

Summary
A variety of extremely challenging biological sequence analyses were conducted on the XSEDE large shared memory resource Blacklight, using current bioinformatics tools and encompassing a wide range of scientific applications. These include genomic sequence assembly, very large metagenomic sequence assembly, transcriptome assembly, and sequencing error correction. The data sets used in these analyses included uncategorized fungal species, reference microbial data, very large soil and human gut microbiome sequence data, and primate transcriptomes, composed of both short-read and long-read sequence data. A new parallel command execution program was developed on the Blacklight resource to handle some of these analyses. These results, initially reported previously at XSEDE13 and expanded here, represent significant advances for their respective scientific communities. The breadth and depth of the results achieved demonstrate the ease of use, versatility, and unique capabilities of the Blacklight XSEDE resource for scientific analysis of genomic and transcriptomic sequence data, and the power of these resources, together with XSEDE support, in meeting the most challenging scientific problems.
Keywords: bioinformatics, genomics, genome, transcriptome, de novo assembly, primates, RNA-seq, NGS, metagenome, large shared memory computing, high-performance computing, data-intensive computing
1. Introduction
High-throughput, next-generation sequencing (NGS) of genomes [1, 2], transcriptomes [3], and epigenomes is currently in a phase of burgeoning growth with each passing development cycle yielding a greater than exponential return in the amount of quality sequence data generated per unit of cost (www.genome.gov/sequencingcosts). This rapid progress in data generation can currently create data sets within weeks, which are computationally intractable [4] for complete scientific analysis because of the large RAM footprint required or the volume of data to be analyzed. This limits their potential use in areas of scientific interest [5] and in translational medicine [6, 7].
The computational requirements of these data sets often exceed the capacity of personal computing systems, server-level infrastructure, distributed high performance computing, and large shared memory high performance computing systems. Hence, many important scientific questions for which the data are or could be available either go unanswered or can only be addressed by a few research groups with a large bioinformatics infrastructure. Here we present science outcomes that highlight the ability of the cache coherent non-uniform memory access architecture of the XSEDE resource Blacklight, housed at the Pittsburgh Supercomputing Center (PSC), to allow efficient genomic analysis of data sets outside the scope of other high performance computing systems, as well as user-friendly high-throughput analysis of standard-sized to large-sized genomic data [8]. With these complementary capabilities, the XSEDE resource Blacklight extends the current technical limits of genomic and transcriptomic assembly for analyses requiring the largest shared memory systems, as well as the scope of genomic research by enabling high-throughput large shared memory analysis.
2. Blacklight
The Blacklight system at the PSC is an SGI Altix UV 1000 (SGI (Silicon Graphics, Inc.), Milpitas, CA 95035, USA) with two partitions, each containing 16 TB of cache coherent shared memory and 2048 cores. This means that a single application running on Blacklight can access up to 16 TB of shared memory using up to ∼2000 cores. The obvious application of this system is for algorithms and problems that benefit from holding large amounts of data in RAM. However, the fast interconnect that facilitates cache coherent non-uniform memory access across the system also enables rapid communication within distributed memory applications. This dual nature of the system allows researchers to run problems across a continuum, from a single, massive shared memory application to many large shared memory applications running in parallel to fully distributed or embarrassingly parallel applications. Because the realm of genomic analysis encompasses all of these modes of computing, this flexibility makes Blacklight convenient and powerful for researchers dealing with diverse genomic analysis pipelines. In addition, because Blacklight is essentially one big system, running a single operating system, it is ideal for rapid prototyping of new serial and parallel algorithms for large data analysis.
