

Abstract
Anticipation is the phenomenon whereby age of onset in genetic disease decreases in successive generations. Three independent reports have claimed anticipation in Creutzfeldt-Jakob disease (CJD) caused by the c.598G>A mutation in PRNP encoding a p.Glu200Lys (E200K) substitution in the prion protein. If confirmed, this finding would carry clear implications for genetic counseling. We analyzed pedigrees with this mutation from four prion centers worldwide (n = 217 individuals with the mutation) to analyze age of onset and death in affected and censored individuals. We show through simulation that selective ascertainment of individuals whose onset falls within the historical window since the mutation’s 1989 discovery is sufficient to create robust false signals both of anticipation and of heritability of age of onset. In our data set, the number of years of anticipation observed depends upon how strictly the data are limited by the ascertainment window. Among individuals whose disease was directly observed at a study center, a 28-year difference between parent and child age of onset is observed (p = 0.002), but including individuals ascertained retrospectively through family history reduces this figure to 7 years (p = 0.005). Applying survival analysis to the most thoroughly ascertained subset of data eliminates the signal of anticipation. Moreover, even non-CJD deaths exhibit 16 years anticipation (p = 0.002), indicating that ascertainment bias can entirely explain observed anticipation. We suggest that reports of anticipation in genetic prion disease are driven entirely by ascertainment bias. Guidelines for future studies claiming statistical evidence for anticipation are suggested.
Introduction
Prion diseases are uniformly fatal, progressive neurodegenerative disorders caused by the conversion of the cellular prion protein, PrPC, to a misfolded conformation known as the prion, or PrPSc, in which Sc stands for scrapie, the prion disease of sheep and goats.1 In humans, prion diseases have an incidence of approximately 1 death per 1 million individuals per year,2 and usually occur as simplex cases in individuals with two wild-type (WT) copies of the prion protein gene (PRNP [MIM 176640]), commonly referred to as sporadic cases. A minority of cases are genetic and, very rarely, prion disease can be environmentally acquired.1 Creutzfeldt-Jakob disease (MIM 123400) caused by the c.598G>A (dbSNP id rs28933385) mutation, which encodes a p.Glu200Lys (E200K) substitution in PrP, is the most common genetic form of prion disease worldwide.3 This point mutation was first identified in 19894 and was established as a dominant Mendelian cause of disease by 1991.5–7 Disease penetrance in mutation heterozygotes appears to reach 80%–100% by age 80.8,9 Reported estimates of the mean age of onset in individuals with this mutation range from 537 to 63,10 and the mean survival after disease onset is 7 months.11
Three reports12,13 (see also Web Resources) have claimed statistical evidence that this genetic prion disease exhibits anticipation, a phenomenon in which successive generations exhibit progressively earlier disease onset or more severe presentation.14 These studies reported a 7 to 14 year younger age of onset or death among children in affected parent-child pairs, suggesting implications for genetic counseling.
The only genetic mechanisms known to cause anticipation are the germline expansion of unstable repeats in disorders such as Huntington’s disease and type 1 myotonic dystrophy,15,16 and telomere shortening in disorders such as dyskeratosis congenita and breast cancer.17,18 Anticipation in a genetic prion disease might raise the question of whether disease in children is accelerated by exposure to infectious material during their parents’ illness, however, the only known routes of human-to-human prion transmission are cannibalism19 and iatrogenic exposure.20,21
Because a variety of sources of ascertainment bias are known to contribute to false statistical signals of anticipation,14,22–25 we set out to determine whether the anticipation reported for Glu200Lys genetic prion disease could be a statistical artifact. An individual’s observed age of onset cannot be greater than the age at interview or ascertainment, and previous studies have modeled the effects of this right truncation of age of onset22 and provided methods of correction.23–26 These methods of correction, however, require either a consistent, known set of ascertainment criteria23 or the use of only a subset of available data.26 In rare diseases, data may be too sparse for subsetting, and may not represent a unified ascertainment effort, but rather consist of a mix of data points ascertained retrospectively (through family histories of varying depth and quality), directly (symptomatic individuals seen clinically), and prospectively (asymptomatic individuals with a mutation, followed for varying amounts of time). We therefore sought to model ascertainment bias due to left- and right-truncation not of the age of onset per se, but of the year of onset. Because the Glu200Lys substitution was discovered only 25 years ago and most prion surveillance programs and clinical centers have been established even more recently, we hypothesized that the selective ascertainment of parents and children whose deaths both occurred within this 25-year window could explain the reported differences in parent and child age of death.
To test this hypothesis, here we combine data from four national prion study centers to assemble the largest Glu200Lys cohort (n = 217 individuals) yet reported. We first create a simulation of the ascertainment of parent-child pairs with a mutation to identify conditions under which naive paired t tests will detect a false signal of anticipation. We explore methods for detecting and controlling for this ascertainment bias. We then apply our analytical framework to our Glu200Lys data set and successfully reproduce the anticipation reported by other groups but demonstrate that this anticipation is a false positive due to ascertainment bias.
Material and Methods
Data Collection
We combined data collected on Glu200Lys individuals and their families from four research centers with data collection practices as follows.
Australian National Creutzfeldt-Jakob Disease Registry
Details of Australian National Creutzfeldt-Jakob Disease Registry (ANCJDR) surveillance mechanisms, as well as data collection and analysis methods, have been reported previously.27,28 In brief, prospective national surveillance of CJD has been undertaken since 1993 with CJD a Notifiable Disease throughout Australia since 2006. ANCJDR collects detailed medico-demographic information on suspect cases, including family histories, and provides diagnostic tests including PRNP genotyping. Year and age of death are primary variables with information on age at onset of first symptom collected if available. Informed, written consent was obtained from participants or legal next of kin. Ethical approval was obtained from the Office of Research Ethics and Integrity at The University of Melbourne.
German CJD Surveillance Unit
Details of German CJD surveillance have been reported previously.29–31 In brief, the Surveillance Unit in Goettingen has collected data on all suspected prion disease cases in Germany since 1993. Diagnostic information is obtained from reporting hospitals and where possible, confirmation by autopsy is sought. The Surveillance Unit also accepts clinical referrals, provides diagnostic tests including PRNP genotyping, and, where possible, collects family history. Age of onset is defined from first symptom of a progressive neuropsychiatric disorder by interview with family members. Informed, written consent was obtained from participants or legal next of kin. Ethical approval was obtained from the Ethical Committee at the University Medical School, Georg-August University Goettingen.
MRC Prion Unit/NHS National Prion Clinic
The UK has had a centralized tertiary clinical referral service for CJD since 1991. Since 2004, all suspected CJD cases from the UK are referred to the NHS National Prion Clinic at the National Hospital for Neurology and Neurosurgery (NHNN) at University College London Hospitals NHS Trust. Age of onset was defined from first symptom of a progressive neuropsychiatric disorder and family history was obtained by interview with family members. Other details of data collection have been described previously.32 Informed, written consent was obtained from participants or legal next of kin. Ethical approval was obtained from the NHNN/Institute of Neurology Joint Research Ethics Committee.
Memory and Aging Center, University of California San Francisco
The UCSF cohort comprises symptomatic and asymptomatic individuals from Glu200Lys families referred from the U.S. and abroad to the rapidly progressive dementia and Prion Disease research program since August 2001.33–35 PRNP genotyping36 was performed at the National Prion Disease Pathology Surveillance Center (Cleveland, OH), or by outside laboratories in some of the individuals who were tested prior to UCSF referral or lived abroad. Symptom onset was determined as previously reported.37 A detailed, usually three generation, family pedigree was made by a neurologist and/or clinical genetic counselor for individuals participating in research. Further data were collected from medical records sent by referring physicians and/or from direct contact with family members (by email or telephone). Informed, written consent was obtained from research participants or legal next of kin. The UCSF data included in this study have been collected through UCSF Institutional Review Board-approved research protocols.
