

Abstract
Whole-genome and exome data sets continue to be produced at a frenetic pace, resulting in massively large catalogs of human genomic variation. However, a clear picture of the characteristics and patterns of neutral and deleterious variation within and between populations has yet to emerge, given that recent large-scale sequencing studies have often emphasized different aspects of the data and sometimes appear to have conflicting conclusions. Here, we comprehensively studied characteristics of protein-coding variation in high-coverage exome sequence data from 6,515 European American (EA) and African American (AA) individuals. We developed an unbiased approach to identify putatively deleterious variants and investigated patterns of neutral and deleterious single-nucleotide variants and alleles between individuals and populations. We show that there are substantial differences in the composition of genotypes between EA and AA populations and that small but statistically significant differences exist in the average number of deleterious alleles carried by EA and AA individuals. Furthermore, we performed extensive simulations to delineate the temporal dynamics of deleterious alleles for a broad range of demographic models and use these data to inform the interpretation of empirical patterns of deleterious variation. Finally, we illustrate that the effects of demographic perturbations, such as bottlenecks and expansions, often manifest in opposing patterns of neutral and deleterious variation depending on whether the focus is on populations or individuals. Our results clarify seemingly disparate empirical characteristics of protein-coding variation and provide substantial insights into how natural selection and demographic history have patterned neutral and deleterious variation within and between populations.
Introduction
Mutations impose a substantial burden on fitness, disease, and longevity through the introduction of deleterious alleles into the population.1–5 A deeper understanding of deleterious variation in humans will have profound implications for disease-mapping studies, personal genomics, and predictive medicine. A considerable amount of theoretical work has been done to inform the dynamics of deleterious variation across a range of demographic models.6–8 Moreover, a large number of empirical studies in humans have been performed to survey patterns of deleterious variation within and between populations.9–13 For example, in a study of 15 African American (AA) and 20 European American (EA) individuals, Lohmueller et al.10 found that the European sample had an excess of putatively deleterious variants and through simulations demonstrated that this was most likely a consequence of the Out-of-Africa bottleneck. The proportional increase in deleterious variation in European versus African populations has also been observed in other studies.8,14–16 Furthermore, Casals et al.17 showed that recent founder effects in the French Canadian Quebec population have led to different patterns of deleterious variation between it and the French Canadian population. Moreover, in a clever study, Szpiech et al.18 found that deleterious alleles were enriched in runs of homozygosity and that variable levels of inbreeding can influence patterns of deleterious variation across populations.
However, not all studies have found a clear relationship between demographic history and empirical patterns of deleterious variation. For example, Tennessen et al.12 noted that characteristics of deleterious variants in EA and AA individuals are sensitive to how deleterious sites are defined. In addition, through detailed simulations and analyses of derived allele frequency (DAF) in a large exome sequencing data set, Simons et al.11 suggested that the deleterious-mutation load is insensitive to recent population history and that the average number of derived alleles per individual at putatively deleterious sites is not significantly different across populations. Similarly, Do et al.19 have recently argued that there are no differences in the per-genome accumulation of deleterious alleles across diverse human populations, which appears to contradict previous claims of differences in the proportion of deleterious variants across populations.10,14,15
Thus, despite the substantial amount of work that has been devoted to documenting and interpreting patterns of neutral and deleterious protein-coding variation in humans, a number of outstanding questions remain. Here, we describe a comprehensive analysis of protein-coding variation in a previously described high-coverage exome sequence data set consisting of 6,515 individuals of European and African ancestry and generated as part of the NHLBI Exome Sequencing Project (ESP).14 Furthermore, we performed extensive simulations of neutral and deleterious variation to help interpret empirical patterns of protein-coding variation. We show that many seemingly disparate observations of neutral and deleterious variation can be accounted for by opposing variation patterns that manifest depending on how variation is summarized and whether the focus is on individuals or populations. Our empirical and simulation results provide insight into how natural selection and demographic history have interacted to influence neutral and deleterious variation within and between populations.
Material and Methods
Analysis of Empirical Data
Analysis of Samples and Exome Sequencing Data
We analyzed the exomes of 6,515 individuals, including 4,298 EA individuals (1,879 males and 2,419 females) and 2,217 AA individuals (582 males and 1,635 females), from the NHLBI ESP.14 Exome data were subjected to standard quality-control filters as previously described.14 We further removed sites whose ancestral inference was inferred with low confidence in the six primate EPO (Enredo, Pecan, Ortheus) alignments.20 The final data set consisted of 1,110,148 single-nucleotide variants (SNVs) in autosomes and X chromosomes.
To avoid biases caused by different sample sizes, for all population-level analyses, such as estimating the site-frequency spectrum (SFS), we randomly sampled 2,217 EA individuals to match the sample size in AA individuals. For all individual-level analyses, we defined SNVs in individuals as sites that are heterozygous or homozygous for the derived allele. We compared the per-individual number of SNVs, heterozygotes, derived homozygotes, and derived alleles between EA and AA individuals with Mann-Whitney tests. Furthermore, to account for heterogeneity in missing data among individuals, we normalized the per-individual number of derived alleles by the per-individual number of total alleles that passed filtering criteria (see Fu et al.14).
We also used an alternative method to account for misidentification of ancestral states. Specifically, we identified the putative ancestral state of each SNV by comparing it to the chimpanzee genome (panTro2), and we corrected ancestral misidentification by using a context-dependent mutation model.21 In brief, this method accounts for the probability of misidentifying the ancestral state of a SNV by modeling the observed number of derived alleles (or derived homozygotes) as a mixture of SNVs whose ancestral states were correctly identified and those that were misidentified under the context-dependent substitution process.22 In total, 1,148,406 SNVs were used in these analyses.
Moreover, we used Fisher’s exact test to compare the average number of derived alleles per individual as a function of allele frequency between deleterious variants and neutral variants within populations, as well as between EA and AA populations for deleterious variants. For example, in the comparison of the enrichment of deleterious rare variants (DAF < 0.05%) in one population, the elements of the 2 × 2 table consisted of the average per-individual number of derived alleles of rare variants (DAF < 0.05%) and of variants with other frequency (DAF ≥ 0.05%) for both the deleterious and neutral sites.
Definition of Deleterious Variants
Quantifying evolutionary constraint through sequence conservation is widely used for identifying genomic regions that have been subject to purifying selection.23,24 We used PhyloP scores25 to identify putatively deleterious variants. PhyloP scores were calculated from 36 eutherian-mammal EPO alignments downloaded from the Ensembl Genome Browser (release 70) in enhanced metafile format (emf). These emf alignments were converted to multiple alignment format (maf) with the script “emf2maf.pl,” also downloaded from Ensembl. Alignment blocks in maf were then sorted with the mafTools package. Finally, sorted maf alignments were converted to SS format with the msa_view program in the PHAST package. To calculate scores, we ran PhyloP (PHAST package) with the following command line option: --msa-format SS --wig-scores --mode CONACC --method LRT.
The calculation of PhyloP scores also requires a neutral model of evolution. For this, we used the phylogenetic tree provided with the 36 mammalian alignments and the substitution-rate matrix and nucleotide frequencies from the placentalMammals.mod file downloaded from the UCSC Genome Browser. PhyloP scores in wiggle (wig) format were converted to bed files with the BEDOPS package.26 PhyloP scores were calculated with and without the human reference sequence (denoted as PhyloPH and PhyloPNH, respectively). Conditional on the 36-way eutherian-mammal phylogeny, simulations were performed with the base_evolve program in the PHAST package.
Population-Genetics Simulations
Forward Population Simulation for Protein-Coding Sequences
We performed forward population simulations with the program SFS_CODE27 under different demographic models and selective regimes. We considered three general demographic models, including a population bottleneck, recent accelerated growth, and a more complicated model, by using previously inferred parameters in the EA and AA samples.12 For the bottleneck model, a bottlenecked population was simulated from a constant population with effective size Ne = 10,000. This population experienced a bottleneck 50 ka ago, where the population size was reduced to 10% (a close approximation of the Out-of-Africa bottleneck)28 and 1% of the original size, and recovered from the bottleneck 25 ka ago. The Out-of-Africa bottleneck has also been modeled as a shorter and more severe bottleneck.29 In this model, a constant population (Ne = 10,000) experienced a bottleneck 118 ka ago and a quick recovery 108 ka ago. During the bottleneck, the population size was reduced to 7.57% of the original. We also simulated data under this model to study how robust our results are to particular implementations of the Out-of-Africa bottleneck.
For the model of recent population growth, a population started expanding from a constant population (Ne = 10,000) 5 ka ago. We considered different growth rates, including 0%, 2.0% (a close approximation for the population with European ancestry),12 and 3.0% per generation.
