI), SUM149PT from Dr. S. Ethier, University of Michigan, Ann Arbor, MI), and 21NT from Dr. A. Pardee (Dana-Farber Cancer Institute, Boston, MA). Cells were cultured in media recommended by the provider, their identity confirmed by STR analysis, and regularly tested for mycoplasma.
Generation of MDA-MB-468 derivate lines (“sub-clones”)
Entry cDNA ORFs in pDONOR223 or pENTR221 were obtained from human ORFeome collection v5.1 or Life Technologies, respectively. Lentiviral expression constructs were generated by Gateway swap into pLenti6.3/V5-Dest vector (Life Technologies) or pHAGE-EF (used for IL11 swap only, vector obtained from S. Elledge lab, Harvard Medical School) destination vectors and sequence verified. Assembling viral particles and transductions were performed following Life Technology protocols. Parental MDA-MB-468 cell lines were transduced with mCherry/Luciferase lentiviral construct (obtained from C. Mitsiades lab, DFCI) prior to derivation of specific sub-clones. Each derivative line was generated from a pool of 1–2×105 transduced cells. Lentiviral-mediated expression was verified by immunoblotting against V5 tag in vitro and further confirmed by immunohistochemistry in vivo. GFP “sub-clone” was derived by lentiviral transduction of pLVX-AcGFP (Life Technologies).
qPCR analysis of clonal composition
The frequency of individual clones within tumors was determined by analyzing the change in qPCR signal from the initial mixture, which was precisely defined through mixing of clones based on cell counts, and the terminal tumor. qPCR was performed using Life Cycler 4800 (Roche) using SYBR green method with reaction mixtures purchased from Kapa Biosystems. Signals from individual clones were determined using a primer anchored in lentiviral backbone (anchor) and a primer specific for the clone-defining factor. As an internal reference we used primers specific for the pericentromeric region of chromosome 12, which does not display copy number alterations in the MDA-MB-468 cell line. Primer sequences are listed in the table below. The primers employed in the quantitation displayed linear amplification with >95% amplification efficiency. Change of frequency relative to the initial mixture was determined from Ct values for clone specific and internal reference qPCR signal based on ddCt method. Clonal proportions in polyclonal tumors were normalized based on total frequency of 1. For calculation of fold expansion, we used the clonality data to infer number of cells, following inferences between tumor mass and cellularity as described in Mathematical Supplement.
| Target | Sequence of primer |
| pLenti6.3/V5-Dest expressed | |
| Anchor | TCCAGTGTGGTGGAATTCTG |
| IL11 | cgtcagctgggaatttgtc |
| SPP1 | CATTCTGTGGGGCTAGGAGA |
| VEGFC | GAGCACTTGCCACTGGTGTA |
| IHH | GGTCTGATGTGGTGATGTCC |
| HGF | CTTTTCCTTTGTCCCTCTGC |
| CCL5 | CTGCTCCTCCAGATCTTTGC |
| VEGFB | CCATGAGCTCCACAGTCAAG |
| FIGF | CTCCACAGCTTCCAGTCCTC |
| CXCL12 | ATCTGAAGGGCACAGTTTGG |
| VCAN | GCGGAGAAATTCACTGGTGT |
| SHH | CCACATTGGGGATAAACTGC |
| VEGFA | GATTCTGCCCTCCTCCTTCT |
| CXCL14 | TTTGGCTTCATTTCCAGCTT |
| LOXL1 | ACTATGAGCCCGAGTTGAGC |
| LOXL3 | GTCTTCGATGTAGGCGGTCT |
| ANGPTL4 | GCGCCAGGACATTCATCT |
| IL6 | GCGGCTACATCTTTGGAATC |
| LACZ | CGGGCCTCTTCGCTATTAC |
| pLVX-AcGFP expressed | |
| GFP F | tcctgggcaataagatggag |
| GFP R | tgggggtattctgctggtag |
| pHAGE-EF-DEST expressed | |
| Anchor | TGGGACGTCGTATGGGTATT |
| IL11 | GGCTGCACCTGACACTTGAC |
| Human-specific centromeric reference locus | |
| F | tttggggccttaacactttg |
| R | aagcaaccagaagcctttca |
Xenograft experiments and doxorubicin treatment
All animal procedures were approved by the DFCI ACUC (DFCI protocol#11-023) and followed NIH guidelines. Tumors were induced by bi-lateral orthotopic injection into 4–5 weeks old female Foxn1nu mice of 1×106 cells re-suspended in 50% Matrigel (BD Biosciences) per transplant. Animals without successful tumor grafting were excluded from the analysis. Tumor volumes were monitored by bi-weekly measurements of tumor diameters with electronic calipers. For Doxorubicin treatment, animals were injected at days 15 and 22 post transplantation with 5 mg/kg Doxorubicin or PBS control. Since tumor sizes distribution of control and treatment groups prior to treatment was similar, no randomization was performed. No blinding was performed during the tumor measurements in live animals.