3. Genome Assembly
A variety of large animal genomes have been assembled on Blacklight using various de novo assembly codes. These include a 1.7-Gbp rattlesnake genome using the Velvet assembler, the 3.4-Gbp Little Skate genome using ABySS, and two species of ∼200-Mbp Drosophila and the ∼3-Gbp Florida Scrub Jay using ALLPATHS-LG. All of these assemblers create large de Bruijn graphs in memory to piece the short DNA reads produced by NGS machines into the large fragments required to assemble a compete genome. Because the assemblers must trace random paths through memory to assemble these fragments, the algorithm is not amenable to distributed programming. Therefore, a large shared memory machine is essential to completing many of these assemblies. Many plant and animal genomes are much larger than these and would require many terabytes of memory to assemble. Researchers have deemed these very large genomes out of reach, but Blacklight's 16 TB of shared memory creates the possibility of doing complete assemblies of these important large genomes, such as Chinese Spring Wheat, with 17 Gbp of DNA.
In addition to doing very large genomes, researchers have used new long-read sequencing technology to obtain assemblies of genomes with complex structures that are difficult to sort out with short reads. Using Blacklight for sequence error correction of long-read Pacific Biosciences single molecule sequence data, researchers were able to employ the CELERA assembler to assemble the genomes of an important group of anaerobic fungal organisms that reside in the rumen of herbivores. These organisms had been previously untenable for genomic analysis because of a highly repetitive and AT-biased genome [9]. This analysis yielded valuable insight into their role in rumen ecology as well as potentially novel enzymes to assist in bioethanol production. These results, which have just been published [10], will greatly contribute to scientists' understanding of these organisms.
4. Metagenomics
Metagenomics, the assembly of multiple genomes using sequence data gathered from a given environment, presents unique challenges due to both the complexity and the amount of data needed to achieve sufficient coverage for assembly. Assembling metagenomes often requires more shared memory than what is needed for genomes of individual organisms or even what is available on high performance computing clusters and typical large memory nodes. Extending the scope of metagenomic analysis and allowing the assembly of very large metagenomes could provide valuable insight into microbial ecology as well as enable the discovery of novel enzymatic pathways with potential to address the emerging biotechnology challenges of the 21st century, including antibiotic resistance, environmentally friendly bio-energy, bioremediation, and sustainable agriculture.
To identify enzymes with potential use in biofuel production, a novel method for metagenomic microbial enzymatic discovery was recently employed that uses a synthetic ecosystem amenable to experimenter control (bioreactor) seeded with a microbial founder community of high diversity allowing for complex higher-order microbial interactions and total de novo metagenomic assembly. In this method, artificial evolutionary pressure is applied to the seeded bioreactor to select for members of the community that contain an experimentally desired feature or metabolic hallmark.
In the experiment reported here, a Brazilian soil sample was selected as the microbial founder community because of soil's extreme species level diversity, the majority of which are resistant to laboratory monoculture. This community was cultured for 8 weeks in an aerobic bioreactor using minimal media and a complex lignocellulotic plant material, sugarcane bagasse, as the sole carbon source with a 90% liquid phase replacement schema conducted every seventh day. Using this method, 300 Gbp of sequence data (Table I) was generated and then assembled on Blacklight using the Velvet code [2]. This assembly required nearly 4 TB of shared RAM on Blacklight (Table II), the only computing platform for open scientific research in the US that is currently configured to provide sufficient shared memory for assemblies of this scale.
Table I.
Next-generation sequencing data and technology type used for assembly of a soil metagenome.
| Sequence library condition | Read count | Technology type | Total sequence |
|---|---|---|---|
| Initial soil sample | 481.9 Million×2 | Illumina HiSeq (paired×100 bp) | 96 Gbp |
| Initial soil sample | 1.1 Million×2 | Roche titanium 454 | 500 Mbp |
| Shaker bioreactor (6 weeks) | 487.1 Million×2 | Illumina HiSeq (paired×100 bp) | 97 Gbp |
| Fermentation bioreactor (8 weeks) | 542.6 Million×2 | Illumina HiSeq (paired×100 bp) | 108 Gbp |
| Sequence total (all data) | ∼3 billion | Illumina/roche | 300 Gbp |
Table II.