Data Annotation
Directly observed individuals were defined as those either seen clinically at one of the four centers or officially reported to one of the centers in its prion disease surveillance role. Indirectly observed individuals were those ascertained through interview with family members. Individuals were considered to have the c.598>A mutation if they (1) had either a genotyping test indicating the presence of the mutation or were related to someone with a positive test and (2) had a diagnosis of CJD or (3) were deemed to have died of CJD based on information obtained from interviewed family members. Individuals were considered to not have the mutation if they (1) had a genotyping test indicating the absence of the c.598G>A mutation or (2) were related to the family only by marriage and thus lacked a blood relationship to any affected individual. The Glu200Lys substitution causes CJD with nearly 100% penetrance,8,9 but individuals with two WT PRNP alleles have a very low disease incidence of only about 1 in 1 million per year.2 This incidence translates into a lifetime risk roughly on the order of 1 in 10,000 for a WT individual. By Bayes’ rule, the high penetrance of the mutation and rarity of nongenetic CJD cases mean that any CJD case in a Glu200Lys pedigree is overwhelmingly likely to be genetic. Therefore, we assumed that all CJD cases in these pedigrees were due to the c.598G>A mutation. Data on four individuals with CJD were flagged as questionable due to uncertain diagnosis, uncertain relatedness to other individuals or uncertain age at death; none of the results reported here differed when the analysis was re-run excluding these individuals. Missing PRNP codon 129 information was imputed from affected family members when possible. For the purpose of assessing rates of predictive genetic testing, “at-risk” individuals were defined as those who were not symptomatic at last follow-up but who did have a parent deemed to have the mutation, per the above criteria. Recursively, individuals were also considered at-risk if they had a parent who qualified as at-risk according to the above definition.
Simulation
We hypothesized (see Introduction) that the selective ascertainment of individuals with disease onset within a specified window (for instance, 1989–2013) might explain reported anticipation in Glu200Lys prion disease. To assess whether such an “ascertainment window” was sufficient to create a false signal of anticipation, we created a simulation model in which parent and child ages of onset are drawn from the same specified distribution but ascertainment of parent-child pairs is selective based on year of onset. For each round of simulation, we generated n = 100,000 parent-child pairs. We only simulated parents and children with the causative mutation. We wished to model a situation in which individuals have been falling ill of prion disease continuously throughout history, and will continue to do so into the future, whereas our data are limited only by an artificial ascertainment window. We therefore generated parents with a year of birth uniformly distributed from 1700 to 2000, in order to ensure that (1) the density of disease onsets over our largest ascertainment window (1880 to 2013) would be uniform and that (2) observed years of onset would effectively be left-truncated only according to our ascertainment criteria and not according to the underlying simulated distribution. We set the child’s birth to occur 28 ± 6 years after the parent’s birth, per the actual distribution from our data. To simulate an age of onset distribution while accounting for censoring due to deaths due to other causes (“competing risks”), we first created a normally distributed 64 ± 10 year age of onset to approximate the distribution in our real data (see Results) and then randomly censored individuals according to the U.S actuarial life table for 2009 (see Web Resources). We did not model changes in the life expectancy over the time periods considered here. Further details of the specific simulations are explained in Results and in the legends of Tables 1 and 2.
Table 1.
Simulations of Ascertainment Bias
| Simulation | Direct ascertainment window | Indirect ascertainment | Mean anticipation (years)a | Anticipation significantb | Mean additive heritabilitya | Heritability significantb | Mean year of birth/age of onset slopea | Slope significantb |
|---|---|---|---|---|---|---|---|---|
| 1 | 1989–2013 | None | 16.4 | 100% | 93% | 100% | −0.67 | 100% |
| 2 | 1950–2013 | None | 4.9 | 98% | 19% | 16% | −0.22 | 100% |
| 3 | 1880–2013 | None | 1.6 | 23% | 3% | 5% | −0.06 | 100% |
| 4 | 1989–2013 | Declining 5%/year | 10.7 | 100% | 59% | 83% | −0.55 | 100% |
| 5 | 1950–2013 | Declining 5%/year | 3.4 | 79% | 14% | 11% | −0.18 | 100% |
| 6 | 1880–2013 | Declining 5%/year | 1.4 | 18% | 3% | 6% | −0.05 | 98% |
| 7 | 1989–2013 | Declining 1%/year | 2.0 | 33% | 6% | 5% | −0.26 | 100% |
| 8 | 1950–2013 | Declining 1%/year | 0.9 | 12% | 4% | 5% | −0.12 | 100% |
| 9 | 1880–2013 | Declining 1%/year | 0.4 | 6% | 1% | 4% | −0.04 | 85% |
| 10 | 1989–2013 | Exhaustive | 0.2 | 5% | −1% | 5% | −0.20 | 99% |
Ascertaining only those individuals with disease onset within a historical window creates false signals of anticipation and heritability (Simulation 1). These false signals are reduced, but not eliminated, by expanding the ascertainment window (Simulations 2 and 3). Probabilistic retrospective ascertainment (see text) reduces these signals further (Simulations 4–9). Only when retrospective ascertainment is 100% exhaustive does anticipation become reliably nonsignificant (Simulation 10).
Averages from 1,000 simulations.
Percentage of 1,000 simulations in which this figure was statistically significant at p < 0.05.
Table 2.
Stratification of Simulated Data
| Cohort Name | Inclusion Criteria | Anticipation | p |
|---|---|---|---|
| Child early birth year | Child born < 1939 | 16 years | 0.009 |
| Child late birth year | Child born ≥ 1939 | 18 years | <0.0001 |
| Child early death year | Child year of onset < 2000 | 26 years | 0.0004 |
| Child late death year | Child year of onset ≥ 2000 | 15 years | <0.0001 |
| Child early death age | Child age onset < 61 | 21 years | <0.0001 |
| Child late death age | Child age onset ≥ 61 | 11 years | 0.003 |
| Parent early death age | Parent age onset < 70 | 12 years | 0.01 |
| Parent late death age | Parent age onset ≥ 70 | 20 years | <0.0001 |
In n = 26 simulated parent-child pairs with independent and identically distributed ages of onset but ascertaining only those pairs whose onsets both fall within 1989–2013, all subsets of data stratified according to the variables defined in Table 3 of Pocchiari et al.13 still show significant anticipation.
Statistical Analyses
All simulations and analyses of Glu200Lys pedigree data were conducted in R 3.0.2. In order to demonstrate the effects of ascertainment bias on the evidence for anticipation reported by previous studies of genetic prion disease, we adopted methods from those studies when possible. For paired t tests, all possible parent-child pairs were generated from Glu200Lys pedigree structures using a SQL join operation. This results in multiple-counting parents of multiple affected children and counting both as parents and as children those individuals in the middle of pedigrees with three or more affected generations. Multiple counting means that pairs are not independent; this mirrors the methods from at least one prior study of prion disease anticipation.13 Based on these paired lists of parents and children, differences in parent and child age of onset were assessed using two-tailed paired t tests (the R t.test function) for naive comparisons of observed age of onset distributions. Correlation between parent and child age of onset was assessed using linear regression (the R lm function). Survival analysis utilized the R survival package. For survival analysis, to avoid multiple-counting and ensure independence of all pairs, we randomly selected one parent-child pair from each pedigree and compared the survival curve for all parents to that of all children using a log rank test (the R survdiff function). Because we randomized which pairs were included, we repeated this analysis for over 1,000 iterations and have reported aggregate statistics. Source code and output are available online (see Web Resources).
Results
Simulated Effects of Ascertainment Window
We generated 100,000 parent-child pairs with all individuals harboring a dominant genetic mutation and ages of onset for all individuals independent and identically distributed, corresponding to a scenario in which no anticipation or heritability of age of onset are present (see Material and Methods and Figure 1A). The mean age of onset without accounting for censoring was 62, whereas the median age of onset in survival analysis accounting for censoring due to intercurrent deaths was 64. When considering all simulated individuals, there was no difference in age of onset (p = 0.91, two-tailed paired t test), no correlation between parent and child age of onset (p = 0.53, linear regression), and no correlation between the year of birth and age of onset for all individuals (p = 0.59, linear regression).
Figure 1.