Immunoblot analysis
2×106 cells per sample were lysed in 100 μl of RIPA buffer. 10 μl of lysate was loaded per well of 4–12% Bis-Tris NuPage Midi gel (Life Technologies). Proteins were transferred to Immobilon PVDF membrane (EMD Millipore, Billerica, USA). Membranes were blocked for 30 minutes in StartingBlock blocking buffer (Thermo Scientific, Waltham, MA), then incubated overnight at 4°C with primary antibodies diluted 1:1000 in PBST in presence of 2.5% BSA. After 3×5 minute washes, membranes were incubated with secondary antibodies at 1:20,000 dilution, washed 2× 5 minutes followed by 20 minute wash. The membranes were developed with Immobilon substrate (EMD Millipore, Billerica, USA). The following antibodies were used: β-actin (Sigma, # A2228), IL11Rα (R&D systems #MAB1977), HRP conjugated anti-mouse and anti-rabbit (Thermo Scientific).
shRNA experiments
shRNA constructs in pLKO lentiviral vectors were obtained from the Broad Institute RNAi consortium. shRNA with the following targeting sequences were used:
| shRNA | sequence |
| IL11Ra shRNA#4 | CGGCAGATTCCACCTATAATT |
| IL11Ra shRNA#5 | TGGGACCATACCAAAGGAGAT |
| IL11Ra shRNA#7 | TGGAGCCAGTACCGGATTAAT |
| IL11Ra shRNA#8 | TGGCGTCTTTGGGAATCCTTT |
| IL11Ra shRNA#9 | ACTGATGAGGGCACCTACATC |
IL11 ELISA
Cells were plated at 1×105 cells per well in a 6-well plate and left overnight at 37°C with 5% CO2. The next morning, the media was replaced and the cells returned to the incubator. After 5 hours of incubation, the cells and the media were collected on ice in order to determine the concentrations of intracellular and secreted IL11 respectively. The harvested cells were counted, re-suspended in PBS and lysed by rapid freeze thaw cycles. The media and cell lysates were used for Human-IL11 ELISA (RayBiotech; ELH-IL11-001) according to manufacturer’s instructions. The values were adjusted for cell numbers as well as final volume to get an estimate of relative concentrations of IL11 in the two vector derivates.
Histological, immunohistochemical and multicolor immunofluorescence analyses
For histological analyses, 5μm sections of formalin fixed paraffin embedded (FFPE) xenografts were stained with hematoxylin and eosin using standard protocols. For analyses of collagen content, the tumor sections were stained with Masson’s trichrome stain kit (American Mastertech) following the manufacturer’s instructions. Immunohistochemical analyses of Bromodeoxyuridine (BrdU, Roche cat#11170376001, clone BMC9318, mouse monoclonal IgG1, 1:100), Ki-67 (Dako M724001, clone MIB-1, mouse monoclonal IgG1, 1:100), CD31 (Neomarkers RB10333, rabbit polyclonal, 1:50) and Smooth muscle actin (SMA, Dako M085101, clone 1A4, mouse monoclonal IgG2a, 1:250) were performed using 5μm sections of FFPE xenografts. The tissues were deparaffinized and rehydrated. After heat-induced antigen retrieval in citrate buffer (pH 6 for BrdU and Ki-67) or Dako target retrieval solution (S2367, pH 9 for CD31 and SMA), the samples were blocked with 3% hydrogen peroxide in methanol followed by goat serum and stained with the primary for 1 hour at room temperature. The samples were then incubated with anti-mouse or anti-rabbit IgG biotinylated antibody (1:100 dilution) for 30 minutes at room temperature followed by the ABC peroxidase System (Vectastain®, ABC System Vector Laboratories). DAB (3,3′-diaminodbenzidine) was used as the colorimetric substrate. The samples were washed twice with PBS-Tween 0.05% between incubations. Finally the slides were counterstained with Harris hematoxylin or 1% methyl green. Scoring for the expression of each marker was done as follows: the percentage of Ki67+ and BrdU+ cells were estimated by counting an average of 1,500–2,000 cells/sample using ImageJ 1.45s software from 4–6 randomly selected regions of the xenografts. Vessel density was scored by counting the number of CD31+ vessels per 20x field for 4–6 randomly selected fields in the tumor and the average was calculated. Blinding was used during key quantification analyses.