In the more realistic demographic model, European and African populations split 51 ka ago, and the European lineage incurred two bottleneck events (the Out-of-Africa bottleneck 51 ka ago and the split of non-African populations 23 ka ago) and an initial population expansion with a growth rate of 0.307% per generation, whereas the African population evolved as a constant population during this period.28 Beginning 5.115 ka ago, accelerated population growth occurred for both European and African populations with growth rates of 1.95% and 1.66%, respectively.12 The simulated AA population is a result of recent admixture from European (20%) and African (80%) populations.
A total of 2,500 individuals were simulated for each parameter combination. For each individual, we simulated 5,000 independent genes, each with four 500 bp exons that are equally spaced with 2,000 bp introns (sequences for the introns were not simulated). The mutation rate was set to 1.5 × 10−8 per base per generation, and the recombination rate was set to 10−8 per base per generation. Additive purifying selection was assumed to act on each nonsynonymous mutation. The selection coefficient |s| follows a gamma distribution Γ(α, β), where the mean selection coefficient for newly arisen deleterious mutations is 0.03 (i.e., ).30 Specifically, in bottleneck models and recent-growth models (Ne = 10,000), the shape parameter α and rate parameter β were set to 0.206 and 0.000343, respectively. In the complicated demographic models for EA and AA samples (Ne = 7,310), α and β were set to 0.206 and 0.00047, respectively. All of the simulations were based on the finite-site model. Mutations that were fixed during the burn-in period were not recorded.
An important measurement of population fitness is mutation load, defined as the proportion by which the population fitness is decreased by deleterious mutations.31–33 Under the assumption of no epistasis, linkage equilibrium, and additive fitness effects across sites, the total mutation load for multiple sites is the sum of mutation load across sites:
For each site l, wAA, wAD, and wDD are the fitness of different genotypes determined by the selection coefficient (s) and the dominance coefficient (h) (i.e., , , and ); the genotype frequency follows , , and under Hardy-Weinberg equilibrium, where p and q are the ancestral allele frequency and DAF, respectively. Thus, mutation load is determined by the number of deleterious variants carried by individuals, their effect size, and the model relating genotypes to fitness (i.e., the value of h). Unless otherwise noted, we considered an additive selection model (h = 0.5).
Results
Reconciling Disparate Empirical Patterns of SNVs and Alleles
We first investigated broad-scale patterns of protein-coding variation within and between populations in 6,515 individuals sequenced as part of the NHLBI ESP.14 Recently, Simons et al.11 reported that the total number of derived alleles per individual was remarkably similar between individuals of European and African ancestry. This observation is striking because, superficially, it appears to contradict other well-known differences in levels of diversity between African and European populations. For example, in the ESP data, nucleotide diversity is significantly higher in AA than in EA individuals (0.030% and 0.023%, respectively; bootstrap, p < 10−5). Similarly, it is well known that the average number of SNVs (defined here as sites that are heterozygous or homozygous for the derived allele) per individual is higher in individuals of African ancestry.9,34 Indeed, the average number of SNVs per individual is significantly greater in AA than in EA individuals (30,124.3 and 28,192.5, respectively; Mann-Whitney test, p < 10−15; Figure 1A) in the ESP data. However, the average number of SNVs per individual masks the underlying opposing patterns of heterozygous and homozygous genotypes between samples, as previously noted by Lohmueller et al.10 Specifically, the average number of heterozygous genotypes per individual is significantly higher in AA than in EA individuals (16,310.1 and 12,334.6, respectively; Mann-Whitney test, p < 10−15; Figure 1A), whereas the average number of derived homozygous genotypes per individual is higher in EA than in AA individuals (16,049.4 and 13,930.6, respectively; Mann-Whitney test, p < 10−15; Figure 1A).
Figure 1.
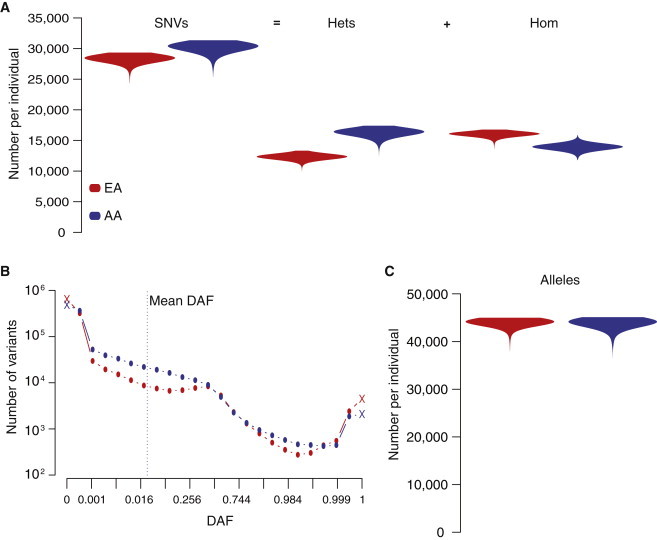
Patterns of Protein-Coding SNVs and Alleles among Populations and Individuals
(A) Violin plot of the number of SNVs per individual in EA and AA populations. Number of SNVs can be decomposed into whether individuals are heterozygous or homozygous for the derived allele.
(B) Modified SFS (see text) in EA and AA populations. The mean DAF (dashed line) in EA and AA populations is nearly identical.
(C) Violin plot of the number of derived alleles per individual in EA and AA populations. Note that the average number of derived alleles per individual is nearly identical in EA and AA populations.
The contrasting patterns of heterozygous and homozygous genotypes in individuals of European and African ancestry are primarily a consequence of the differences in demographic history, most notably the Out-of-Africa bottleneck, between populations. To illustrate this, we constructed a modified SFS of all variant sites, including sites where the derived allele is absent in one population and polymorphic in the other, as well as sites where the derived allele is fixed in one population and polymorphic in the other (Figure 1B). The modified SFS differs between EA and AA populations—the EA sample has more nearly fixed and fixed variants, and the AA sample contains more rare variants (Figure 1B). The modified SFS shows that the higher number of derived homozygous genotypes in EA individuals is largely due to the increased number of fixed or nearly fixed variants, as expected in bottlenecked populations because of stronger genetic drift. Similarly, the higher number of heterozygous genotypes in AA individuals is primarily attributable to the number of variants that had either been lost in EA individuals because of drift or arose after population splitting (Figure 1B). However, these two opposing patterns effectively cancel each other out such that the mean DAFs of the modified SFS in the EA and AA samples are nearly identical (0.02014 for EA and 0.02018 for AA populations). Thus, the total number of derived alleles per individual is approximately equal between EA and AA individuals (44,050.4 and 43,938.6, respectively; Mann-Whitney test, p > 0.05; Figure 1C).
In short, genetic diversity of protein-coding sequences is higher in AA than in EA individuals,35 and AA individuals have on average more SNVs than EA individuals; however, the total number of derived alleles per individual is nearly identical. The differences in empirical patterns of SNVs and derived alleles between EA and AA individuals reflect the unique demographic history of each population, which has influenced the SFS and composition of genotypes among individuals.
Unbiased Identification of Putatively Deleterious SNVs
Many different approaches have been developed for identifying putatively deleterious variation, particularly in protein-coding sequences.25,36–41 The most common functional-prediction methods were recently found to have a strong bias in identifying deleterious variants depending on whether the reference human genome sequence contains the ancestral or derived allele.11 In particular, the probability of calling a variant as deleterious is much lower for sites where the reference sequence carries the derived allele than for sites where the reference sequence carries the ancestral allele.
We hypothesized that because conservation is an integral component of nearly all functional-prediction methods, the strong reference bias is caused by assessing levels of conservation with alignments that contain the human reference sequence; thus, reference-derived sites appear to be less conserved. To test this hypothesis, we first simulated 1 Mb sequence alignments by conditioning on the 36-way eutherian-mammal phylogeny. We then calculated PhyloP with and without the simulated human sequence (denoted PhyloPH and PhyloPNH, respectively). The distribution of PhyloPH scores was significantly different between human ancestral and derived sites (Mann-Whitney test, p < 10-15; Figure 2A). However, this bias was completely absent in the PhyloPNH scores (Mann-Whitney test, p = 0.87; Figure 2A), and therefore removing the human sequence before calculating conservation is an effective strategy for mitigating reference-sequence bias.
Figure 2.