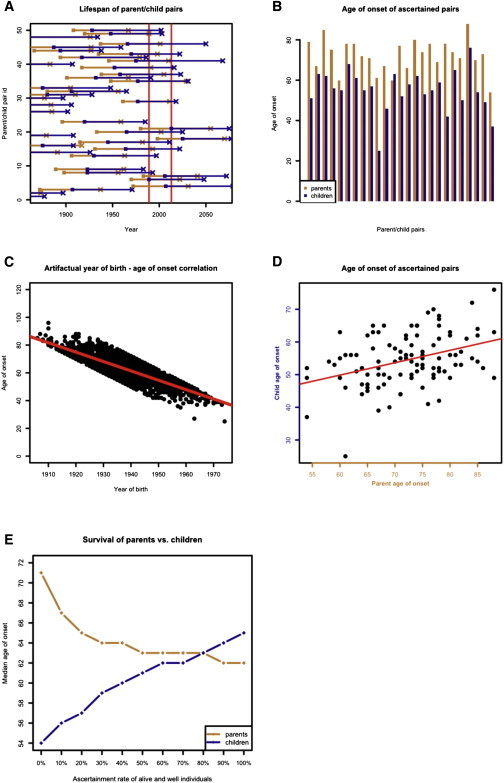
Simulation of Ascertainment Bias Due to Year of Onset Windowing
(A) Visual representation of a subset of simulated data. Parent-child pairs are arranged along the y axis. Parent is born on orange square and has disease onset on orange X; child is born on blue square and has disease onset on blue X. Age of onset distributions are identical, but in Simulation 1, only those pairs in which both individuals have onset between 1989 and 2013 inclusive (red vertical lines) can be ascertained. In the above example, only one pair (#49) meets these criteria.
(B) Among ascertained pairs in Simulation 1, children have almost categorically younger onset than their parents, leading to 17 years of observed anticipation (p < 0.0001, two-tailed paired t test). For visibility, a subset of simulated points is shown.
(C) Ascertainment windowing introduces an artifactual correlation between year of birth and age of onset, slope = −0.67 (p < 0.0001, linear regression).
(D) Ascertainment windowing also leads to a correlation between parent and child age of onset and thus a false signal of heritability (slope = 0.48, p < 0.0001, linear regression).
(E) Ascertained pairs are supplemented with those pairs where one individual’s onset occurs in 1989–2013 and the other is alive and well as of 2013. The plot shows that the median survival of parents and children in Kaplan-Meier curves depends upon what proportion of alive and well individuals are included. If 0% are included, then 17 years anticipation are observed, just as in (B). As ascertainment increases, the anticipation is reduced. As inclusion of alive and well individuals approaches 100%, children have longer survival than parents. This is because more of the children are censored, so their age of onset distribution better reflects the hypothetical distribution of age of onset without the influence of competing risks.
Next we considered the effects of selectively ascertaining individuals whose disease onset occurs within an “ascertainment window,” which we define as a range of years in which disease onsets can be observed. It is impossible to ascertain ages of onset after 2013, the last year in which our data were collected. Right-truncating the year of onset at 2013 and considering all affected pairs introduced only a very small difference between parent and child age of onset (0.37 years, p < 0.0001, two-tailed paired t test). Because we had generated parent-child pairs with years of birth as early as 1700, only a minority of pairs was affected by this right truncation. Spreading the simulated pairs out over an even greater range of years of parent birth (year 0–2000) made this anticipation cease to be significant (p = 0.31, two-tailed paired t test), indicating that right truncation of year of onset is not sufficient cause a false signal of anticipation. In practice, any real data set will be both left-truncated (when researchers began to study the disease) and right-truncated (at the present year or when the study stopped, whichever is earlier). For Glu200Lys CJD, we reasoned that ascertainment rates would be higher after the discovery of the disease’s causal mutation in 1989,4 so we next considered the effects of ascertaining only those individuals whose year of onset occurs between 1989 and 2013 inclusive.
When only those simulated pairs in which parent and child disease onset both occurred within this window were considered (Figure 1A), we observed 17 years of anticipation (p < 0.0001, two-tailed paired t test, Figure 1B). Because parents were born, on average, 28 years earlier than their children, yet the window of observation was only 25 years long, the ascertained pairs were vastly enriched for those in which the child dies at least 3 years younger than the parent. Under these conditions, we also observed an artifactual correlation between year of birth and age of onset, with slope of −0.67, (p < 0.0001, linear regression, Figure 1C). This relationship is intuitive because an individual born in 1970, for instance, can only be included in the data set if onset occurs by age 43 (year 2013), and an individual born in 1940 can only be included if onset occurs at age 49 or later (year 1989). Year of birth/age of onset correlation is therefore a consequence of ascertaining only individuals with the year of onset within a particular window. This problem has previously been noted in Huntington’s disease pedigrees.38 Among the ascertained pairs, parent and child age of onset were also correlated with a slope of 0.48 (p < 0.0001, linear regression, Figure 1D). In single parent-offspring regression, the slope can be doubled to obtain an estimate of phenotypic variance explained by additive genetic heritability plus environmental effects.39 Applying this formula to the ascertained data would suggest that age of onset is up to 96% heritable. Because the true distributions of parent and child ages of onset in our simulation were neither different nor correlated, this shows that the year of onset “windowing” simulated here is sufficient to create false signals of both anticipation and heritability. We confirmed this finding through 1,000 iterations with the same set of simulation conditions and n = 100 ascertained pairs to simulate a realistic sample size for a rare disease (Table 1, Simulation 1).
Because we knew the anticipation and heritability identified in our above simulation to be false signals, we asked whether improved ascertainment or simple analytical methods could disprove them. We considered a two-step ascertainment model; in the first step, we ascertained all individuals with disease onset between 1989 and 2013 (simulating clinical visits), and in the second step we ascertained any and all individuals still alive and well as of 2013 who were parents or children of the individuals ascertained in step 1 (simulating genetic testing and prospective follow-up of individuals with the mutation). When we compared parent and child survival curves under this two-step ascertainment model in Kaplan-Meier survival analysis, the median age of onset of children was greater than that of parents (64 versus 62 years, p < 0.0001, log rank test). This counter-intuitive finding of later child onset can be explained as follows. Because parents are born earlier than children, pairs in this two-step ascertainment model are predominantly ones in which the parent dies of genetic prion disease between 1989 and 2013 while the child is alive and well in 2013. In such pairs, the parent’s age of onset distribution is truncated by competing risks (parents who would have had older disease onsets were never ascertained because they died of other causes first, thus making disease onset appear younger). The child’s age of onset distribution, however, is less affected by competing risks (children who would have older disease onsets are ascertained through genetic testing even if they will never develop the disease in their lifetimes).
Recall that in the two-step ascertainment model above, an affected individual is ascertained in step 1, and if that individual has a parent or child alive and well in 2013, then that parent or child is always ascertained. In practice, however, it is difficult to ascertain 100% of asymptomatic relatives of an affected individual. We therefore also modeled incomplete ascertainment of asymptomatic individuals. When we did so, we saw that the false signal of anticipation from naive t tests was not fully corrected (Figure 1E). The difference in median survival according to the log rank test was 17 years when no censored individuals were included, and this difference shrunk to 7 years when 20% of individuals alive and well in 2013 were included, and 2 years when 50% of such individuals were included. This indicates that accounting for censored observations is not sufficient to remove false signals of anticipation unless ascertainment of censored individuals is exhaustive. In long-term studies of predictive genetic testing for neurodegenerative diseases, it has been reported that only 3% to 24% of individuals with risk of having inherited a mutation pursue predictive genetic testing.40–45 Survival analysis including asymptomatic individuals with a mutation is therefore unlikely to mitigate false signals of anticipation and heritability in this disease setting.
We also asked whether including year of birth as a covariate could eliminate the false signal of heritability we observed in our simulation. When both parent age of onset and child year of birth were used in a linear model to explain child age of onset, only the child’s year of birth proved to be correlated (slope = −0.72, p < 0.0001, linear regression), whereas parent age of onset was no longer significant (p = 0.73, linear regression). When we simulated an age of onset that truly was heritable, this method reduced, but did not eliminate, the estimate of heritability (see Table S1 available online).