Multicolor immunofluorescence for cleaved Caspase 3 (Cell Signaling cat#9661, rabbit monoclonal IgG, 1:50) and/or V5 (Invitrogen R960-25, mouse monoclonal IgG2a, 1:100 was performed similarly as above. After heat-induced antigen retrieval at pH 6, the samples were blocked with goat serum and stained with the primary overnight at 4°C followed by incubation with goat anti-rabbit IgG Alexa 488-conjugated (Life Technologies, 1:100 dilution, for detection of cleaved Caspase 3) and goat anti-mouse IgG2a Alexa 555-conjugate (Life Technologies, 1:100 dilution, for detection of V5) for 45 minutes at room temperature. The samples were protected for long-term storage with VECTASHIELD HardSet Mounting Medium with DAPI (Vector laboratories, cat #H-1500). Before image analysis, the samples were stored at −20°C for at least 48 hours. Different immunofluorescence images from multiple areas of each sample were acquired with a Nikon Ti microscope attached to a Yokogawa spinning-disk confocal unit using a 60x plain apo objective, and OrcaER camera controlled by Andor iQ software. The montage images were created using the stitching plugin1 in (Fiji Is Just) ImageJ 1.48f software.
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay
FFPE sections of the xenografts were deparaffinized and rehydrated. Sections were then treated with 60μg/ml proteinase K (20mg/ml, Invitrogen, DNase and RNase free) in PBS for 15 min at room temperature. Protease digestion was stopped by consecutive washes in PBS and TdT buffer (Thermo Scientific®). The sections were blocked with 3% hydrogen peroxide in methanol to inhibit endogenous peroxidase activity. TUNEL assays were performed at 37°C for 1 hour in TdT buffer, 150 mM NaCl, 2 μM biotin 16-dUTP (Roche) and 80 U/ ml TdT (Thermo Scientific ®; EP0162). Following washing in PBS, labeled cells were visualized with the ABC peroxidase System (Vectastain®, ABC System Vector Laboratories) using DAB (3,3′-diaminodbenzidine) as the colorimetric substrate. The slides were counterstained with Harris hematoxylin. The percentage of TUNEL+ cells were estimated by counting an average of 600–1000 cells/sample using ImageJ 1.45s software from 4–6 randomly selected regions of the tumors.
Statistical analysis
Sample size was determined based on pilot experiments followed by larger scale studies to obtain significant differences (including the animal experiments). Estimation of variation within experimental group, normality test and statistical analyses indicated in figure legends were performed with Prism software (Graph Pad). Unless otherwise specified, p values refer to the results of the 2-tailed t-test.
Extended Data
Extended Data Figure 1. Proliferation, apoptosis and vascularization in selected groups.
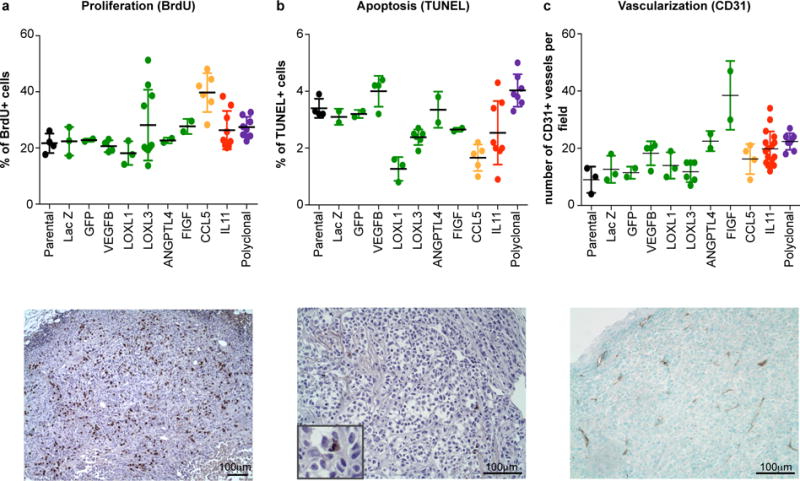
Quantitation and representative pictures of immunohistochemical analysis for markers of a, proliferation, b, apoptosis, and c, vascularization. Each dot represents individual tumor, error bars indicate SD.