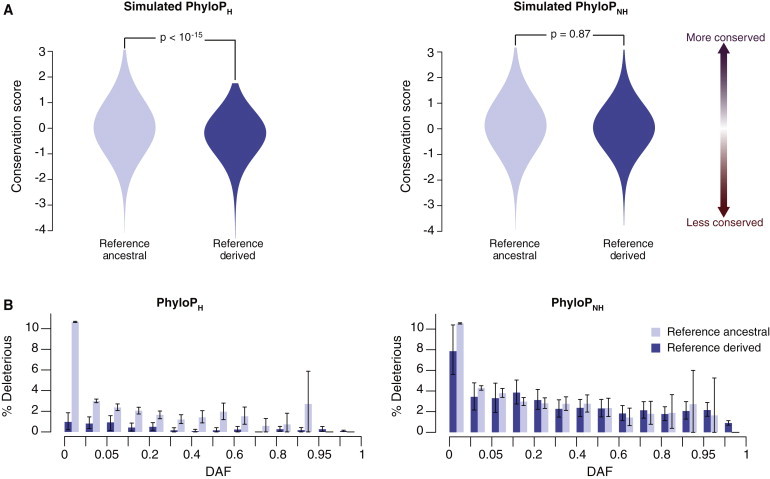
Identifying and Correcting Reference Bias of PhyloP
(A) Distribution of PhyloP scores calculated from sequence data that we simulated by conditioning on the branch lengths and topology of the 36-way eutherian-mammal phylogeny. PhyloPH (left) and PhyloPNH (right) were calculated on alignments including and excluding the human sequence, respectively. In each plot, the distribution of conservation scores is shown for ancestral and derived sites from the human reference sequence. Note that removing the simulated human sequence before calculating PhyloP mitigates the strong reference-bias effect.
(B) Proportion of deleterious variants in the observed data for reference ancestral and derived sites when conservation scores are calculated on alignments including (PhyloPH, left) or excluding (PhyloPNH, right) the human sequence as a function of DAF. Error bars indicate approximate 95% confidence intervals.
Next, we recalculated PhyloP scores in the real data by using the 36-way eutherian-mammal alignments with and without the human reference sequence. We defined deleterious variants as those with PhyloP scores exceeding the 90% percentile of the empirical distribution of all PhyloP scores. As expected, we observed a strong reference bias for PhyloPH, such that the proportion of variants called deleterious was markedly different for reference ancestral and derived sites as a function of DAF (Figure 2B). PhyloPNH largely eliminated this bias (Figure 2B), as predicted by the simulation results. All of the analyses described below were based on the 107,736 sites identified as putatively deleterious with the use of PhyloPNH, and the vast majority (85%) were nonsynonymous.
Patterns of Deleterious SNVs among Individuals and Populations
We next considered the average number of deleterious SNVs per individual in the EA and AA samples. Overall, patterns of deleterious SNVs among individuals followed the trends observed for all SNVs (Figure 3A). Specifically, AA individuals had on average more deleterious SNVs per individual than did EA individuals (686.2 and 643.8, respectively; Mann-Whitney test, p < 10−15; Figure 3A). Similarly, when deleterious SNVs were decomposed into genotypes, AA individuals had on average more heterozygous deleterious genotypes than did EA individuals (479.1 and 381.9, respectively; Mann-Whitney test, p < 10−15; Figure 3A), whereas EA individuals had on average more deleterious derived homozygous genotypes than did AA individuals (262.2 and 207.3, respectively; Mann-Whitney test, p < 10−15; Figure 3A).
Figure 3.
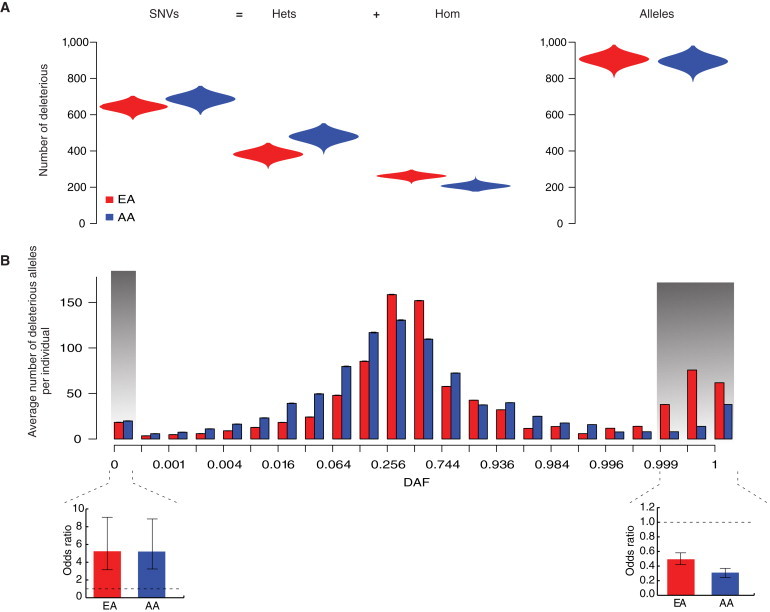
Empirical Patterns of Deleterious Protein-Coding Variants Carried by Individuals
(A) The average number of deleterious SNVs, heterozygous genotypes, homozygous genotypes, and derived alleles per individual. The average number of deleterious alleles per individual is small but significantly different between EA and AA individuals.
(B) The average number of deleterious alleles per individual in EA and AA samples as a function of population DAF. The inset bar plots compare the odds that a derived allele is deleterious to the odds that a derived allele is neutral per individual for variants with a DAF ≤ 0.05% (left) and ≥ 99.9% (right) in EA and AA individuals, respectively. Error bars denote the 95% confidence interval of the odds ratio. Note that on average, EA individuals carry significantly more nearly fixed or fixed deleterious alleles than do AA individuals (p = 8.63 × 10−16).
Small but Significant Increase in the Number of Deleterious Alleles in EA Individuals
Recently, Simons et al.11 studied patterns of deleterious variation in the ESP data and found that the mean frequency of derived deleterious alleles was not significantly different between EA and AA samples. We first replicated this observation and found no significant difference in the mean frequency of derived deleterious alleles between EA and AA individuals (0.00423 and 0.00419, respectively; t test, p = 0.82).
To explore patterns of deleterious alleles more directly, we next compared the distributions of the number of derived deleterious alleles per individual for the EA and AA samples (Figure 3A). We found that the average number of derived deleterious alleles was slightly but significantly higher in EA individuals than in AA individuals (905.7 and 893.2, respectively; Mann-Whitney test, p < 10−15). This result was robust to (1) different PhyloPNH thresholds used to define deleterious SNVs, (2) correction for heterogeneity in levels of missing data among individuals, (3) removal of hypermutable CpG sites, (4) recalculation of PhyloPNH by removal of SNVs within 10 bp of a gap in the eutherian-mammal alignment (to mitigate potential subtle biases caused by indels), and (5) sequence errors by removal of derived singletons. Furthermore, we also used a context-dependent mutation-rate model to correct for ancestral misidentification (see Material and Methods), which also resulted in small but statistically significant differences in the average number of derived deleterious alleles carried by EA and AA individuals (994.8 and 978.0, respectively; Mann-Whitney test, p < 10−15).
To help interpret this observation, we analyzed the average number of derived deleterious alleles per individual as a function of DAF (Figure 3B). Although most deleterious SNVs (and all SNVs) were rare in frequency at the population level, most deleterious SNVs (and all SNVs)12 carried by individuals were common (Figure 3B). Both EA and AA individuals had on average significantly more rare (DAF < 0.05%) deleterious alleles than putatively neutral alleles (Fisher’s exact test, p = 2.02 × 10−8 and 3.61 × 10−9 for EA and AA individuals, respectively; Figure 3B). Similarly, both EA and AA individuals had on average significantly fewer nearly fixed or fixed deleterious alleles (DAF ≥ 99.9%) than neutral alleles (Fisher’s exact test, p = 2.59 × 10−21 and 2.29 × 10−28 for EA and AA individuals, respectively; Figure 3B). However, on average, EA individuals had significantly more nearly fixed or fixed deleterious alleles than did AA individuals (Fisher’s exact test, p = 8.63 × 10−16; Figure 3B). Importantly, this small increase in the number of nearly fixed or fixed deleterious alleles was responsible for the higher average number of deleterious derived alleles in EA individuals.
Proportionally More Deleterious Variation in EA Individuals
Previously, it was observed that European populations possessed proportionally more deleterious SNVs than did African populations.10,34,42 However, this observation might have been influenced by the previously unrecognized reference bias in methods of predicting deleterious variants. To this end, we used PhyloPNH to calculate the proportion of deleterious SNVs (while correcting for sample size) in the EA and AA populations. Consistent with previous studies, the proportion of deleterious SNVs in EA individuals was significantly higher than that in AA individuals (9.36% and 8.28%, respectively, Fisher’s exact test, p = 1.06 × 10−85), and therefore this finding was not an artifact of reference bias. We also found a small but statistically significant increase in the average proportion of deleterious alleles per individual in the EA and AA samples (2.06% and 2.03%, respectively; Mann-Whitney test, p < 10−15).