Simulated Effects of Different Ascertainment Criteria
In Simulation 1 (Table 1) we assumed that it was only possible to include individuals directly ascertained within the window from 1989 to 2013. In reality, some Glu200Lys families have been followed since as early as 19637 and it is also possible to obtain information on deceased individuals retrospectively through interview with family members, though it can be more difficult for individuals whose year of death is long past.46 We therefore asked whether false anticipation and heritability would still be observed with larger or more flexible ascertainment windows. In this analysis, we limited the number of ascertained pairs to 100 in order to simulate a realistic sample size for a rare disease. Expanding the size of the direct ascertainment window back to 1950 or 1880 (but still not adding indirect ascertainment) decreased the strength of the false signals quantitatively, but did not reliably eliminate them, even when the window was longer than a human lifespan (Table 1, Simulations 1–3).
Next, we considered including some indirectly ascertained individuals. To simulate retrospective ascertainment, we first directly ascertained individuals with onset within the window, then ascertained any parents or children thereof whose onset had already occurred prior to the beginning of the window. We hypothesized that it might be more difficult to ascertain individuals who died long ago, so for indirectly ascertained individuals we applied a linear reduction in ascertainment probability according to how many years prior to the beginning of the ascertainment window the individual had disease onset. In Simulation 4 (Table 1), for instance, direct ascertainment from 1989-2013 is exhaustive, but the probability of indirect ascertainment declines by 5% for each year prior to 1989 that an individual has onset so that, for instance, an individual with onset in 1988 can be retrospectively ascertained with 95% probability, in 1987 with 90% probability, and so on. Including some indirectly ascertained individuals in this manner further reduced the magnitude of anticipation but did not completely eliminate it in most scenarios tested (Simulations 4–9). The false signal of heritability was weaker than that of anticipation and could be reliably reduced to statistical insignificance in the most extreme scenarios tested (Simulations 6–9). Only when indirect ascertainment was 100% exhaustive and independent of the year of onset (Simulation 10) did a signal of anticipation cease to be detected, though a year of birth/age of onset correlation still existed.
Together, our simulations demonstrate that ascertaining individuals over a larger time window or including some indirect ascertainment (retrospective phenotyping) can quantitatively reduce the false signals of anticipation and heritability but that these false signals are still likely to be observed as statistically significant under anything other than almost complete ascertainment. Our results indicate that whenever retrospective ascertainment is less than exhaustive, there is a risk of observing false signals of anticipation and heritability of age of onset.
Stratification of Simulated Data
One recent report of anticipation in Glu200Lys genetic prion disease13 argued that the observed anticipation must be real because anticipation was observed, using paired t tests, within every subset of 26 parent-child pairs when these were stratified by age of death, year of birth, or year of death. To determine whether such an analysis could indeed rule out a false positive due to ascertainment bias, we stratified 26 simulated parent-child pairs with onset between 1989 and 2013 in the same way described in the previous report (Table 2). In our simulation, anticipation is not real, yet a strong signal of anticipation is nevertheless observed in all eight strata. Therefore, these methods of stratification do not eliminate spurious signals of anticipation due to ascertainment bias.
Characteristics of Glu200Lys Pedigree Data
We combined data on Glu200Lys families from four independent study centers worldwide (Table 3). Each individual data set reflected a different method of ascertainment (see Material and Methods) and accordingly, the proportion of individuals ascertained indirectly or asymptomatic as of last follow-up varied considerably (Table 3).
Table 3.
Characteristics of Glu200Lys Data Sets
| Study Center | In Operation Since | n c.598G>A Individuals | n c.598G>A Parent-Child Pairs | Proportion of c.598G>A Individuals Ascertained Indirectly | Proportion of c.598G>A Individuals Who Are Asymptomatic |
|---|---|---|---|---|---|
| ANCJDR | 1993 | 24 | 7 | 42% | 8% |
| German CJD Surveillance | 1993 | 32 | 3 | 38% | 3% |
| MRC Prion Unit | 1991 | 57 | 29 | 25% | 23% |
| UCSF | 2001 | 104 | 58 | 73% | 37% |
| All | 217 | 97 | 52% | 25% |
Data from the four study centers varied in terms of the length of time for which data had been collected, the degree of indirect ascertainment, and the ascertainment of asymptomatic individuals with the mutation. See Material and Methods for details.
Of individuals whose disease onset or death was directly observed by one of the study centers, 65% had no reported family history of prion disease. This figure might reflect some combination of (1) incomplete reporting of family history, (2) underdiagnosis of affected individuals in earlier generations, (3) censoring of asymptomatic individuals with the mutation upon death due to other causes, and (4) de novo mutations. Similarly, we knew the genotypes of only 22% of at-risk individuals (see Material and Methods) in our data sets. Together, these figures suggest that neither retrospective ascertainment nor prospective following of individuals with the mutation are exhaustive in our data set.
Disease Duration and Genotypic Influence
Disease duration was defined as time from first symptom (see Data Collection in Material and Methods) to death. Disease duration followed a nonnormal distribution (p < 0.0001, Shapiro-Wilk normality test) with a median of 162 days (n = 61, interquartile range 205 days), similar to the figure reported elsewhere.11 Disease duration did not differ by study center (p = 0.35, Kruskal-Wallis test, n = 61) or between directly and indirectly ascertained individuals (p = 0.08, Kruskal-Wallis test, n = 38 and 23 respectively). PRNP codon 129 information was available for a subset of individuals with known disease duration. Among individuals with a haplotype encoding Glu200Lys cis 129Met, disease duration was shorter for individuals with a trans allele encoding 129Met (median of 137 days, n = 25) than a trans allele encoding 129Val (median of 426 days, n = 7) and this difference was significant (p = 0.02, Kolmogorov-Smirnov test), consistent with previously reported data.11 Disease duration appeared to differ between haplotypes encoding Glu200Lys cis 129Met (n = 42) and Glu200Lys cis 129Val (n = 6) proteins, with a median duration of 137 versus 331 days respectively (p = 0.04, Kolmogorov-Smirnov test), although trans codon 129 information was not available for all of these individuals, so it is possible that the longer duration among cis 129Val individuals might be due to higher rates of codon 129 heterozygosity. Because disease duration was generally a year or less and our data included age of onset for some individuals but age of death for others, we decided to consider age of onset and death interchangeably in the anticipation analysis, preferring age of death when both were available.
Age of Onset or Death and Genotypic Influence
Overall, the mean age of onset or death in affected individuals was 62 ± 10 years (±SD, n = 158) and was approximately normally distributed (p = 0.36, Shapiro-Wilk normality test; Figure 2A). The median observed age of onset or death was 63 years (n = 158). The median rose to 64 years when we included asymptomatic individuals with the mutation, censored at date of last follow-up or death due to other causes, and applied a survival analysis (n = 207). This survival analysis indicated 93% disease penetrance by age 80, within the range of previously reported estimates,8,9 but we note that this figure is biased upward by the ascertainment of affected families and incomplete genotyping of unaffected individuals. To our knowledge, 64 years is older than any other measure of central tendency of age of onset for this mutation yet reported, probably because our use of survival analysis accounts for censoring due to competing risks, whereas other published estimates have been based simply on observed ages of onset.3,7,10,47 Even our estimate is likely biased downward due to incomplete genotyping of unaffected individuals.
Figure 2.
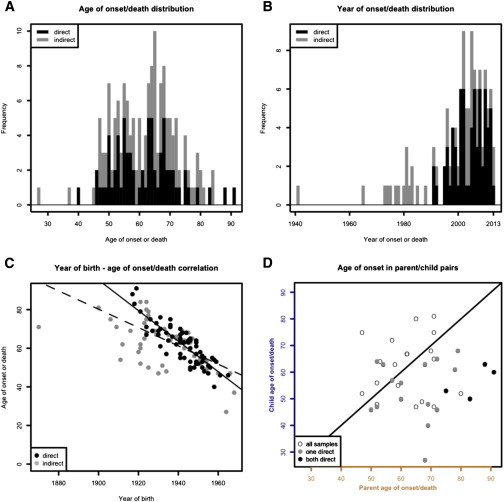
Ascertainment Bias and Anticipation in Glu200Lys Pedigree Data
(A) Distribution of age of onset or death by ascertainment mode.