Extended Data Figure 2. Estimations of clonal frequencies.
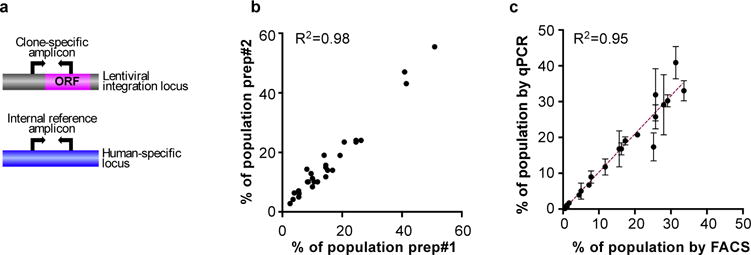
a, Schematic outline of the quantification of clonal composition based on qPCR. Changes in clonal frequencies are determined based on changes in the ratios of clone-specific and a human-specific reference amplicon between initial mixtures and the resulting tumors. b, Reproducibility of clonality analysis between two different DNA preparations/qPCR from same tumor. c, Correlation between the results obtained using fluorescent cell sorting (FACS) and qPCR based determination of clonal frequency after 6 weeks in vitro culture. Green Fluorescent Protein (GFP) labeled parental cells were mixed with individual sub-clones at initial ratios of 20:1. R2 indicates goodness of fit of linear regression.
Extended Data Figure 3. Mathematical model.
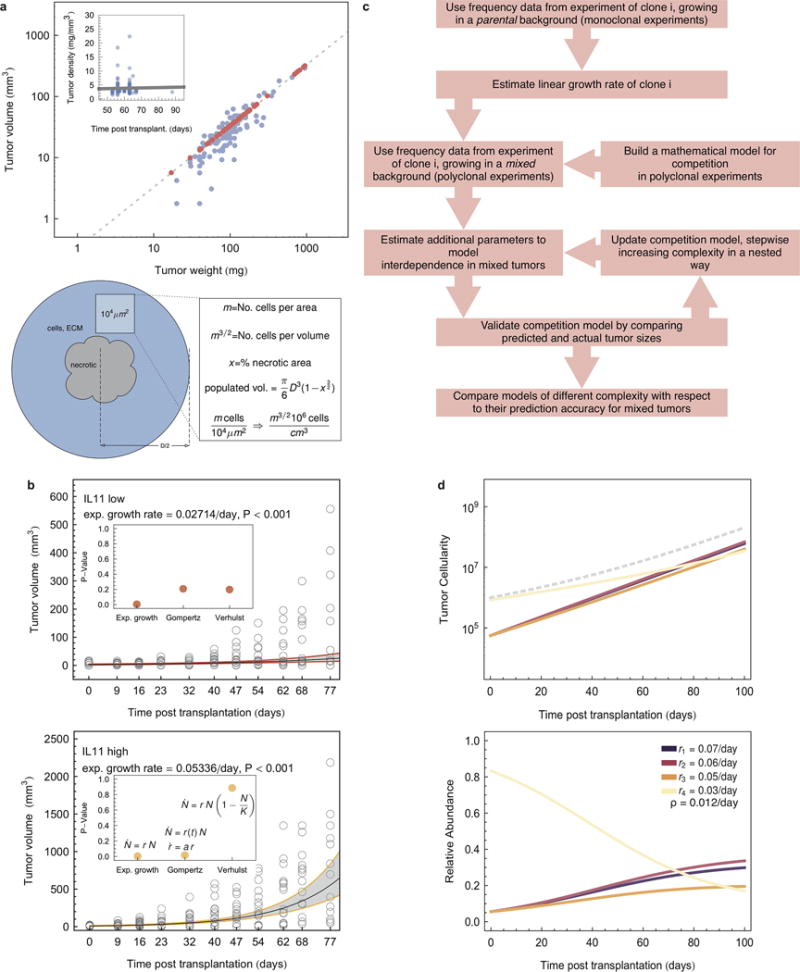
a, Upper panel: estimation of tumor volume-density relation. The line represents a linear regression with slope 0.33 (P<0.01). Red dots are predictions for which one value of the pair was missing. Inset: tumor density over time from clone-vs-parental competition experiments (dots), The line represents the linear regression. Tumor density did not correlate with the time of harvest (slope 0.012, P=0.68). Lower panel: schematic of estimation of cell numbers in tumor samples from two dimensional slices. b, tumor volume over time from experimens (empty circles) and linear regression (exponential tumor growth law, black lines), with 0.95 confidence intervals (gray areas). Inset: comparison of P-values using differnt growth laws. c, flow chart of mathematical modeling approach. d, upper panel: growth dynamics under non-cell autonomous driving, according to mathematical model (Model B, see SI), estimated additional effect of IL11 was 0.012/day. Example of four individual sub-clones (e.g. IL11, LOXL3, slow growing CCL5, LACZ), total tumor size indicated by dashed line, lower panel: frequency dynamics for the same set.