Walltime and peak RAM usage for soil metagenome assembly.
| Assembly characteristic | Value |
|---|---|
| Velveth peak RAM usage | 3.6 TB (3600 GB) |
| Velveth walltime hours | 60 |
| Velvetg peak RAM usage | 1.2 TB (1200 GB) |
| Velvetg walltime hours | 83 |
The use of the standard malloc library with Velvet resulted in extreme memory fragmentation as memory usage passed 3 TB. The frequent use of the memory management routines malloc and free on such a large memory space eventually resulted in there not being 500 KB of contiguous memory in 6 TB. To overcome this memory fragmentation problem, which prevented the assembly from completing, the Hoard memory allocation package was used as a drop-in replacement for the standard malloc library [11]. Use of the more uniform Hoard allocation structures significantly reduced memory fragmentation. This increased overall runtime and RAM usage slightly but allowed the assemblies to complete without error.
After assembly, further analysis was performed to assess the quality of the assembly and identify potential enzymes of interest. These downstream analyses only required minimal computing resources and hence were performed on the researchers' local hardware or on specialized public resources. Taxonomic analysis of the resulting assembly was performed with on the public MG-RAST server [12] and yielded the expected distribution of species found in soil environments dominated by the taxon Proteobacteria, the predominant phylotype found in soil (Table III, Figure 1). In addition, this analysis revealed the presence of 70 phyla in the metagenome. This extensive diversity shows that this methodology can be used to analyze diverse communities, which are often needed to examine functions that would be absent in smaller more homogeneous samples.
Table III.
Domain-level phylogenetic distribution of metagenomic contigs.
| Domain | Percent of total assembled contigs (%) |
|---|---|
| Bacteria | 92.30 |
| Eukaryota | 6.48 |
| Archaea | 0.95 |
| Viruses | 0.15 |
| Other | 0.12 |
Figure 1.
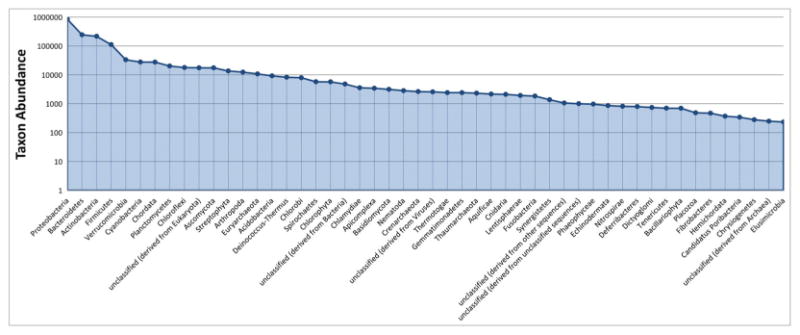
Species-level phylogenetic distribution of metagenomic contigs.
Further analysis of the assembled data resulted in the identification of a large number of enzymes related to the breakdown of plant cell walls, a highly desired group of enzymes that have the potential to accelerate bio-ethanol production [13], demonstrating the effectiveness of these methods for the identification of novel enzymes of bio-industrial importance from prokaryota. To accomplish this analysis, MetaGeneMark [14] was used to predict protein gene models (Table IV). These protein gene models were used as input to hmmscan [15], which uses Markov domain profiling to scan these gene models against protein databases to identify enzymes of interest. In this case, hmmscan was used to identify protein domains that are responsible for lignocellulosic degradation in putative carbohydrate active enzymes (CAZy) [16] using the CAZy database (dbCAN) [17]. This method, which uses mathematical modeling of domain structure for protein function prediction in place of traditional homology-based searches, allows for detection of remote homologs and has been used successfully for the prediction of CAZy members in other metagenomes [18]. The hmmscan search, employing a conservative sequence identification statistical significance e-value of e-4, revealed a large number of CAZy enzymes (56,626) that could aid in the production of lignocellulosic biofuel (Table V).
Table IV.