(B) Years of onset or death, when known, occur overwhelmingly in the 25-year window since the mutation’s discovery in 1989.
(C) Year of birth and age of onset are artifactually correlated. This correlation is strongest in directly ascertained individuals (black line) but remains highly significant when indirectly ascertained individuals (gray points) are included in addition (dashed black line).
(D) Parent versus child age of onset or death. Points below the diagonal line indicate pairs in which the child dies younger than the parent. Directly ascertained pairs (black points) fall categorically below the line, pairs with one directly ascertained member are largely below the line (gray points), and pairs in which both individuals are indirectly ascertained fall on both sides (white points).
Age of onset or death did not differ by study center (p = 0.18, Model I ANOVA, n = 158), sex (p = 0.74, unpaired t test, n = 132), or mode of ascertainment (direct versus indirect, p = 0.66, unpaired t test, n = 158). Among individuals with a haplotype encoding Glu200Lys cis 129Met, age of onset or death did not differ between trans 129Met (n = 46) and trans 129Val (n = 10) genotypes (p = 0.71, two-tailed unpaired t test; p = 0.39, log rank test, n = 64 Met-Met versus 17 Met-Val including censored individuals), consistent with a previous report.48 It was unclear whether age of onset or death might differ between individuals with haplotypes encoding Glu200Lys cis 129Met versus Glu200Lys cis 129Val, because only a weak trend could be detected whether censored individuals were included (censored median 64 versus 57 years, p = 0.05, log rank test, n = 142 Met versus 13 Val) or excluded (mean 63 versus 56 years, p = 0.11, two-tailed unpaired t test, n = 105 Met versus 10 Val). Because trans codon 129 did not influence age of onset or death and because a child’s cis codon 129 is inherited from the affected parent, we deemed it unnecessary to consider codon 129 genotype in our anticipation analyses.
Ascertainment Bias in Glu200Lys Pedigree Data
Excluding asymptomatic individuals, the year of disease onset or death was known for 94% of directly ascertained individuals (68 of 72) and 55% of indirectly ascertained individuals (50 of 91). Year of disease onset or death occurred between 1989 and 2013 in 92% of all individuals for whom this variable was known (Figure 2B). This 25-year window was shorter than the typical difference between parent and child year of birth, which was 28 ± 6 years (mean ± SD, n = 150 including unaffected individuals), thus partially limiting ascertainment to parent-child pairs in which the child died at least 3 years younger than the parent. For individuals ascertained directly, year of birth and age of onset were even more strongly correlated than in our simulation (slope = −0.79, p < 0.0001, linear regression, Figure 2C, black line). The correlation weakened only slightly when indirectly ascertained individuals were included as well (slope = −0.48, p < 0.0001, linear regression, Figure 2C, dashed line).
Our simulation (see above) indicated that ascertainment bias could introduce false signals of heritability. We did not, however, observe any correlation between parent and child age of onset (p = 0.99, linear regression, n = 39 pairs) nor in sibling pair ages of onset (p = 0.63, linear regression, n = 26 pairs). Thus we do not find any signal, whether real or artifactual, that age of onset in this disease is heritable. This is not surprising because our simulation indicated that false signals of heritability are weaker than false signals of anticipation (Table 1), and the small size of our data set might also leave us underpowered to detect any true heritability of age of onset that might exist.
Strength of Anticipation Signal Depends upon Degree of Ascertainment Bias
Because signatures of ascertainment bias were strongly evident in our data, we expected to see a difference between parent and child age of onset in observed pairs. On the basis of our simulation results (Table 1), we hypothesized that the strength of the anticipation signal from naive paired t tests would vary depending on how flexible we were able to make our ascertainment window, within the constraints of our available data. When we considered only pairs in which both parent and child were ascertained directly, we observed 28 years of anticipation (p = 0.002, two-sided paired t test, n = 4 pairs). When we additionally included pairs with one indirectly ascertained individual, this figure dropped to 15 years (p = 0.0002, two-sided paired t test, n = 20 pairs). When we also included pairs in which both individuals were ascertained indirectly, this dropped further to 7 years (p = 0.005, two-sided paired t test, n = 39 pairs). Thus, the number of years of anticipation observed depends upon how narrow the ascertainment window is and the mechanism of case reporting.
The strength of the anticipation signal also differed by study center according to the extent of ascertainment bias in each individual data set. The UCSF data set, which was the largest and had both the highest percentage of indirectly ascertained individuals (73%) and the least correlation between year of birth and age of onset or death (slope = −0.41), showed the least evidence of anticipation (1 year, p = 0.68, two-sided paired t test). The MRC Prion Unit showed a marginal paired difference (7 years, p = 0.10, two-sided paired t test), whereas the Australian and German cohorts showed larger differences (18 years, p = 0.05 and 31 years, p = 0.03 respectively, two-sided paired t test).
Application of Survival Analysis to Glu200Lys Anticipation
The foregoing analysis indicated that our data exhibited a nominal signal of anticipation when analyzed with naive paired t tests. Because this signal depended upon the degree of ascertainment bias, and because our data exhibited signatures of ascertainment bias similar to what we had observed in our simulation, we suspected that the anticipation we observed was an artifact. We next set out to determine whether a survival analysis including asymptomatic individuals with the c.598G>A mutation would correct this artifact. Based on our simulation results (Figure 1E) and the fact that our ascertainment of asymptomatic individuals with the mutation was not exhaustive, we expected that survival analysis would not fully eliminate the signal of anticipation in our Glu200Lys data. For the survival analysis, we considered all parent-child pairs in which both individuals harbored the mutation, regardless of censored status. To ensure independence of tested pairs and avoid multiple counting, we randomly selected one such pair from each pedigree and compared parent and child survival curves. Across 1,000 iterations, parent and child survival curve medians differed by an average of 7 years (log rank test p < 0.05 in 82% of iterations). Because our simulation indicated that more thorough ascertainment of censored individuals reduces artifactual anticipation signals (Figure 1E), we repeated the same analysis on only the UCSF and MRC Prion Unit cohorts, which had the highest proportion of asymptomatic individuals with the mutation (Table 3). Within these cohorts, the median difference in parent and child survival curves was only 1 year and was usually not significant (p < 0.05 in only 8% of 1,000 iterations). This is consistent with the hypothesis that observed parent-child age of onset differences in Glu200Lys prion disease are due to incomplete ascertainment and not due to true anticipation.
Anticipation in Age at Death Due to Other, Non-Prion, Causes
The foregoing analyses suggested that the anticipation we observed in Glu200Lys families could be attributed entirely to ascertainment bias. We reasoned that if this were true, then anticipation in age of death might be observed in deaths attributable to other causes as well, with these unrelated deaths providing a kind of negative control. When we compared age at death for all parent-child pairs in our data set in which neither individual had CJD listed as cause of death, we observed 14 years of anticipation (p = 0.002, two-sided paired t test, n = 37 pairs). In many cases, the cause of death was not listed or was ambiguous, so we could not rule out the possibility that some of those individuals might actually have died of undiagnosed or unreported prion disease. When we therefore considered only those pairs in which each individual was known not to harbor the mutation (see Material and Methods) and/or died of a known cause clearly distinct from prion disease (cancer, heart attack, accident, etc.), the anticipation grew to 35 years (p = 0.001, two-sided paired t test, n = 9 pairs). This set included some individuals who had died much younger than the typical age of onset for Glu200Lys prion disease (Figure 2A), introducing a potential new source of ascertainment bias as individuals who die young might be counted as children but are unlikely to ever become parents.14 When we further filtered our data to include only individuals with age at death greater than 40, 16 years of anticipation were still observed (p = 0.002, two-sided paired t test, n = 5 pairs).