Extended Data Figure 4. Reproducibility and frequency-independence of tumor-growth promoting effects of IL11.
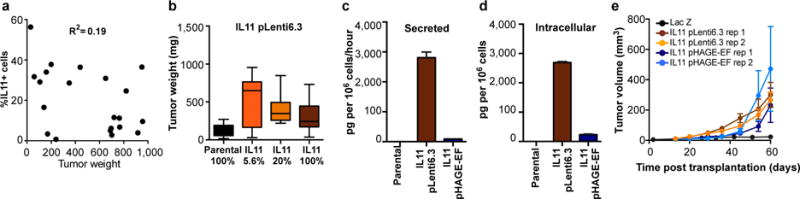
a, Relation between tumor weight and fraction of IL11 sub-clone cells upon tumor harvest. b, Final weights of tumors initiated from the indicated mixtures of IL11 expressing and parental cells using pLenti6.3 backbone; n=21 for the 5.6% IL11, n=10 for the other groups. c, Secreted (pg per 106 cells per hour) and d, Intracellular (pg per 106 cells) levels of IL11 protein determined by ELISA in parental cells and in the IL11-expressing clones derived using the indicated lentiviral constructs. e, Growth kinetics of tumors initiated by transplantation of mixtures containing IL11-expressing cells from the indicated backbones competing with the parental cells.
Extended Data Figure 5. IL11 in clonal cooperation.
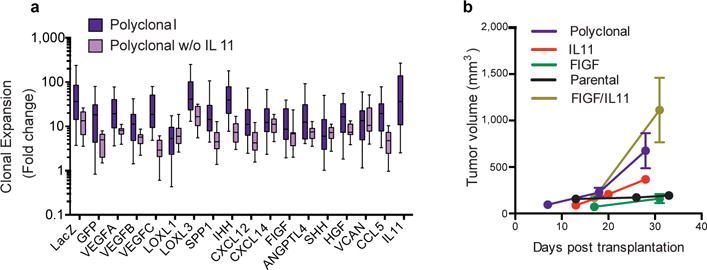
a, Expansion (fold change over initial number of cells) of indicated sub-clones in the polyclonal tumors initiated with/without IL11 sub-clone, n=10/group. b, Growth curves of the tumors initiated by transplantation of the indicated groups, IL11+FIGF indicates tumors initiated by 1:1 mixtures of IL11 and FIGF sub-clones.
Extended Data Figure 6. The effects of doxorubicin on tumor growth and clonal composition.