MetaGeneMark gene prediction numbers for metagenomes.
| Conditions | MeteGeneMark protein gene models |
|---|---|
| Semi-aerobic enrichment | 1,307,802 |
| Aerobic bioreactor | 1,286,826 |
| Total assembly (all reads, 300 Gbp) | 2,250,504 |
Table V.
Carbohydrate active enzyme (CAZy) members predicted in the metagenomes.
| Conditions | Glycoside hydrolases (GH) | Accessory enzymes (AA) | Pectin lyase (PL) | Carbohydrate esterase (CE) |
|---|---|---|---|---|
| Semi-aerobic enrichment | 18,450 | 4014 | 1801 | 8775 |
| Aerobic bioreactor | 17,394 | 3084 | 1303 | 6743 |
| Total assembly (all reads, 300 Gbp) | 32,143 | 6662 | 3414 | 14,407 |
To explore which of the identified CAZy enzymes were active in the metagenomic community, mass spectrometry proteomic analysis Orbitrap LC/MS Thermo Scientific (Thermo Fisher Scientific Inc, Waltham, MA, USA) was conducted on the metagenome reactor using the CAZy peptide database. Interpolated CAZy-identified peptides were then aligned to the metagenomic assembly protein model database using the BLAST algorithm, and only exact ungapped matches of nine or more peptides were considered valid. This validation yielded 469 proteomically confirmed enzymes (Figure 2) in the metagenome assemblies. These targets will be prioritized for future studies of cloning and expression to assess their specific role in lignocellulose degradation.
Figure 2.
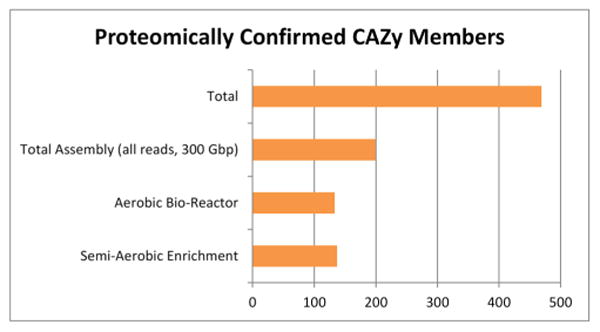
Orbitrap LC/MS metagenomic peptide confirmation results.
This experiment demonstrates the utility of Blacklight's large shared memory architecture for studying metagenomic communities at previously impractical resolutions. By using this methodology with a different bioreactor schema and varied evolutionary pressure, alternate microbial ecosystems can be evolved containing other metabolic hallmarks of experimental interest. These methods, together with other ‘omics’ data and the right computational resources, form an effective high throughput, high resolution platform for gene discovery.
5. The Non-Human Primate Reference Transcriptome Resource
5.1. Data generation with massive RNA-seq
In 2010, a committee of researchers set out to create a non-human primate reference transcriptome resource (NHPRTR) to help establish the genetic basis for phenotypic differences observed between primates, including differences between humans and non-human primates (NHPs). Such a resource can provide valuable information regarding evolutionary processes, as well as insight into human health and disease from the pharmacogenomics work performed on the animal models for infectious disease and novel treatments. To provide a comprehensive resource, a committee of experts chose 13 primate species. Tissues samples were then taken from 21 tissues and next-generation sequencing of RNA (RNA-seq) was performed using three different approaches. The result was 40.5 billion 100 nucleotide reads that needed to be assembled into transcriptomes for each species and RNA-seq method used (13 species×3 methods= 39 assemblies). Because most of these species do not have any reference genome, the transcriptomes must be assembled de novo. The details behind the motivation for this resource and the generation of the RNA-seq data are described in detail in the NHPRTR paper [19].
5.2. Enabling large-scale de novo transcriptome asembly with trinity on Blacklight
As described in the NHPRTR paper, investigators found that assembling the nearly 2 billion reads required as input for these de novo transcriptome assemblies was beyond the capabilities of their local systems and even beyond the capacity of the programs' initial estimates of large data inputs. At this point, they applied for an XSEDE allocation on Blacklight at the PSC, along with Extended Collaborative Support Services (ECSS) from XSEDE to help them perform these transcriptome assemblies of unprecedented size and scale using Trinity [3]. Through XSEDE's ECSS, PSC worked closely with the Trinity developers to harden Trinity on Blacklight and find the best way to run these massive assemblies.