Overview of Simulations
To better understand the empirical patterns of neutral and deleterious protein-coding variation described above, we performed extensive simulations. As above, all the variants that segregated in the combined simulated samples were included in the analysis. We first focused on demographic models with either population bottlenecks or recent accelerated growth to delineate how each of these processes influences patterns of deleterious variation. We then considered a more complicated and realistic model of human history that features both bottlenecks and recent growth. For each demographic model and parameter combination, we simulated 2,500 individuals, and for each individual we simulated 5,000 independent genes with an exonic length of 2 kb by using previously inferred parameters for the distribution of fitness effects of protein-coding variation30 and assuming an additive model of selection (see Material and Methods). We used the simulated data to investigate characteristics of present-day deleterious SNVs in individuals and populations, when they arose, and the distribution of their fitness effects. Unless otherwise noted, we considered variants in the simulations to be deleterious if their effect size was |s| ≥ 10−4 (i.e., 2Ne|s| >> 1) because effects less than this are effectively neutral.
Temporal Dynamics of Deleterious Alleles in Bottleneck Models
We simulated protein-coding variation in bottleneck models where population size decreased to 0.1× and 0.01× 50 ka ago and then recovered to prebottleneck levels 25 ka ago (Figure 4A). Note that the decrease in size to 0.1× closely approximates the estimated reduction in size during the Out-of-Africa bottleneck.28 We summarized levels of neutral and deleterious variation as the mean number of derived alleles per individual per kilobase and compared the density of mutations found in contemporary individuals in bottlenecked and constant-sized populations as a function of DAF, when they arose, and their selection coefficient (Figure 4).
Figure 4.
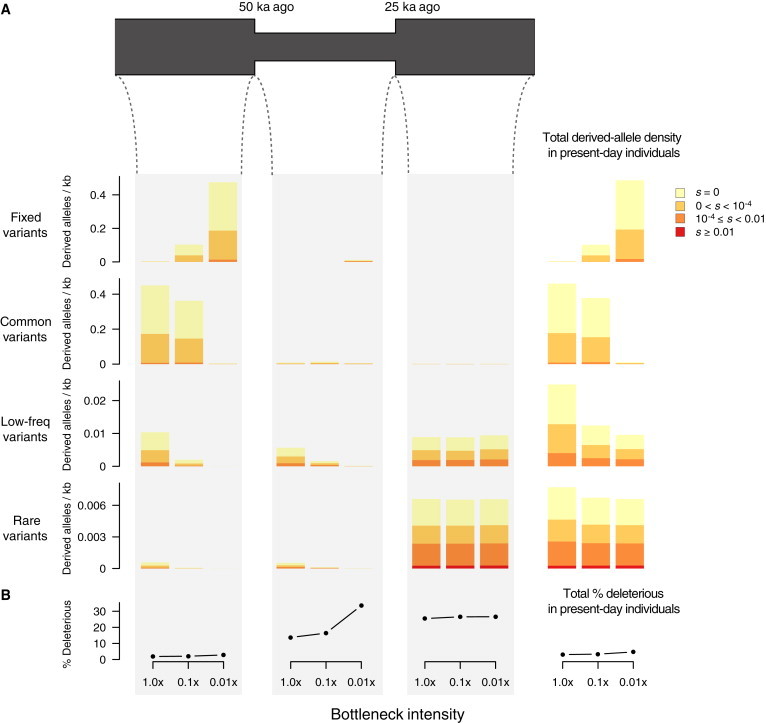
Temporal Decomposition of Neutral and Deleterious Variation among Present-Day Individuals in Bottleneck Models
(A) Diagram of the simulated bottleneck model, in which population size decreased to 0.1× or 0.01× 50 ka ago and then recovered to prebottleneck levels with a size of 10,000 25 ka ago. Bar plots on the far right show the average number of derived alleles per individual per kilobase in present-day individuals as a function of DAF (fixed DAF = 1, common DAF ≥ 0.05, low-frequency 0.01 ≤ DAF < 0.05, and rare DAF < 0.01) and selection coefficients. We decomposed variant density in present-day individuals according to when the mutation arose (before, during, or after the bottleneck), as shown in the bar plots under the demographic model. Thus, we can obtain variant density of present-day individuals for each DAF category (rows) by simply adding variant density across the three time epochs considered.
(B) The proportion of derived deleterious alleles (|s| ≥ 10−4) per individual in present-day individuals and as a function of when the mutation arose.
Several salient points emerge from Figure 4A, in which the density of protein-coding variants in contemporary individuals is decomposed into the time in which the variants arose. First, deleterious variants that occurred before or during the bottleneck constitute a small proportion of deleterious variants in the population (9.5% for constant-sized populations and 3.9% and 1.4% for 0.1× and 0.01× bottlenecked populations, respectively). However, almost all fixed and the majority of common deleterious variants in present-day individuals arose during this time period (Figure 4A), and as a result the number of derived deleterious alleles carried by these individuals was slightly higher than that carried by individuals from constant-sized populations (Figure 4A). For example, the mean density of derived deleterious alleles per individual in the bottleneck model with intensity 0.1× was 0.0167/kb, whereas in individuals from constant-sized populations, it was 0.0151/kb (1.1-fold increase). Second, although individuals from a bottlenecked population carried more fixed deleterious mutations, they were nearly always of weak effect (|s| ∼10−4; Figure 4A). Third, strongly deleterious (|s| > 0.01) mutations carried by present-day individuals always arose recently and were rare in the population (Figure 4A).
The proportion of deleterious alleles in individuals sampled from the current generation was modestly affected by bottlenecks (3.05% for individuals from constant-sized populations and 3.34% and 4.67% for 0.1× and 0.01× bottlenecked populations, respectively; Figure 4B). However, the relative accumulation of deleterious and neutral variants exhibited different patterns depending on when mutations arose. Specifically, deleterious variants that arose before the bottleneck tended to be lost because of drift, but those that survived tended to increase in frequency; as a result, the proportion of derived deleterious alleles per individual slightly increased for variants that arose in this period (Figure 4B). Deleterious variants that occurred during the bottleneck increased both in number and in frequency because of less efficient purging, so that proportionally more derived deleterious alleles arising in this epoch accumulated in present-day individuals (Figure 4B). In contrast, and as expected, the accumulation of derived deleterious alleles that arose after the bottleneck did not change as a function of bottleneck intensity (Figure 4B).
We also considered a different parameterization of the Out-of-Africa bottleneck.29 In this model, a constant population (Ne = 10,000) reduced its size to 7.57% 118 ka ago and recovered to the original population size after 10 ka. Patterns of neutral and deleterious variation in this bottleneck model with a quick recovery were similar to that described above. For instance, fewer than 3% of deleterious variants in the contemporary population arose before or during the bottleneck, but they comprised 47.8% of derived deleterious alleles carried by individuals and were of modest effect size (10−4 ≤ |s| < 0.01). The mean density (proportion) of derived deleterious alleles per individual in this bottleneck model was 0.0176/kb (3.29%), compared to 0.0158/kb (3.00%) in constant-sized populations.
Temporal Dynamics of Deleterious Alleles in Recent-Growth Models
To better understand how recent growth has influenced the burden of deleterious alleles among individuals, we simulated protein-coding variation in models where populations started expanding 5 ka ago (with growth rates of 2% or 3%) and compared patterns of neutral and deleterious derived alleles per individual to those of constant-size population models (Figure 5A). Note that a growth rate of 2% approximately corresponds to that estimated by Tennessen et al.12 for European populations. We also considered a higher growth rate because it is likely that recent studies have underestimated this parameter.43–46
Figure 5.
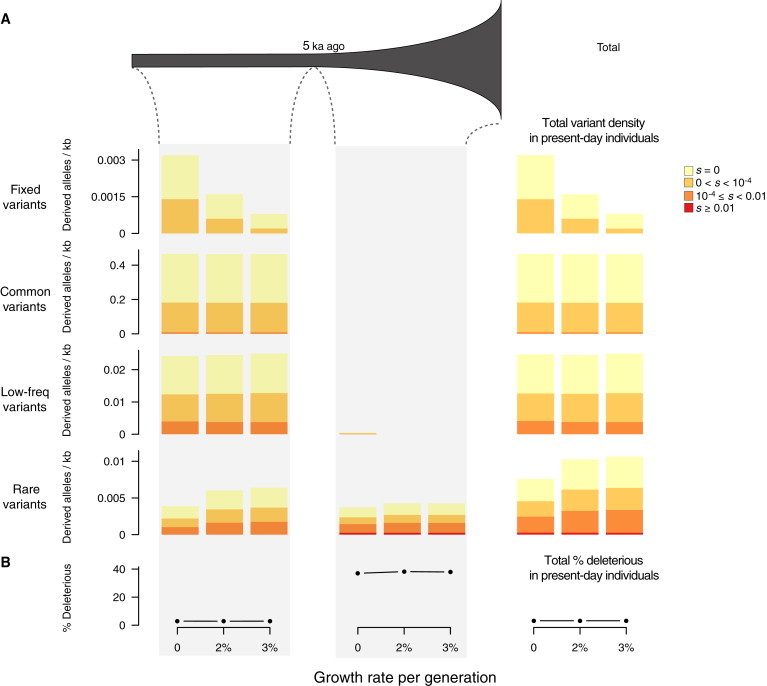
Temporal Decomposition of Neutral and Deleterious Variation among Present-Day Individuals in Recent-Growth Models
(A) Diagram of simulated models of recent accelerated growth. Here, populations started growing at a rate of 2% or 3% per generation from a constant population size of 10,000 individuals 5 ka ago. As in Figure 4, the far right column shows the mean number of derived alleles per individual per kilobase in present-day individuals as a function of DAF (fixed DAF = 1, common DAF ≥ 0.05, low-frequency 0.01 ≤ DAF < 0.05, and rare DAF < 0.01) and selection coefficients. Similarly, we decomposed variant density in present-day individuals according to when the mutation arose (before or during growth).