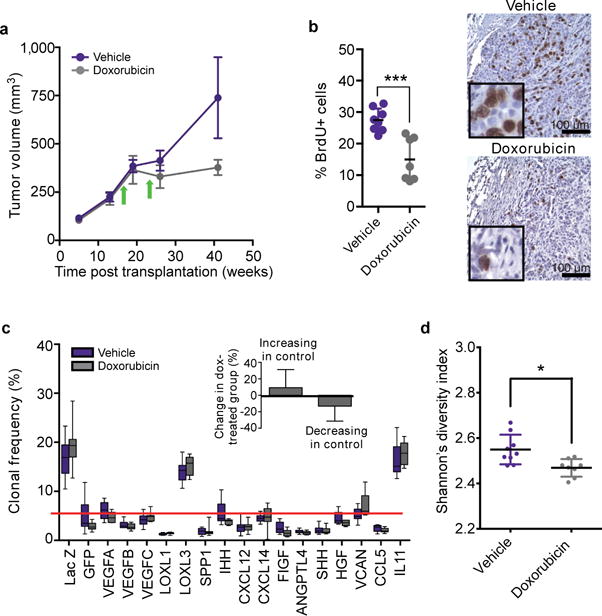
a, Tumor growth. b, assessment of cell proliferation by BrdU staining, and c, clonal composition of tumors initiated by polyclonal mixtures followed by treatment of the animals bearing established tumors with vehicle control or doxorubicin, arrows mark intraperitoneal injections of doxorubicin (5mg/kg) or vehicle. The insert in c quantifies changes in frequency of clones expanding and shrinking compared to the initial frequencies. Interaction factor for 2-way ANOVA between control and doxorubicin groups is statistically significant (p=0.0059). d, Shannon index for clonal diversity of vehicle and doxorubicin treated tumors, * indicates p<0.05 in two-sample Kolmogorov-Smirnov test.
Extended Data Figure 7. Validation of IL11Rα shRNA.
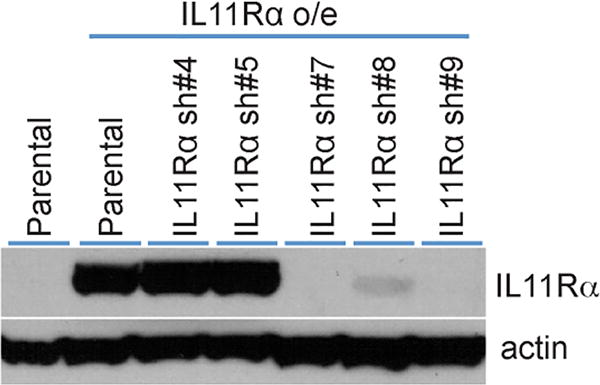
Since the commercially available IL11Rα antibodies are not sufficiently sensitive to detect endogenous IL11Rα protein in the MDA-MB-468 cells, we tested the ability of shRNA to down-regulate the expression of exogenously expressed IL11Rα. Cells overexpressing IL11Rα were stably transduced with IL11Rα-targeting shRNAs and the expression of IL11Rα and -actin (loading control) were analyzed by immunoblotting.
Extended Data Figure 8. The effects of IL11 on the tumor microenvironment.
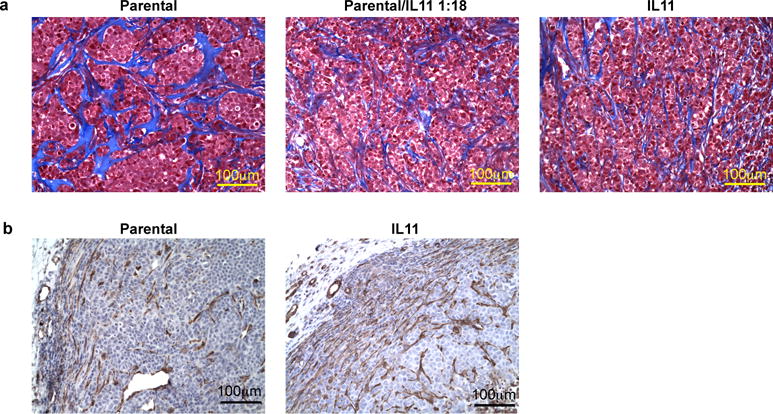
a, Collagen organization in parental and IL11 expressing tumors. Representative images of collagen structure (blue) in the indicated tumors as determined by tri-chrome staining. b, Smooth muscle actin positive (SMA) stromal cells in control and Il11 expressing tumors. Representative images of immunohistochemical staining for SMA.
Extended Data Figure 9. IL11 cells are not specifically eliminated in IL11/LOXL3 tumors.