Discussion
The prediction of age of onset in those carrying mutations causal of neurodegenerative diseases is important for genetic counseling, clinical trial design,49,50 and understanding of fundamental disease mechanisms. We chose to investigate bias in a specific genetic prion disease as it has been reported12,13 that Glu200Lys genetic prion disease exhibits anticipation, with children succumbing to disease 7 to 14 years younger than their parents on average. We approached this claim with skepticism; the causal mutation is not a repeat expansion and human prion strains are not transmitted in childbirth or through casual contact between people,51–53 leaving no obvious mechanism for anticipation. Such a large decrease in age of onset in germline transmission ought to lead to juvenile onset cases within a few generations, as observed in repeat expansion disorders,15 yet no such cases are observed in Glu200Lys families (Figure 2A). In the present study, we considered the possibility that reported evidence of anticipation in Glu200Lys prion disease arises instead from ascertainment bias.
We tested for anticipation in our data using naive paired t tests, and we did observe a statistically significant difference between parent and child age of onset, but four separate lines of evidence argue that this difference is artifactual rather than biological. First, 92% of known years of onset or death for CJD-affected individuals in our data set fell within the 25-year window from the mutation’s discovery in 1989 to the time of our study in 2013, whereas parents were born on average 28 years earlier than their children. This enriches for pairs in which children die at least 3 years younger than their affected parent. Pairs in which children die older than their parents are much less likely to be observed during this time window. Second, we observed an artifactual correlation between year of birth and age of onset reflective of this “windowing” effect because individuals born later in time are only captured in this data set if they have earlier onset. The distribution of ages of onset is therefore shifted older for a parent’s year of birth (for instance, 1920) than for their child’s year of birth (for instance, 1950). Third, the strength of the anticipation signal we observed depended upon how strictly the data were limited by the ascertainment window. When we required that both parent and child had to be directly observed as patients by the study centers contributing data, we observed 28 years of anticipation. When all indirectly ascertained individuals were also included, this figure dropped to 7 years, and when we performed survival analysis on only the data from two centers most active in performing predictive genetic testing, we saw no difference in age of onset at all. Fourth, when using naive paired t tests, we observed anticipation even among individuals in our pedigrees who did not harbor the mutation and/or died of causes unrelated to prion disease.
None of the three previous studies reporting Glu200Lys anticipation12,13 (see also Web Resources) presented data on the distribution of year of onset, tested for correlation between year of birth and age of onset, or carried out a negative control by assessing anticipation in unrelated deaths as we have done here. One of these studies12 found a stronger anticipation (14 years rather than 7) when only directly genotyped individuals were included, similar to our finding that the strength of anticipation depends upon the duration of the ascertainment window. We showed that the stratification analyses, as performed in one study,13 do not remove the effects of ascertainment bias. Because anticipation was observed in certain analyses of our data as well, we consider it likely that the differences between our study and the previous studies arise from differences in methodology rather than differences in whether anticipation is truly present in the study population.
In assessing the evidence for anticipation in Glu200Lys prion disease, we have created a simulation model with relevance to other diseases as well. In contrast to previous studies of anticipation, we consider ascertainment to be limited by year of onset, not by age of onset per se. In general we find that ascertaining only those individuals with onset within a restricted time period is sufficient to create robust false signals of anticipation. This is of general concern because the genetic causes of many dominant Mendelian disorders were identified within the past 25 years, leaving a relatively narrow window for ascertainment. Our simulation shows that even high rates of retrospective ascertainment are unlikely to completely remove the resulting false signal of anticipation. Although certain statistical methods can help to correct for this bias when ascertainment criteria are consistent or data sets large enough to allow subsetting, we believe our own data may be typical of some rare diseases in representing a mixture of ascertainment modes, with both retrospective and prospective phenotyping present but nonexhaustive. For such data sets, we agree with the view54 that statistical tests alone might be inadequate to discriminate between situations with and without anticipation. We believe that one route toward preventing spurious claims of anticipation such as seen in Glu200Lys genetic prion disease might lie not in the requirement of more rigorous statistical tests but in an expectation of thorough and transparent assessment of a data set’s degree of bias. Toward that end, we propose that future studies’ reporting factors that determine age of onset in adult-onset dominant conditions should be expected to provide (1) a histogram of year of onset or death, (2) a test for correlation between year of birth and age of onset, (3) descriptive statistics on the extent of retrospective ascertainment and predictive genetic testing or prospective follow-up, and (4) a test for anticipation in deaths of other causes. Our results also have implications for study design in adult-onset genetic diseases; year of birth information should be collected, asymptomatic individuals with a mutation should be tracked, and retrospective ascertainment should be as thorough as possible. These measures will make it easier to quantify and reduce ascertainment bias.
Though heritability of age of onset was not observed in our Glu200Lys data, we noticed in our simulation that windowed ascertainment can also create a false signal that age of onset is heritable. This false signal could easily be disproven by including child year of birth as a covariate in parent-offspring regression. Our result suggests that estimates of the heritability of age of onset in genetic disease made based on parent-offspring regression ought to be scrutinized carefully and accepted only if the parent-child age of onset correlation remains after controlling for year of birth.
In summary, the phenomenon of anticipation previously reported in Glu200Lys prion disease is likely an artifact. Ascertainment bias is a pervasive problem in age of onset in genetic disease but can be reduced through appropriate data collection methods and recognized with simple analytical tools.
Acknowledgments