To begin, PSC installed the latest, optimized version of Trinity contributed by the National Center for Genome Analysis Support (NCGAS) at IU, without which these assemblies would have taken several times longer to complete [20]. Even with this optimized version, challenges appeared immediately. While Blacklight had plenty of shared memory to handle the assemblies, at one point in the assembly, the Chrysalis phase [3], Trinity was creating and working on hundreds of thousands of files. Even very large assemblies of say, 600 million reads, while still producing tens of thousands of files, had no problem executing on the default Lustre filesystem, but the 2 billion read assemblies being attempted here produced too many files to be handled efficiently by this filesystem. To work around this, PSC established a local filesystem attached directly to Blacklight. This alleviated the problem for a single massive Trinity assembly, but I/O-related slowdowns occurred with many massive assemblies running at once.
Finally, an ideal workflow was devised (Figure 3), utilizing Blacklight's RAM disk at the right points in the workflow to speed up the calculations, run many assemblies at once, and avoid problematic I/O issues but also avoid wastefully using RAM disk for large files where it was less beneficial. First, preprocessing of the data, a primarily serial task, was performed on the research group's local resources. The preprocessed data were then moved to Blacklight's Lustre filesystem, and the initial Inchworm stage of the assembly was performed entirely on the Lustre filesystem using 64 cores. For the Chrysalis stage, we introduced a modification to the Trinity code that allowed the Chrysalis directory to be given a different path from the rest of the Trinity working directory. This allowed the Chrysalis files to be created on RAM disk, while large files that did not need RAM disk remained on the Lustre filesystem. This phase generally required 128 cores (1 TB RAM) to provide extra memory resources to store files on the RAM disk associated with the job. After the Chrysalis phase was complete, the job script would back up the Chrysalis directory to the Lustre filesystem but then continue to operate on those files in RAM disk for the final QuantifyGraph and Butterfly stages of Trinity. We found that using 64 threads on 64 cores for the QuantifyGraph and Butterfly stages and running from RAM disk provided optimal performance, reducing runtime of those steps from a total of 250 h (when running from Lustre using 32 threads) to 50 h. Even after these optimizations, a significant amount of resources were needed, with a typical de novo assembly for one primate species with ∼1.8 billion RNA-seq reads taking around 550 compute hours using 64–128 cores (35,200–70,400 service units).
Figure 3.
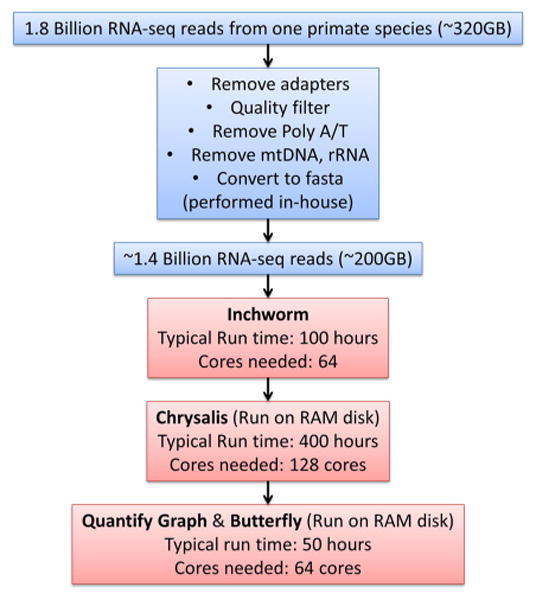
Walltime and core counts for various stages of the Trinity pipeline to assemble a single primate transcriptome. See Reference [3] for descriptions of the phases of Trinity.