(B) The proportion of deleterious alleles (|s| ≥ 10−4) per individual in present-day individuals and as a function of when the mutation arose.
As expected, population expansions led to a significant skew toward rare variation in the population—61.3%, 85.4%, and 87.7% of all SNVs were rare in constant-size, 2% growth, and 3% growth models, respectively. In population-expansion models, a significant amount of rare variation arose during the growth epoch. At the level of individuals, however, recent population expansions had much more subtle consequences. For example, the average density of rare derived alleles per individual per kilobase in constant-size, 2% growth, and 3% growth models was 0.0076, 0.0103, and 0.0106, respectively (Figure 5A). Furthermore, the average density of rare deleterious (|s| ≥ 10−4) alleles per individual in constant-size, 2% growth, and 3% growth models was 0.0024, 0.0032, and 0.0034, respectively (Figure 5A). Note that in both constant-sized and growth models, nearly all strongly deleterious variants (|s| > 0.01) arose recently (Figure 5A).
The proportion of derived deleterious alleles per individual was slightly lower in expanding populations than in constant-sized populations (3.20%, 3.15%, and 3.18% for constant-sized, 2% growth, and 3% growth models, respectively; Figure 5B). Thus, as predicted by population-genetics theory,47 population growth allows deleterious alleles to be purged more efficiently. Deleterious mutations have a lower probability of fixation in an exponentially growing population than in a constant-sized population; also, most deleterious mutations will eventually be lost,48 predicting a proportional reduction of deleterious mutations at the individual level. However, for mutations that reach fixation, the expected fixation time increases with population size. Weakly deleterious mutations can segregate longer at lower frequency in the population and result in a proportional increase in deleterious variants at the population level. For example, in the simulated data, the proportion of deleterious variants in the population increased from 30.2% in populations with no growth to 36.4% and 35.8% in the 2% and 3% growth models, respectively, consistent with previous observations.49
We note that the patterns of deleterious variation among individuals and populations in our simulations did not change monotonously as a function of growth rate. This was most likely a consequence of sampling variation, given that the effects of the growth rates considered here (2% versus 3%) on patterns of variation were quantitatively similar in the short amount of time (∼200 generations) that elapsed since the onset of population growth.
Temporal Dynamics of Deleterious Alleles in Complicated Demographic Models
The results described above allowed us to isolate the effects of bottlenecks and population expansions on patterns of deleterious variation. Here, we consider the combined effects of bottlenecks, recent accelerated growth, population structure, and admixture (Figure 6A) by using previously inferred parameters for the EA and AA samples by Tennessen et al.12
Figure 6.
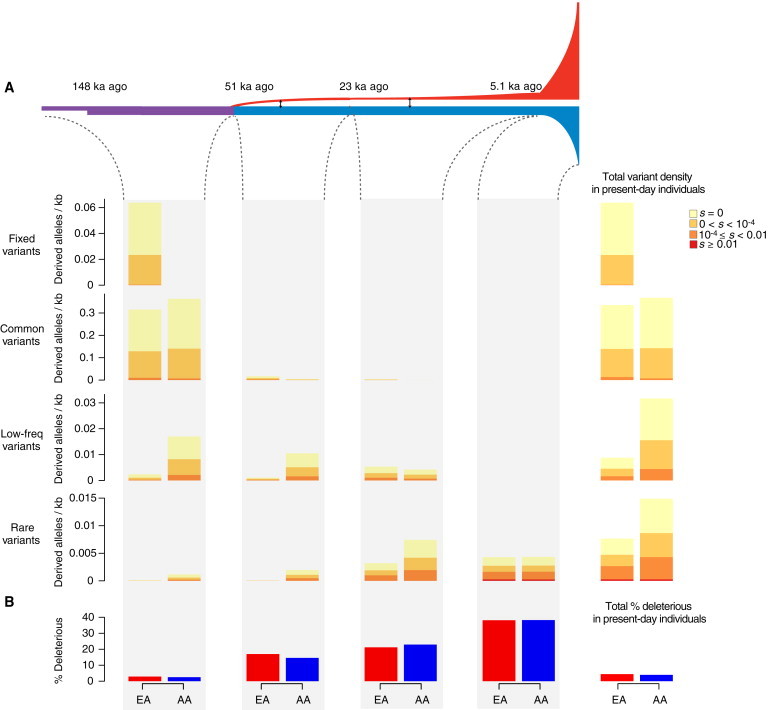
Temporal Decomposition of Neutral and Deleterious Variation among Present-Day Individuals in a More Realistic Demographic Model
(A) Diagram of a more realistic demographic model for EA and AA populations. This model involves multiple bottlenecks in the EA lineage, recent accelerated growth, and admixture as inferred by Tennessen et al.12 As in Figure 4, the far right column shows the mean number of derived alleles per individual per kilobase in present-day individuals as a function of DAF (fixed DAF = 1, common DAF ≥ 0.05, low-frequency 0.01 ≤ DAF < 0.05, and rare DAF < 0.01) and selection coefficients. Similarly, we decomposed variant density in present-day individuals according to when the mutation arose (before the Out-of-Africa bottleneck, during the bottleneck, during the initial period of growth in EA individuals, or during the recent accelerated growth in both EA and AA individuals).
(B) The proportion of deleterious alleles (|s| ≥ 10−4) per individual in present-day individuals and as a function of when the mutation arose.
On average, EA individuals carried more fixed and common deleterious alleles than did AA individuals (0.0140/kb and 0.0076/kb in EA and AA individuals, respectively; Figure 6A); all of them were of modest effect size (i.e., 10−4 ≤ |s| < 0.01), and over 95% arose ≥ 23 ka ago. In contrast, AA individuals carried on average more rare and low-frequency derived deleterious alleles than did EA individuals (0.0043/kb and 0.0087/kb in EA and AA individuals, respectively; Figure 6A) because of their larger effective population size between 5.1 and 51 ka ago. In both EA and AA individuals, all of the deleterious mutations with strong effect (|s| ≥ 0.01) were rare in the population, and the majority (96.1% in EA and 95.8% in AA) arose in the last 5.1 ka (Figure 6A), consistent with previous simulation results.50 Overall, EA individuals carried on average more deleterious alleles per individual than did AA individuals (0.0182/kb and 0.0164/kb in EA and AA individuals, respectively; Figure 6A), in agreement with empirical observations in the empirical data (Figure 3).
Consistent with the expected effects of bottlenecks and recent accelerated growth as described above, the proportion of derived deleterious alleles per individual exhibited differences between simulated EA and AA individuals in different time epochs (Figure 6B). Specifically, for variants that arose before or during the European bottlenecks, the proportion of derived deleterious alleles per individual was higher in EA than in AA individuals (Figure 6B). However, for variants that arose during recent accelerated population growth, the proportion of derived deleterious alleles per individual was slightly lower in EA than in AA individuals (Figure 6B). Overall, the average proportion of derived deleterious alleles in present-day individuals in the simulated data was 4.39% for EA individuals and 3.96% for AA individuals (Figure 6B).
Opposing Patterns of Variation in Populations and Individuals
The temporal decomposition of present-day variation into different time epochs (Figures 4, 5, and 6) intimates that patterns of deleterious variation manifest differently depending on whether the focus is on populations or individuals. To more clearly articulate this important point, we calculated the density and proportion of deleterious variants and derived alleles in the simulated data for populations and individuals.
At the population level, the density of deleterious variants in bottleneck models decreased from 1.01/kb in constant-size models to 0.97/kb and 0.94/kb in bottlenecks of intensity 0.1× and 0.01×, respectively (Figure 7A). However, in individuals, the density of derived deleterious alleles increased from 0.0151/kb in constant-size models to 0.0167/kb and 0.0240/kb in bottlenecks of intensity 0.1× and 0.01×, respectively (Figure 7B). Note that in the bottleneck model with a 0.1× decrease in size, this corresponds to approximately 10.2% more derived deleterious alleles per individual than in constant-size models. This increase was largely due to the bottleneck-mediated shift in the SFS toward higher allele frequencies and, in some cases, fixation.