We would like to thank all affected individuals and their families for contributing to this research; physicians who referred or reported cases to surveillance and provided pertinent clinical, neuroradiological, and neuropathological data; the staff of the Goettingen research unit and the Center for Neuropathology and Prion Research at the Ludwig-Maximilian University Munich; the U.S. National Prion Disease Pathology and Surveillance Center for providing genotyping services; and Michael E. Talkowski, Benjamin M. Neale, Mark J. Daly, and Daniel G. MacArthur for their contributions to this study. The Australian National Creutzfeldt-Jakob Disease Registry is funded by the Commonwealth Department of Health. German CJD Surveillance is supported by grants from the Robert Koch Institute by funds of the Federal Ministry of Health (grant no 1369-341). MRC Prion Unit is funded by Medical Research Council UK; the UK clinical studies were supported in part by the National Institute for Health Research’s Biomedical Research Centre at UCLH. This work from UCSF was supported by NIH through NIH/NIA R01 AG-031189, K23 AG021989, P50AG023501, NIH/NCRR Grant Number UL1 RR024131, NIH/NIA AG021601, NIH/NINDS Contract N01-NS-0-2328, and Michael J. Homer Family Fund. E.V.M. received no specific funding for this work and volunteers his time on behalf of Prion Alliance. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. M.D.G. has served as a consultant for Lundbeck, MedaCorp, The Council of Advisors, and Neurophage.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Mitrova, E. (2012). The role of anticipation in the incidence of CJD with the mutation E200K. Presented at the CJD Family Foundation Conference, Washington, D.C., July 14, 2012. Archived on August 5, 2014: http://web.archive.org/web/20140805095200/http://www.cjdfoundation.org/shared/cjdfoundation/files/webfm/admin/2012-conference/Eva-Mitrova.pdf
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
U.S. Social Security Administration 2009 Life Tables. Archived on June 2, 2014: http://web.archive.org/web/20140602090427/http://www.ssa.gov/oact/STATS/table4c6.html
Source code and output for this study: https://github.com/ericminikel/e200k-anticipation
References
- 1.Prusiner S.B. Prions. Proc. Natl. Acad. Sci. USA. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Holman R.C., Belay E.D., Christensen K.Y., Maddox R.A., Minino A.M., Folkema A.M., Haberling D.L., Hammett T.A., Kochanek K.D., Sejvar J.J., Schonberger L.B. Human prion diseases in the United States. PLoS ONE. 2010;5:e8521. doi: 10.1371/journal.pone.0008521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kong Q., Surewicz W.K., Petersen R.B., Chen S.G., Gambetti P., Parchi P., Capellari S., Goldfarb L., Montagna P., Lugaresi E. Prion Biology and Diseases. Cold Spring Harbor Laboratory Press; 2004. Inherited Prion Diseases. [Google Scholar]
- 4.Goldgaber D., Goldfarb L.G., Brown P., Asher D.M., Brown W.T., Lin S., Teener J.W., Feinstone S.M., Rubenstein R., Kascsak R.J. Mutations in familial Creutzfeldt-Jakob disease and Gerstmann-Sträussler-Scheinker’s syndrome. Exp. Neurol. 1989;106:204–206. doi: 10.1016/0014-4886(89)90095-2. [DOI] [PubMed] [Google Scholar]
- 5.Goldfarb L.G., Korczyn A.D., Brown P., Chapman J., Gajdusek D.C. Mutation in codon 200 of scrapie amyloid precursor gene linked to Creutzfeldt-Jakob disease in Sephardic Jews of Libyan and non-Libyan origin. Lancet. 1990;336:637–638. doi: 10.1016/0140-6736(90)93443-s. [DOI] [PubMed] [Google Scholar]
- 6.Goldfarb L.G., Mitrová E., Brown P., Toh B.K., Gajdusek D.C. Mutation in codon 200 of scrapie amyloid protein gene in two clusters of Creutzfeldt-Jakob disease in Slovakia. Lancet. 1990;336:514–515. doi: 10.1016/0140-6736(90)92073-q. [DOI] [PubMed] [Google Scholar]
- 7.Hsiao K., Meiner Z., Kahana E., Cass C., Kahana I., Avrahami D., Scarlato G., Abramsky O., Prusiner S.B., Gabizon R. Mutation of the prion protein in Libyan Jews with Creutzfeldt-Jakob disease. N. Engl. J. Med. 1991;324:1091–1097. doi: 10.1056/NEJM199104183241604. [DOI] [PubMed] [Google Scholar]
- 8.Chapman J., Ben-Israel J., Goldhammer Y., Korczyn A.D. The risk of developing Creutzfeldt-Jakob disease in subjects with the PRNP gene codon 200 point mutation. Neurology. 1994;44:1683–1686. doi: 10.1212/wnl.44.9.1683. [DOI] [PubMed] [Google Scholar]
- 9.Spudich S., Mastrianni J.A., Wrensch M., Gabizon R., Meiner Z., Kahana I., Rosenmann H., Kahana E., Prusiner S.B. Complete penetrance of Creutzfeldt-Jakob disease in Libyan Jews carrying the E200K mutation in the prion protein gene. Mol. Med. 1995;1:607–613. [PMC free article] [PubMed] [Google Scholar]
- 10.Schelzke G., Kretzschmar H.A., Zerr I. Clinical aspects of common genetic Creutzfeldt-Jakob disease. Eur. J. Epidemiol. 2012;27:147–149. doi: 10.1007/s10654-012-9660-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pocchiari M., Puopolo M., Croes E.A., Budka H., Gelpi E., Collins S., Lewis V., Sutcliffe T., Guilivi A., Delasnerie-Laupretre N. Predictors of survival in sporadic Creutzfeldt-Jakob disease and other human transmissible spongiform encephalopathies. Brain. 2004;127:2348–2359. doi: 10.1093/brain/awh249. [DOI] [PubMed] [Google Scholar]
- 12.Rosenmann H., Kahana E., Korczyn A.D., Kahana I., Chapman J., Gabizon R. Preliminary evidence for anticipation in genetic E200K Creutzfeldt-Jakob disease. Neurology. 1999;53:1328–1329. doi: 10.1212/wnl.53.6.1328. [DOI] [PubMed] [Google Scholar]
- 13.Pocchiari M., Poleggi A., Puopolo M., D’Alessandro M., Tiple D., Ladogana A. Age at Death of Creutzfeldt-Jakob disease in subsequent family generation carrying the E200K mutation of the prion protein gene. PLoS ONE. 2013;8:e60376. doi: 10.1371/journal.pone.0060376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Penrose L.S. The problem of anticipation in pedigrees of dystrophia myotonica. Ann. Eugen. 1948;14:125–132. doi: 10.1111/j.1469-1809.1947.tb02384.x. [DOI] [PubMed] [Google Scholar]
- 15.Orr H.T., Zoghbi H.Y. Trinucleotide repeat disorders. Annu. Rev. Neurosci. 2007;30:575–621. doi: 10.1146/annurev.neuro.29.051605.113042. [DOI] [PubMed] [Google Scholar]
- 16.La Spada A.R., Taylor J.P. Repeat expansion disease: progress and puzzles in disease pathogenesis. Nat. Rev. Genet. 2010;11:247–258. doi: 10.1038/nrg2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vulliamy T., Marrone A., Szydlo R., Walne A., Mason P.J., Dokal I. Disease anticipation is associated with progressive telomere shortening in families with dyskeratosis congenita due to mutations in TERC. Nat. Genet. 2004;36:447–449. doi: 10.1038/ng1346. [DOI] [PubMed] [Google Scholar]
- 18.Martinez-Delgado B., Yanowsky K., Inglada-Perez L., Domingo S., Urioste M., Osorio A., Benitez J. Genetic anticipation is associated with telomere shortening in hereditary breast cancer. PLoS Genet. 2011;7:e1002182. doi: 10.1371/journal.pgen.1002182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gajdusek D.C. Unconventional viruses and the origin and disappearance of kuru. Science. 1977;197:943–960. doi: 10.1126/science.142303. [DOI] [PubMed] [Google Scholar]
- 20.Brown P., Preece M.A., Will R.G. “Friendly fire” in medicine: hormones, homografts, and Creutzfeldt-Jakob disease. Lancet. 1992;340:24–27. doi: 10.1016/0140-6736(92)92431-e. [DOI] [PubMed] [Google Scholar]
- 21.Hewitt P.E., Llewelyn C.A., Mackenzie J., Will R.G. Creutzfeldt-Jakob disease and blood transfusion: results of the UK Transfusion Medicine Epidemiological Review study. Vox Sang. 2006;91:221–230. doi: 10.1111/j.1423-0410.2006.00833.x. [DOI] [PubMed] [Google Scholar]
- 22.Heiman G.A., Hodge S.E., Wickramaratne P., Hsu H. Age-at-interview bias in anticipation studies: computer simulations and an example with panic disorder. Psychiatr. Genet. 1996;6:61–66. doi: 10.1097/00041444-199622000-00005. [DOI] [PubMed] [Google Scholar]
- 23.Vieland V.J., Huang J. Statistical evaluation of age-at-onset anticipation: a new test and evaluation of its behavior in realistic applications. Am. J. Hum. Genet. 1998;62:1212–1227. doi: 10.1086/301823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsai W.Y., Heiman G.A., Hodge S.E. New simple tests for age-at-onset anticipation: application to panic disorder. Genet. Epidemiol. 2005;28:256–260. doi: 10.1002/gepi.20057. [DOI] [PubMed] [Google Scholar]
- 25.Boonstra P.S., Gruber S.B., Raymond V.M., Huang S.-C., Timshel S., Nilbert M., Mukherjee B. A review of statistical methods for testing genetic anticipation: looking for an answer in Lynch syndrome. Genet. Epidemiol. 2010;34:756–768. doi: 10.1002/gepi.20534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rabinowitz D., Yang Q. Testing for age-at-onset anticipation with affected parent-child pairs. Biometrics. 1999;55:834–838. doi: 10.1111/j.0006-341x.1999.00834.x. [DOI] [PubMed] [Google Scholar]