This de novo assembly method has proven successful, generating transcriptomes with a mean average size >2 KB for most RNA-seq methods used. Out of the 39 total assemblies required, 20 of the largest assemblies were performed on Blacklight over a period of a few weeks (Table VI).
Table VI.
Summaries of 20 primate transcriptome assemblies completed on Blacklight.
| Species | Library | Number of input sequences | Number of contigs | Total length (bp) | N25 (bp) | N50 (bp) | N75 (bp) | Longest contig contig (bp) |
|---|---|---|---|---|---|---|---|---|
| Baboon | TOT | 149,018,017 | 658,581 | 280,530,426 | 1029 | 434 | 276 | 35,170 |
| Baboon | UDG | 1,543,247,564 | 1,131,951 | 844,127,360 | 3534 | 1368 | 479 | 131,395 |
| Chimpanzee | UDG | 1,465,013,009 | 987,615 | 1,433,298,968 | 6356 | 3806 | 1666 | 47,873 |
| Cynomolgus macaque (Chinese) | RNA | 1,675,591,113 | 911,282 | 864,011,637 | 4769 | 2316 | 706 | 59,032 |
| Cynomolgus macaque (Chinese) | UDG | 1,629,828,914 | 990,604 | 1,055,391,869 | 5479 | 2822 | 875 | 122,916 |
| Cynomolgus macaque (Mauritian) | RNA | 1,078,143,527 | 1,142,531 | 929,071,752 | 4368 | 1813 | 525 | 30,364 |
| Cynomolgus macaque (Mauritian) | TOT | 166,350,980 | 526,723 | 199,771,360 | 657 | 377 | 266 | 36,976 |
| Cynomolgus macaque (Mauritian) | UDG | 1,177,348,077 | 1,015,657 | 834,198,307 | 4360 | 1927 | 532 | 22,239 |
| Gorilla | UDG | 1,256,121,406 | 732,336 | 1,122,255,357 | 5878 | 3680 | 1804 | 33,526 |
| Japanese macaque (Indonesian) | RNA | 1,863,420,069 | 703,246 | 737,890,249 | 5010 | 2687 | 898 | 21,620 |
| Marmoset | TOT | 253,098,348 | 332,782 | 118,141,291 | 545 | 357 | 261 | 14,561 |
| Marmoset | UDG | 1,659,423,714 | 814,235 | 475,863,605 | 2206 | 785 | 359 | 122,518 |
| Pig-tailed macaque | RNA | 1,725,523,068 | 969,993 | 1,044,146,568 | 5189 | 2807 | 916 | 25,056 |
| Pig-tailed macaque | UDG | 1,573,381,596 | 1,301,087 | 1,222,337,940 | 5015 | 2265 | 668 | 32,637 |
| Rhesus macaque (Chinese) | RNA | 1,209,350,072 | 923,017 | 836,358,971 | 4694 | 2230 | 639 | 32,796 |
| Rhesus macaque (Chinese) | UDG | 1,310,236,599 | 969,421 | 850,250,785 | 4638 | 2114 | 594 | 34,020 |
| Rhesus macaque (Indian) | RNA | 3,200,476,713 | 703,246 | 737,890,249 | 5010 | 2687 | 898 | 21,620 |
| Rhesus macaque (Indian) | UDG | 1,410,322,373 | 1,051,149 | 832,770,332 | 3966 | 1644 | 519 | 68,269 |
| Ring-tailed lemur | UDG | 1,403,229,556 | 611,678 | 822,247,525 | 6169 | 3630 | 1468 | 33,401 |
| Sooty mangabey | UDG | 1,635,074,685 | 1,188,472 | 1,465,648,107 | 6431 | 3483 | 1116 | 30,331 |
Geographic distinction for some species are indicated in parentheses.
RNA, standard mRNA-seq; TOT, total RNA; UDG, standard mRNA-seq with uracil DNA glycosylase.