Figure 7.
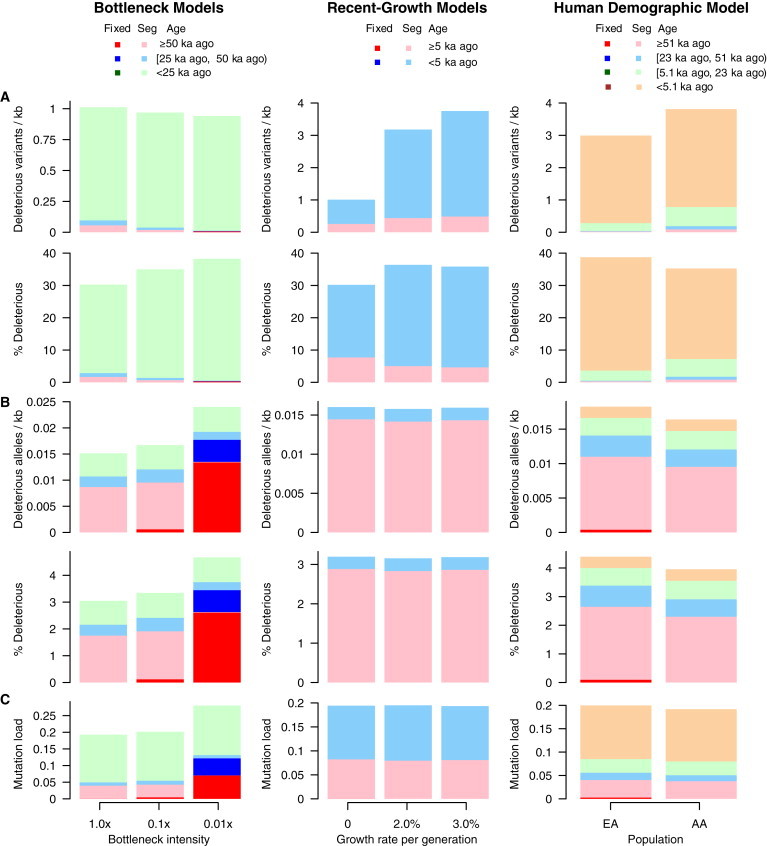
Opposing Patterns of Deleterious SNVs and Alleles in Individuals and Populations
(A) The number and proportion of deleterious variants per kilobase in populations as a function of mutation age and DAF.
(B) The mean number and proportion of derived deleterious alleles per individual per kilobase as a function of mutation age and DAF.
(C) The mutation load as a function of mutation age and DAF. Note that the Tennessen et al.12 model refers to the demographic model shown in Figure 6.
Variants are colored on the basis of whether they are fixed or segregating (“seg”) and when they arose (“age”).
Similarly, recent population growth led to opposing density patterns of derived deleterious alleles in populations and individuals. Recent growth led to a substantial increase in the density of deleterious variants in populations, largely because of SNVs arising during the period of recent growth (Figure 7A). For example, the density of deleterious variants in populations increased from 1.01/kb in constant-size models to 3.18/kb and 3.75/kb in models with 2% and 3% growth, respectively (Figure 7A). Conversely, the average density of derived deleterious alleles in individuals decreased slightly from 0.0160/kb in constant-sized populations to 0.0158/kb and 0.0159/kb in models with 2% and 3% growth, respectively (Figure 7B). In the 2% growth model, this corresponds to approximately 1.5% fewer deleterious alleles per individual than in constant-size models. These results are consistent with Gazave et al.,51 who found the same trend of growth decreasing the density of deleterious alleles per individual for variants with a selection coefficient of |s| ≥ 10−4.
We further compared the load of deleterious mutations in constant, bottleneck, and recent-growth models (Figure 7C) by assuming additive and independent effects among deleterious alleles. In the bottleneck model, load was modestly higher than in constant-size populations. For example, in bottlenecks with a 0.1× intensity (corresponding to the Out-of-Africa event), load was on average 4.5% higher than in constant-size populations. It is important to note that the accumulation of fixed deleterious variants contributed to 2.2% of mutation load in this model but only 0.1% in the constant-sized model. In models with a bottleneck intensity of 0.01×, fixed deleterious variants accounted for 43.2% of the mutation load. Thus, although the increase in load was modest, it had significant evolutionary implications, given that fixed variants cannot be purged from the population. In contrast, recent population expansions had negligible effects on load (Figure 7C), consistent with previous studies.11,51
Finally, for comparison, we also summarized the average density of deleterious variants and alleles at the population and individual levels, as well as mutation load, for the more realistic demographic model of EA and AA populations. As expected, for equal sample sizes, more variants were observed in the simulated AA population than in the EA population (Figure 7A). The average density of derived deleterious alleles in EA individuals was 0.0182/kb, which is approximately 11.3% higher than that for AA individuals (0.0164/kb; Figure 7B). Notably, 21.4% of the difference in the density of derived deleterious alleles was attributable to the increase in fixed deleterious mutations in EA individuals (Figure 7B). As a result, mutation load is predicted to be slightly inflated in EA individuals (20.0% in EA individuals versus 19.2% in AA individuals; Figure 7C).
Discussion
Recent large-scale sequencing studies have afforded unique insights into the spectrum of human genomic variation.9–12,19,45,51 However, these studies have emphasized different aspects of the data and, in some cases, appear to have come to conflicting conclusions, particularly for deleterious variants.10–12,19 To better understand empirical patterns of neutral and deleterious variation, we performed a comprehensive analysis of a large exome sequencing data set. Furthermore, we leveraged extensive simulations on the dynamics of neutral and deleterious alleles in nonequilibrium populations to facilitate interpretations of the exome data. Importantly, we developed an approach to mitigate the previously described strong reference-bias effects observed in nearly all methods of defining deleterious variation, which should be useful in a wide variety of applications.
Our empirical and simulation results show that characteristics of neutral and deleterious variation can result in opposing patterns depending on how variation is summarized. For example, the total number of SNVs in the AA population is significantly higher than that in the EA population, and AA individuals carry significantly more SNVs on average than do EA individuals. However, in EA individuals, the strongly skewed SFS due to population bottlenecks caused some SNVs to be lost and others to drift to higher frequency. Thus, the composition of genotypes in the EA and AA samples is considerably different (Figures 1 and 3)—EA individuals have on average more homozygous derived sites, and AA individuals have more heterozygous sites. Therefore, although there can be large differences in the average number of SNVs per individual between populations, the average number of derived alleles is approximately equal.
Furthermore, opposing patterns of variation can also manifest depending on whether the focus is on individuals or populations, which is the primary factor accounting for seemingly disparate empirical characteristics of protein-coding variation in recent studies. For instance, although recent accelerated growth has profoundly influenced protein-coding variation in populations, it has thus far had limited impact on individuals. For example, 80.8% and 73.4% of deleterious SNVs in the EA and AA populations, respectively, have a population DAF < 0.05%, but they comprise only 2.0% and 2.2% of derived deleterious alleles carried by EA and AA individuals, respectively. In contrast, although only 0.20% and 0.06% of deleterious variants are nearly fixed or fixed (DAF ≥ 99.9%) in the EA and AA populations, respectively, they comprise 19.4% and 6.7% of derived deleterious alleles carried by EA individuals and AA individuals, respectively. Thus, although most SNVs are rare in the population (i.e., have a DAF < 0.05%), most of the variants carried by individuals are common.
Our simulation results also show that population bottlenecks and expansions have opposing effects on patterns of variation in populations and individuals. Compared to populations of constant size, bottlenecks reduce, whereas recent growth increases, the number of neutral and deleterious variants in the population. Similarly, bottlenecks and expansions have opposite effects on the SFS—the former leads to a skew toward common variation, and the latter results in a skew toward rare variation.47,48,52 As a result, compared with a constant-size population, individuals from populations that have experienced bottlenecks tend to carry more deleterious alleles, whereas individuals from expanding populations carry slightly fewer derived deleterious alleles.