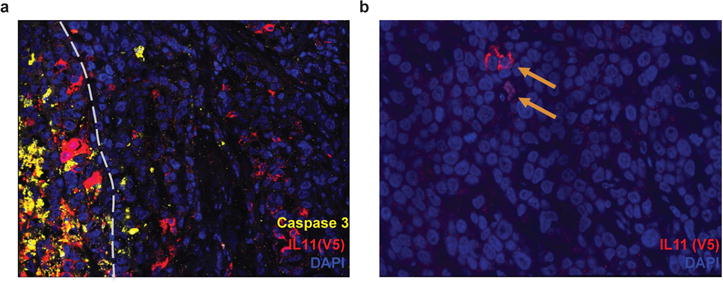
a, Immunofluorescence analysis of apoptosis in 1:1 IL11/LOXL3 tumors. Apoptotic marker cleaved Caspase 3 (yellow) indicates lack of increase in apoptosis in IL11 (red, V5+) cells bordering LOXL3 (V5−, as LOXL3 cDNA has a stop codon prior to the tag). Grey dashed line demarcates the border of the necrotic area, where most of cell death occurs. b, Occasional IL11+ cells (indicated by arrows) could still be detected in the remnants of 1:18 IL11/LOXL3 tumors.
Extended Data Table 1.
List of factors employed in sub-clonal derivations.
| Official gene symbol | Official gene name | Rationale for picking |
|---|---|---|
| LACZ | beta-D-galactosidase | Control |
| GFP | green fluorescent protein | Control |
| VEGFA | vascular endothelial growth factor A | Angiogenesis |
| VEGFB | vascular endothelial growth factor B | Lymphangiogenesis, metastasis |
| VEGFC | vascular endothelial growth factor C | Angiogenesis, ymphangiogenesis, and metastasis |
| LOXL1 | Iysyl oxidase-like 1 | Invasion and metastasis |
| LOXL3 | Iysyl oxidase-like 3 | Invasion and metastasis |
| SPP1 | secreted phosphoprotein 1 | Promotion of tumor growth through recruitment of bone marrow-derived cells |
| IHH | indian hedgehog | Activation of stroma |
| FIGF | c-fos induced growth factor | Lymphangiogenesis, metastasis |
| CXCL12 | chemokine (C-X-C motif) ligand 12 | Leukocyte infiltration, proliferation, metastasis |
| CXCL14 | chemokine (C-X-C motif) ligand 14 | Increased motility and invasiveness |
| SHH | sonic hedgehog | Promotion of tumor growth |
| VCAN | versican | Invasion, metastasis and growth |
| HGF | hepatocyte grow1h factor | Migration, adhesion and angiogenesis |
| CCL5 | chemokine (C-C motif) ligand 5 | Recruitment of monocytes |
| IL11 | interleukin 11 | Bone metastasis |
| ANGPTL4 | angiopoietin-like 4 | Angiogenesis and metastasis |
| IL6* | interleukin 6 | Survival, proliferation |
IL6 expressing sub-clone was generated and tested in the pilot experiments but was excluded due to high systemic toxicity.
Supplementary Material
Acknowledgments
We thank Drs. James DeGregori, Aaron Goldman, Andrii Rozhok, Mithat Gonen, and members of the Polyak and Michor laboratories for their critical reading of this manuscript and useful discussions. We thank Lisa Cameron in the DFCI Confocal Microscopy for her technical support. This work was supported by the Dana-Farber Cancer Institute Physical Sciences-Oncology Center (U54CA143798 to F.M.), CDRMP Breast Cancer Research Program W81XWH-09-1-0561 (A.M.), Cellex Foundation (V.A.), Deutsche Akademie der Naturforscher Leopoldina LPDS 2012-12 (P.M.A) and the Breast Cancer Research Foundation (K.P.).
Footnotes
AUTHOR CONTRIBUTIONS: A.M. developed experimental model, performed xenograft experiments and data analyses. D.T. performed immunohistochemical analyses and quantitations, and assisted with animal experiments. P.M.A. performed mathematical modeling and data analysis. V.A. assisted with image acquisition and analyses. K.P. supervised with help from F.M. All authors helped to design the study and write the manuscript.