5.3. Characterizing the de novo assembled transcriptomes
In order to evaluate the accuracy of the de novo transcriptome assemblies, we determined the percentage that our assembly reconstituted the publicly available genome annotations (Table VII). Currently, only five NHP species from our data set have reference genomes: chimpanzee (panTro4), gorilla (gorGor3), rhesus macaque (rheMac3), marmoset (calJac3), and mouse lemur (micMur1). For species without reference genomes, we mapped to the nearest genome. For the gene models, we used annotations that were generated from native mRNA (RefSeq) and annotations that were computationally predicted (ENSEMBL). In most cases, the genes were either assembled >80% of their gene model or not assembled at all. In every NHP species with a reference genome, there was an improvement in assembling RefSeq genes compared with assembling ENSEMBL gene predictions. Most notably, we were able to recover >90% of RefSeq genes in rhesus macaque. The lack of coverage of genes in mouse lemur may represent the incompleteness of the draft genome, which lies entirely in scaffolds.
Table VII.
Percentage of known (RefSeq) and predicted genes (ENSEMBL) covered by de novo assembled transcriptomes.
| Species (genome) | RefSeq genes Percentage of all covered >80% | RefSeq genes (%) | ENSEMBL gene predictions covered >80% | Percentage of all ENSEMBL gene predictions (%) |
|---|---|---|---|---|
| Chimpanzee (panTro4) | 1888 | 77% | 18,928 | 68 |
| Gorilla (gorGor3) | N/A | N/A | 17,142 | 59 |
| Rhesus Macaque (rheMac3) | 5519 | 91% | 14,290 | 57 |
| Marmoset (calJac3) | 124 | 73% | 18,247 | 56 |
| Mouse Lemur (micMur1) | N/A | N/A | 3693 | 15 |
Each species was aligned to its nearest genome indicated in parentheses. If greater than 80% of the full gene model was reconstituted by the de novo assembly, the gene model was counted. Annotations were downloaded from UCSC Genome Browser Tracks. ‘N/A’ refers to unavailable annotation files.
The ability of Trinity to recover most of the known gene models shows that the de novo assemblies built for NHP species without a reference genome are good representations of the actual annotations. Because many studies are designed to be dependent on certain reference genomes, we provide transcriptome assemblies for many additional NHP species without reference genomes and also show that it is possible to accurately rebuild any species' transcriptome with high-coverage RNA-seq data.
In addition to the known genes that were built by Trinity, we were interested in the assembled transcripts that were not currently annotated. We looked for evidence of novel putative noncoding RNAs by filtering the assemblies for sequences that were in the current annotation and/or contained open reading frames that were 90 bp or longer. Noncoding RNA genes are RNA molecules that are transcribed but are not translated into proteins. Noncoding RNA genes have been implicated in many biological roles ranging from necessary components of protein translation (transfer RNAs) to major effectors of X inactivation (Xist) [21]. The abundance of long noncoding RNAs in NHP genomes remains unclear. In chimpanzee, we identified 4489 possible novel noncoding RNAs. Figure 4 shows a putative novel noncoding RNA in chimpanzee that contains an exon from a human long intergenic non-protein coding RNA (LINC RNA), LINC00457. LINC00457 is primarily expressed in the human brain [22].
Figure 4.
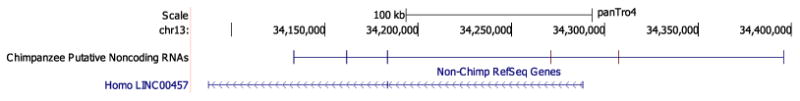
Putative novel noncoding RNA gene in chimpanzee on chromosome 13. This putative gene overlaps an exon from human long intergenic noncoding RNA gene 457.