- 27.Collins S., Law M.G., Fletcher A., Boyd A., Kaldor J., Masters C.L. Surgical treatment and risk of sporadic Creutzfeldt-Jakob disease: a case-control study. Lancet. 1999;353:693–697. doi: 10.1016/s0140-6736(98)08138-0. [DOI] [PubMed] [Google Scholar]
- 28.Collins S., Boyd A., Lee J.S., Lewis V., Fletcher A., McLean C.A., Law M., Kaldor J., Smith M.J., Masters C.L. Creutzfeldt-Jakob disease in Australia 1970-1999. Neurology. 2002;59:1365–1371. doi: 10.1212/01.wnl.0000031793.11602.8c. [DOI] [PubMed] [Google Scholar]
- 29.Windl O., Giese A., Schulz-Schaeffer W., Zerr I., Skworc K., Arendt S., Oberdieck C., Bodemer M., Poser S., Kretzschmar H.A. Molecular genetics of human prion diseases in Germany. Hum. Genet. 1999;105:244–252. doi: 10.1007/s004399900124. [DOI] [PubMed] [Google Scholar]
- 30.Grasbon-Frodl E., Lorenz H., Mann U., Nitsch R.M., Windl O., Kretzschmar H.A. Loss of glycosylation associated with the T183A mutation in human prion disease. Acta Neuropathol. 2004;108:476–484. doi: 10.1007/s00401-004-0913-4. [DOI] [PubMed] [Google Scholar]
- 31.Zerr I., Kallenberg K., Summers D.M., Romero C., Taratuto A., Heinemann U., Breithaupt M., Varges D., Meissner B., Ladogana A. Updated clinical diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Brain. 2009;132:2659–2668. doi: 10.1093/brain/awp191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beck J.A., Poulter M., Campbell T.A., Adamson G., Uphill J.B., Guerreiro R., Jackson G.S., Stevens J.C., Manji H., Collinge J., Mead S. PRNP allelic series from 19 years of prion protein gene sequencing at the MRC Prion Unit. Hum. Mutat. 2010;31:E1551–E1563. doi: 10.1002/humu.21281. [DOI] [PubMed] [Google Scholar]
- 33.Geschwind M.D., Haman A., Miller B.L. Rapidly progressive dementia. Neurol. Clin. 2007;25:783–807. doi: 10.1016/j.ncl.2007.04.001. vii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Geschwind M.D., Shu H., Haman A., Sejvar J.J., Miller B.L. Rapidly progressive dementia. Ann. Neurol. 2008;64:97–108. doi: 10.1002/ana.21430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paterson R.W., Torres-Chae C.C., Kuo A.L., Ando T., Nguyen E.A., Wong K., DeArmond S.J., Haman A., Garcia P., Johnson D.Y. Differential diagnosis of Jakob-Creutzfeldt disease. Arch. Neurol. 2012;69:1578–1582. doi: 10.1001/2013.jamaneurol.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Parchi P., Giese A., Capellari S., Brown P., Schulz-Schaeffer W., Windl O., Zerr I., Budka H., Kopp N., Piccardo P. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann. Neurol. 1999;46:224–233. [PubMed] [Google Scholar]
- 37.Rabinovici G.D., Wang P.N., Levin J., Cook L., Pravdin M., Davis J., DeArmond S.J., Barbaro N.M., Martindale J., Miller B.L., Geschwind M.D. First symptom in sporadic Creutzfeldt-Jakob disease. Neurology. 2006;66:286–287. doi: 10.1212/01.wnl.0000196440.00297.67. [DOI] [PubMed] [Google Scholar]
- 38.Squitieri F., Sabbadini G., Mandich P., Gellera C., Di Maria E., Bellone E., Castellotti B., Nargi E., de Grazia U., Frontali M., Novelletto A. Family and molecular data for a fine analysis of age at onset in Huntington disease. Am. J. Med. Genet. 2000;95:366–373. doi: 10.1002/1096-8628(20001211)95:4<366::aid-ajmg13>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 39.Visscher P.M., Hill W.G., Wray N.R. Heritability in the genomics era—concepts and misconceptions. Nat. Rev. Genet. 2008;9:255–266. doi: 10.1038/nrg2322. [DOI] [PubMed] [Google Scholar]
- 40.Owen J., Beck J., Campbell T., Adamson G., Gorham M., Thompson A., Smithson S., Rosser E., Rudge P., Collinge J. Predictive testing for inherited prion disease: report of 22 years experience. Eur. J. Hum. Genet. 2014 doi: 10.1038/ejhg.2014.42. Published online April 9, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goizet C., Lesca G., Dürr A., French Group for Presymptomatic Testing in Neurogenetic Disorders Presymptomatic testing in Huntington’s disease and autosomal dominant cerebellar ataxias. Neurology. 2002;59:1330–1336. doi: 10.1212/01.wnl.0000032255.75650.c2. [DOI] [PubMed] [Google Scholar]
- 42.Laccone F., Engel U., Holinski-Feder E., Weigell-Weber M., Marczinek K., Nolte D., Morris-Rosendahl D.J., Zühlke C., Fuchs K., Weirich-Schwaiger H. DNA analysis of Huntington’s disease: five years of experience in Germany, Austria, and Switzerland. Neurology. 1999;53:801–806. doi: 10.1212/wnl.53.4.801. [DOI] [PubMed] [Google Scholar]
- 43.Harper P.S., Lim C., Craufurd D. Ten years of presymptomatic testing for Huntington’s disease: the experience of the UK Huntington’s Disease Prediction Consortium. J. Med. Genet. 2000;37:567–571. doi: 10.1136/jmg.37.8.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tassicker R.J., Marshall P.K., Liebeck T.A., Keville M.A., Singaram B.M., Richards F.H. Predictive and pre-natal testing for Huntington Disease in Australia: results and challenges encountered during a 10-year period (1994-2003) Clin. Genet. 2006;70:480–489. doi: 10.1111/j.1399-0004.2006.00701.x. [DOI] [PubMed] [Google Scholar]
- 45.Morrison P.J., Harding-Lester S., Bradley A. Uptake of Huntington disease predictive testing in a complete population. Clin. Genet. 2011;80:281–286. doi: 10.1111/j.1399-0004.2010.01538.x. [DOI] [PubMed] [Google Scholar]
- 46.Baker H.E., Poulter M., Crow T.J., Frith C.D., Lofthouse R., Ridley R.M. Aminoacid polymorphism in human prion protein and age at death in inherited prion disease. Lancet. 1991;337:1286. doi: 10.1016/0140-6736(91)92953-y. [DOI] [PubMed] [Google Scholar]
- 47.Kovács G.G., Puopolo M., Ladogana A., Pocchiari M., Budka H., van Duijn C., Collins S.J., Boyd A., Giulivi A., Coulthart M., EUROCJD Genetic prion disease: the EUROCJD experience. Hum. Genet. 2005;118:166–174. doi: 10.1007/s00439-005-0020-1. [DOI] [PubMed] [Google Scholar]
- 48.Gabizon R., Rosenmann H., Meiner Z., Kahana I., Kahana E., Shugart Y., Ott J., Prusiner S.B. Mutation and polymorphism of the prion protein gene in Libyan Jews with Creutzfeldt-Jakob disease (CJD) Am. J. Hum. Genet. 1993;53:828–835. [PMC free article] [PubMed] [Google Scholar]
- 49.Ryman D.C., Acosta-Baena N., Aisen P.S., Bird T., Danek A., Fox N.C., Goate A., Frommelt P., Ghetti B., Langbaum J.B.S., Dominantly Inherited Alzheimer Network Symptom onset in autosomal dominant Alzheimer disease: a systematic review and meta-analysis. Neurology. 2014;83:253–260. doi: 10.1212/WNL.0000000000000596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bateman R.J., Xiong C., Benzinger T.L.S., Fagan A.M., Goate A., Fox N.C., Marcus D.S., Cairns N.J., Xie X., Blazey T.M., Dominantly Inherited Alzheimer Network Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med. 2012;367:795–804. doi: 10.1056/NEJMoa1202753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Belay E.D. Transmissible spongiform encephalopathies in humans. Annu. Rev. Microbiol. 1999;53:283–314. doi: 10.1146/annurev.micro.53.1.283. [DOI] [PubMed] [Google Scholar]
- 52.Xiao X., Miravalle L., Yuan J., McGeehan J., Dong Z., Wyza R., MacLennan G.T., Golichowski A.M., Kneale G., King N. Failure to detect the presence of prions in the uterine and gestational tissues from a Gravida with Creutzfeldt-Jakob disease. Am. J. Pathol. 2009;174:1602–1608. doi: 10.2353/ajpath.2009.081045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Murray K., Peters J., Stellitano L., Winstone A.M., Verity C., Will R.G. Is there evidence of vertical transmission of variant Creutzfeldt-Jakob disease? J. Neurol. Neurosurg. Psychiatry. 2011;82:729–731. doi: 10.1136/jnnp.2009.172148. [DOI] [PubMed] [Google Scholar]
- 54.Hodge S.E., Wickramaratne P. Statistical pitfalls in detecting age-of-onset anticipation: the role of correlation in studying anticipation and detecting ascertainment bias. Psychiatr. Genet. 1995;5:43–47. doi: 10.1097/00041444-199521000-00007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.