It is of interest to note that in the bottleneck simulations, we also considered a severe reduction of population size to 0.01× during the bottleneck. In this case, 0.93% of deleterious variants were fixed in the population, which contributed to a substantial proportion (73.3%) of derived deleterious alleles carried by individuals. Although this is more extreme than the estimated intensity of the Out-of-Africa bottleneck, it might be relevant for some populations that experienced stronger founder effects.53–56
Our simulation results recapitulate many of the qualitative patterns of neutral and deleterious variation observed in the empirical data. For example, the Tennessen et al.12 demographic model (Figure 6) predicts that EA individuals will on average carry more derived deleterious alleles than will AA individuals. Furthermore, the simulations also show that the proportion of derived deleterious alleles per individual is predicted to be higher in EA than in AA individuals. Despite the qualitative agreement between the empirical and simulated data, we do observe quantitative differences. Specifically, simulations suggest that the average density of derived deleterious alleles in EA individuals is ∼11.3% higher than in AA individuals, whereas in the empirical data, it is only ∼1.4% higher in EA individuals. A number of factors most likely contribute to this quantitative difference, including differences in demography and distribution of mutational effect sizes in real and simulated data. Another important difference is that in simulations, deleterious variants can be identified precisely, whereas in empirical data they are inferred with considerable error. Indeed, a substantial amount of the higher density of deleterious alleles in EA individuals in the simulated data is attributable to weakly deleterious mutations (|s| ≈10−4), and it is probably more difficult to identify these in empirical data. Thus, developing methods for predicting the functional and evolutionary significance of human genetic variation remains an important endeavor.
Recently, Simons et al.11 used allele-frequency data from the same data set and found that the average DAF of deleterious variants was not significantly different between EA and AA populations. Overall, our empirical results are qualitatively similar and in broad agreement with their findings. However, we observed that the average number of deleterious alleles was slightly but significantly higher in EA individuals than in AA individuals (905.7 and 893.2 for EA and AA individuals, respectively; Mann-Whitney test, p < 10−15; Figure 3). These differences in the average number of deleterious alleles per individual and distribution of effect sizes observed in the simulated models suggest that there might be subtle differences in mutation load between EA and AA populations and that they might be governed by mutations with small fitness effects. Simons et al. also observed that mutation load was slightly higher in EA populations than in AA populations (see their Figure S1011) as a result of mutations of weak effect. However, we note that the distinct genotypic composition between EA and AA individuals suggests that differences in mutation load might be larger if deleterious alleles are dominant or recessive.11 Furthermore, variability in mutation load might be greater in populations that experienced more intense bottlenecks (Figures 4 and 7). More generally, as eloquently noted by Lohmueller,57 homogeneity of load between populations does not imply homogeneity in patterns of deleterious variation, as illustrated in our empirical and simulation data (Figure 7). Finally, although our simulation and empirical results demonstrate that patterns of deleterious SNVs and alleles vary among populations, these differences are small in relation to the magnitude of variability among individuals. Furthermore, the majority of deleterious alleles carried by individuals are predicted to have selection coefficients on the order of |s| ≈ 10−4. Although such weakly deleterious alleles are important over evolutionary timescales, their contribution to the burden of human disease is largely unknown. Thus, it would be imprudent to directly relate differences in patterns of evolutionarily defined deleterious variation and mutation load to differences in disease burden. Nonetheless, recent demographic history does influence the characteristics and spectrum of rare and common variation within and between populations, which has important implications in the design and interpretation of disease-mapping studies.49 More generally, the continued development of experimental and computational methods of identifying and predicting the functional consequences of genetic variation is ultimately the most direct and accurate means of assessing individual disease risk.
Acknowledgments
We thank members of the J.M.A. laboratory, Kirk E. Lohmueller, Jonathan K. Pritchard, Guy Sella, Carlos D. Bustamante, Shamil R. Sunyaev, and David Reich for helpful feedback and discussions related to this work.
Contributor Information
Wenqing Fu, Email: wqfu@uw.edu.
Joshua M. Akey, Email: akeyj@uw.edu.
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://www.1000genomes.org/
Ensembl Genome Browser, http://useast.ensembl.org/index.html
mafTools, https://github.com/dentearl/mafTools
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
SFS_CODE, http://sfscode.sourceforge.net/SFS_CODE/index/index.html
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Haldane J.B.S. The effect of variation on fitness. Am. Nat. 1937;71:337–349. [Google Scholar]
- 2.Crow J.F. Genetic Loads and the Cost of Natural Selection. In: Kojima K.-i., editor. Mathematical Topics in Population Genetics. Springer-Verlag; New York: 1970. pp. 128–177. [Google Scholar]
- 3.Crow J.F. The origins, patterns and implications of human spontaneous mutation. Nat. Rev. Genet. 2000;1:40–47. doi: 10.1038/35049558. [DOI] [PubMed] [Google Scholar]
- 4.Gavrilov L.A., Gavrilova N.S., Kroutko V.N., Evdokushkina G.N., Semyonova V.G., Gavrilova A.L., Lapshin E.V., Evdokushkina N.N., Kushnareva Y.E. Mutation load and human longevity. Mutat. Res. 1997;377:61–62. doi: 10.1016/s0027-5107(97)00058-4. [DOI] [PubMed] [Google Scholar]
- 5.Lynch M. Rate, molecular spectrum, and consequences of human mutation. Proc. Natl. Acad. Sci. USA. 2010;107:961–968. doi: 10.1073/pnas.0912629107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kimura M., Maruyama T., Crow J.F. The Mutation Load in Small Populations. Genetics. 1963;48:1303–1312. doi: 10.1093/genetics/48.10.1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ohta T. Slightly deleterious mutant substitutions in evolution. Nature. 1973;246:96–98. doi: 10.1038/246096a0. [DOI] [PubMed] [Google Scholar]
- 8.Peischl S., Dupanloup I., Kirkpatrick M., Excoffier L. On the accumulation of deleterious mutations during range expansions. Mol. Ecol. 2013;22:5972–5982. doi: 10.1111/mec.12524. [DOI] [PubMed] [Google Scholar]
- 9.Abecasis G.R., Auton A., Brooks L.D., DePristo M.A., Durbin R.M., Handsaker R.E., Kang H.M., Marth G.T., McVean G.A., 1000 Genomes Project Consortium An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lohmueller K.E., Indap A.R., Schmidt S., Boyko A.R., Hernandez R.D., Hubisz M.J., Sninsky J.J., White T.J., Sunyaev S.R., Nielsen R. Proportionally more deleterious genetic variation in European than in African populations. Nature. 2008;451:994–997. doi: 10.1038/nature06611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Simons Y.B., Turchin M.C., Pritchard J.K., Sella G. The deleterious mutation load is insensitive to recent population history. Nat. Genet. 2014;46:220–224. doi: 10.1038/ng.2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tennessen J.A., Bigham A.W., O’Connor T.D., Fu W., Kenny E.E., Gravel S., McGee S., Do R., Liu X., Jun G., Broad GO. Seattle GO. NHLBI Exome Sequencing Project Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337:64–69. doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hodgkinson A., Casals F., Idaghdour Y., Grenier J.C., Hernandez R.D., Awadalla P. Selective constraint, background selection, and mutation accumulation variability within and between human populations. BMC Genomics. 2013;14:495. doi: 10.1186/1471-2164-14-495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fu W., O’Connor T.D., Jun G., Kang H.M., Abecasis G., Leal S.M., Gabriel S., Rieder M.J., Altshuler D., Shendure J., NHLBI Exome Sequencing Project Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature. 2013;493:216–220. doi: 10.1038/nature11690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kidd J.M., Gravel S., Byrnes J., Moreno-Estrada A., Musharoff S., Bryc K., Degenhardt J.D., Brisbin A., Sheth V., Chen R. Population genetic inference from personal genome data: impact of ancestry and admixture on human genomic variation. Am. J. Hum. Genet. 2012;91:660–671. doi: 10.1016/j.ajhg.2012.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Subramanian S. The abundance of deleterious polymorphisms in humans. Genetics. 2012;190:1579–1583. doi: 10.1534/genetics.111.137893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Casals F., Hodgkinson A., Hussin J., Idaghdour Y., Bruat V., de Maillard T., Grenier J.C., Gbeha E., Hamdan F.F., Girard S. Whole-exome sequencing reveals a rapid change in the frequency of rare functional variants in a founding population of humans. PLoS Genet. 2013;9:e1003815. doi: 10.1371/journal.pgen.1003815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Szpiech Z.A., Xu J., Pemberton T.J., Peng W., Zöllner S., Rosenberg N.A., Li J.Z. Long runs of homozygosity are enriched for deleterious variation. Am. J. Hum. Genet. 2013;93:90–102. doi: 10.1016/j.ajhg.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Do, R., Balick, D., Li, H., Adzhubei, I., Sunyaev, S., and Reich, D. (2014). No evidence that natural selection has been less effective at removing deleterious mutations in Europeans than in West Africans. arXiv, arXiv:1402.4896, http://arxiv.org/abs/1402.4896. [DOI] [PMC free article] [PubMed]