References
- 1.Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012;481:306–313. doi: 10.1038/nature10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23–28. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 3.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 4.Gerlinger M, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Landau DA, et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell. 2013;152:714–726. doi: 10.1016/j.cell.2013.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ding L, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012 doi: 10.1038/nature10738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burrell RA, McGranahan N, Bartek J, Swanton C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature. 2013;501:338–345. doi: 10.1038/nature12625. [DOI] [PubMed] [Google Scholar]
- 8.Anderson K, et al. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature. 2011;469:356–361. doi: 10.1038/nature09650. [DOI] [PubMed] [Google Scholar]
- 9.Marusyk A, Polyak K. Tumor heterogeneity: causes and consequences. Biochim Biophys Acta. 2010;1805:105–117. doi: 10.1016/j.bbcan.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Merlo LM, Pepper JW, Reid BJ, Maley CC. Cancer as an evolutionary and ecological process. Nat Rev Cancer. 2006;6:924–935. doi: 10.1038/nrc2013. [DOI] [PubMed] [Google Scholar]
- 11.Weinberg RA. The biology of cancer. Garland Science; 2007. [Google Scholar]
- 12.Bissell MJ, Hines WC. Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat Med. 2011;17:320–329. doi: 10.1038/nm.2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gatenby RA, Gillies RJ. A microenvironmental model of carcinogenesis. Nat Rev Cancer. 2008;8:56–61. doi: 10.1038/nrc2255. [DOI] [PubMed] [Google Scholar]
- 14.DeGregori J. Challenging the axiom: does the occurrence of oncogenic mutations truly limit cancer development with age? Oncogene. 2013;32:1869–1875. doi: 10.1038/onc.2012.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kreso A, et al. Variable clonal repopulation dynamics influence chemotherapy response in colorectal cancer. Science. 2013;339:543–548. doi: 10.1126/science.1227670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gerrish PJ, Lenski RE. The fate of competing beneficial mutations in an asexual population. Genetica. 1998;102–103:127–144. [PubMed] [Google Scholar]
- 17.Putoczki T, Ernst M. More than a sidekick: the IL-6 family cytokine IL-11 links inflammation to cancer. J Leukoc Biol. 2010;88:1109–1117. doi: 10.1189/jlb.0410226. [DOI] [PubMed] [Google Scholar]
- 18.Ernst M, et al. STAT3 and STAT1 mediate IL-11-dependent and inflammation-associated gastric tumorigenesis in gp130 receptor mutant mice. J Clin Invest. 2008;118:1727–1738. doi: 10.1172/JCI34944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Putoczki TL, et al. Interleukin-11 is the dominant IL-6 family cytokine during gastrointestinal tumorigenesis and can be targeted therapeutically. Cancer Cell. 2013;24:257–271. doi: 10.1016/j.ccr.2013.06.017. [DOI] [PubMed] [Google Scholar]
- 20.Inda MM, et al. Tumor heterogeneity is an active process maintained by a mutant EGFR-induced cytokine circuit in glioblastoma. Genes Dev. 2010;24:1731–1745. doi: 10.1101/gad.1890510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bissell MJ, Radisky D. Putting tumours in context. Nat Rev Cancer. 2001;1:46–54. doi: 10.1038/35094059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med. 1995;1:27–31. doi: 10.1038/nm0195-27. [DOI] [PubMed] [Google Scholar]
- 23.Levayer R, Moreno E. Mechanisms of cell competition: themes and variations. J Cell Biol. 2013;200:689–698. doi: 10.1083/jcb.201301051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maley CC, Reid BJ, Forrest S. Cancer prevention strategies that address the evolutionary dynamics of neoplastic cells: simulating benign cell boosters and selection for chemosensitivity. Cancer Epidemiol Biomarkers Prev. 2004;13:1375–1384. [PubMed] [Google Scholar]
- 25.Nagy JD. Competition and natural selection in a mathematical model of cancer. Bulletin of mathematical biology. 2004;66:663–687. doi: 10.1016/j.bulm.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 26.Wu M, Pastor-Pareja JC, Xu T. Interaction between Ras(V12) and scribbled clones induces tumour growth and invasion. Nature. 2010;463:545–548. doi: 10.1038/nature08702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cleary AS, Leonard TL, Gestl SA, Gunther EJ. Tumour cell heterogeneity maintained by cooperating subclones in Wnt-driven mammary cancers. Nature. 2014;508:113–117. doi: 10.1038/nature13187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Calbo J, et al. A functional role for tumor cell heterogeneity in a mouse model of small cell lung cancer. Cancer Cell. 2011;19:244–256. doi: 10.1016/j.ccr.2010.12.021. [DOI] [PubMed] [Google Scholar]
- 29.Navin NE, Hicks J. Tracing the tumor lineage. Mol Oncol. 2010;4:267–283. doi: 10.1016/j.molonc.2010.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Preibisch S, Saalfeld S, Tomancak P. Globally optimal stitching of tiled 3D microscopic image acquisitions. Bioinformatics. 2009;25:1463–1465. doi: 10.1093/bioinformatics/btp184. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.