- 20.Abecasis G.R., Altshuler D., Auton A., Brooks L.D., Durbin R.M., Gibbs R.A., Hurles M.E., McVean G.A., 1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hernandez R.D., Williamson S.H., Bustamante C.D. Context dependence, ancestral misidentification, and spurious signatures of natural selection. Mol. Biol. Evol. 2007;24:1792–1800. doi: 10.1093/molbev/msm108. [DOI] [PubMed] [Google Scholar]
- 22.Hwang D.G., Green P. Bayesian Markov chain Monte Carlo sequence analysis reveals varying neutral substitution patterns in mammalian evolution. Proc. Natl. Acad. Sci. USA. 2004;101:13994–14001. doi: 10.1073/pnas.0404142101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pennacchio L.A., Olivier M., Hubacek J.A., Cohen J.C., Cox D.R., Fruchart J.C., Krauss R.M., Rubin E.M. An apolipoprotein influencing triglycerides in humans and mice revealed by comparative sequencing. Science. 2001;294:169–173. doi: 10.1126/science.1064852. [DOI] [PubMed] [Google Scholar]
- 24.Boffelli D., McAuliffe J., Ovcharenko D., Lewis K.D., Ovcharenko I., Pachter L., Rubin E.M. Phylogenetic shadowing of primate sequences to find functional regions of the human genome. Science. 2003;299:1391–1394. doi: 10.1126/science.1081331. [DOI] [PubMed] [Google Scholar]
- 25.Pollard K.S., Hubisz M.J., Rosenbloom K.R., Siepel A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010;20:110–121. doi: 10.1101/gr.097857.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Neph S., Kuehn M.S., Reynolds A.P., Haugen E., Thurman R.E., Johnson A.K., Rynes E., Maurano M.T., Vierstra J., Thomas S. BEDOPS: high-performance genomic feature operations. Bioinformatics. 2012;28:1919–1920. doi: 10.1093/bioinformatics/bts277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hernandez R.D. A flexible forward simulator for populations subject to selection and demography. Bioinformatics. 2008;24:2786–2787. doi: 10.1093/bioinformatics/btn522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gravel S., Henn B.M., Gutenkunst R.N., Indap A.R., Marth G.T., Clark A.G., Yu F., Gibbs R.A., Bustamante C.D., 1000 Genomes Project Demographic history and rare allele sharing among human populations. Proc. Natl. Acad. Sci. USA. 2011;108:11983–11988. doi: 10.1073/pnas.1019276108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Keinan A., Mullikin J.C., Patterson N., Reich D. Measurement of the human allele frequency spectrum demonstrates greater genetic drift in East Asians than in Europeans. Nat. Genet. 2007;39:1251–1255. doi: 10.1038/ng2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boyko A.R., Williamson S.H., Indap A.R., Degenhardt J.D., Hernandez R.D., Lohmueller K.E., Adams M.D., Schmidt S., Sninsky J.J., Sunyaev S.R. Assessing the evolutionary impact of amino acid mutations in the human genome. PLoS Genet. 2008;4:e1000083. doi: 10.1371/journal.pgen.1000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morton N.E., Crow J.F., Muller H.J. An Estimate of the Mutational Damage in Man from Data on Consanguineous Marriages. Proc. Natl. Acad. Sci. USA. 1956;42:855–863. doi: 10.1073/pnas.42.11.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Crow J.F. Some possibilities for measuring selection intensities in man. Hum. Biol. 1958;30:1–13. [PubMed] [Google Scholar]
- 33.Muller H.J. Our load of mutations. Am. J. Hum. Genet. 1950;2:111–176. [PMC free article] [PubMed] [Google Scholar]
- 34.Nickerson D.A., Taylor S.L., Weiss K.M., Clark A.G., Hutchinson R.G., Stengård J., Salomaa V., Vartiainen E., Boerwinkle E., Sing C.F. DNA sequence diversity in a 9.7-kb region of the human lipoprotein lipase gene. Nat. Genet. 1998;19:233–240. doi: 10.1038/907. [DOI] [PubMed] [Google Scholar]
- 35.Campbell M.C., Tishkoff S.A. African genetic diversity: implications for human demographic history, modern human origins, and complex disease mapping. Annu. Rev. Genomics Hum. Genet. 2008;9:403–433. doi: 10.1146/annurev.genom.9.081307.164258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kircher M., Witten D.M., Jain P., O’Roak B.J., Cooper G.M., Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014;46:310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chun S., Fay J.C. Identification of deleterious mutations within three human genomes. Genome Res. 2009;19:1553–1561. doi: 10.1101/gr.092619.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Davydov E.V., Goode D.L., Sirota M., Cooper G.M., Sidow A., Batzoglou S. Identifying a high fraction of the human genome to be under selective constraint using GERP++ PLoS Comput. Biol. 2010;6:e1001025. doi: 10.1371/journal.pcbi.1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schwarz J.M., Rödelsperger C., Schuelke M., Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods. 2010;7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 41.Kumar P., Henikoff S., Ng P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 42.Lachance J., Vernot B., Elbers C.C., Ferwerda B., Froment A., Bodo J.M., Lema G., Fu W., Nyambo T.B., Rebbeck T.R. Evolutionary history and adaptation from high-coverage whole-genome sequences of diverse African hunter-gatherers. Cell. 2012;150:457–469. doi: 10.1016/j.cell.2012.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Coventry A., Bull-Otterson L.M., Liu X., Clark A.G., Maxwell T.J., Crosby J., Hixson J.E., Rea T.J., Muzny D.M., Lewis L.R. Deep resequencing reveals excess rare recent variants consistent with explosive population growth. Nat. Commun. 2010;1:131. doi: 10.1038/ncomms1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Keinan A., Clark A.G. Recent explosive human population growth has resulted in an excess of rare genetic variants. Science. 2012;336:740–743. doi: 10.1126/science.1217283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nelson M.R., Wegmann D., Ehm M.G., Kessner D., St Jean P., Verzilli C., Shen J., Tang Z., Bacanu S.A., Fraser D. An abundance of rare functional variants in 202 drug target genes sequenced in 14,002 people. Science. 2012;337:100–104. doi: 10.1126/science.1217876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.United Nations, Department of Economic and Social Affairs, Population Division (2011). World Population Prospects: The 2010 Revision, Volume I: Comprehensive Tables. ST/ESA/SERA/313. http://esa.un.org/wpp/documentation/pdf/WPP2010_Volume-I_Comprehensive-Tables.pdf.
- 47.Kimura M. Stochastic processes and distribution of gene frequencies under natural selection. Cold Spring Harb. Symp. Quant. Biol. 1955;20:33–53. doi: 10.1101/sqb.1955.020.01.006. [DOI] [PubMed] [Google Scholar]
- 48.Waxman D. A unified treatment of the probability of fixation when population size and the strength of selection change over time. Genetics. 2011;188:907–913. doi: 10.1534/genetics.111.129288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lohmueller K.E. The impact of population demography and selection on the genetic architecture of complex traits. PLoS Genet. 2014;10:e1004379. doi: 10.1371/journal.pgen.1004379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Maher M.C., Uricchio L.H., Torgerson D.G., Hernandez R.D. Population genetics of rare variants and complex diseases. Hum. Hered. 2012;74:118–128. doi: 10.1159/000346826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gazave E., Chang D., Clark A.G., Keinan A. Population growth inflates the per-individual number of deleterious mutations and reduces their mean effect. Genetics. 2013;195:969–978. doi: 10.1534/genetics.113.153973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nei M., Maruyama T., Chakraborty R. The bottleneck effect and genetic variability in populations. Evolution. 1975;29:1–10. doi: 10.1111/j.1558-5646.1975.tb00807.x. [DOI] [PubMed] [Google Scholar]
- 53.Atzmon G., Hao L., Pe’er I., Velez C., Pearlman A., Palamara P.F., Morrow B., Friedman E., Oddoux C., Burns E., Ostrer H. Abraham’s children in the genome era: major Jewish diaspora populations comprise distinct genetic clusters with shared Middle Eastern Ancestry. Am. J. Hum. Genet. 2010;86:850–859. doi: 10.1016/j.ajhg.2010.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Palamara P.F., Lencz T., Darvasi A., Pe’er I. Length distributions of identity by descent reveal fine-scale demographic history. Am. J. Hum. Genet. 2012;91:809–822. doi: 10.1016/j.ajhg.2012.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sajantila A., Salem A.H., Savolainen P., Bauer K., Gierig C., Pääbo S. Paternal and maternal DNA lineages reveal a bottleneck in the founding of the Finnish population. Proc. Natl. Acad. Sci. USA. 1996;93:12035–12039. doi: 10.1073/pnas.93.21.12035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang S.R., Agarwala V., Flannick J., Chiang C.W., Altshuler D., Hirschhorn J.N., GoT2D Consortium Simulation of Finnish population history, guided by empirical genetic data, to assess power of rare-variant tests in Finland. Am. J. Hum. Genet. 2014;94:710–720. doi: 10.1016/j.ajhg.2014.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lohmueller K.E. The distribution of deleterious genetic variation in human populations. bioRxiv. 2014 doi: 10.1016/j.gde.2014.09.005. [DOI] [PubMed] [Google Scholar]