5.4. Hosting a community resource
Now that the initial set of the massive de novo assemblies have been completed, the finished transcriptomes are being hosted on storage resources at the PSC so that the community of researchers interested in these data can apply for an XSEDE allocation and use Blacklight or other XSEDE resources to further work with and analyze the data. The researchers working with NHPRTR are planning additional assemblies and cross-transcriptome alignments, for which the ready availability of these data on XSEDE will be most useful [19]. Lastly, because they have been encouraged by these results, the NHPRTR group of researchers has started a larger set of brain and region-specific deep RNA-sequencing of the 21 tissues across all the primates, which will create an additional 11 billion reads and an expanded resource for research in the NHPs, including evolutionary models, improved genome annotation across all primates (humans included), and improved models for infectious disease like HIV (using SIV) and AIDS.
6. Rapid Algorithm Development
One of the potential advantages of Blacklight's massive shared memory architecture is to enable scientists and developers to quickly prototype new parallel solutions to their research problems. As a simple example, when working with very large genomic data sets or other very large volume data, researchers often encounter circumstances where a heterogeneous group of large memory commands need to be executed expeditiously. While working with Trinity on Blacklight, a researcher and Trinity developer was able to quickly design a program to efficiently execute parallel commands that require large amounts of shared memory during the QuantifyGraph and Butterfly stages of Trinity. This program, Parafly, uses C++ and OpenMP to launch a large set of jobs with varying memory requirements, filling the need for a versatile parallel execution program within Trinity. Parafly accepts a flat file with the group of commands that a user wishes to execute for input, placing minimal requirements on the end user for operation. The program operates by loading all commands to be executed into an array data structure, assigning thread conditions to each command to be executed, then executes each command in parallel while logging the exit status of each command. Parafly was incorporated into the main Trinity code, and since then has been spun off as a separate project and extended to efficiently execute any group of tasks that require a large amount of shared memory per system thread. The Parafly resource can be found for download at http://parafly.sourceforge.net/.
7. Conclusion
Results have been presented comprising large single organism genome assembly, massive 300 Gbp metagenomic assembly of thousands of microorganisms, requiring 3.5 TB of RAM, and high-throughput, high-memory assemblies of 20 primate transcriptomes. These advances are breaking new ground in their respective fields, and some, like the metagenome assembly and development of the NHPRTR would have been extremely difficult or impossible to do on any other system. While these diverse accomplishments highlight the power and flexibility of Blacklight's architecture for the assembly of NGS data, the research community is still becoming aware of these capabilities. As a result, Blacklight's potential to assemble the largest single organism genomes, or even larger metagenomic samples, has not yet been fully tested. As more groups engage with researchers who have benefitted from this resource, and engage with XSEDE through its ECSS and novel and innovative project programs, we expect demand to continue to grow, along with our ability to harness the full potential of available NGS data to solve the most challenging problems in computational biology.
Acknowledgments
This work was supported with funding from the Oklahoma Bioenergy Center and the Department of Energy, Awards 06103-OKL and ZDJ-7-77608-01 (Brian Couger and Rolf Prade); the National Institutes of Health (NIH), including R01HG006798, R01NS076465, R24RR032341, and the Starr Cancer Consortium grant number I7-A765 (Chris Mason); the Tri-Institutional Training Program in Computational Biology and Medicine and the National Science Foundation Graduate Research Fellowship Program under Grant No. NSF DGE-1144153 (Lenore Pipes). This work also used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant number OCI-1053575. Specifically, it used the Blacklight system at the Pittsburgh Supercomputing Center (PSC).
References
- 1.Myers EW, et al. A whole-genome assembly of Drosophila. Science. 2000;287(5461):2196–2204. doi: 10.1126/science.287.5461.2196. [DOI] [PubMed] [Google Scholar]
- 2.Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Research. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grabherr MG, et al. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nature Biotechnol. 2011;29(7):644–52. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stein LD. The case for cloud computing. Genome Biology. 2010;11(5):207. doi: 10.1186/gb-2010-11-5-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nekrutenko A, Taylor J. Next-generation sequencing data interpretation: enhancing reproducibility and accessibility. Nature Reviews Genetics. 2012;13:667–672. doi: 10.1038/nrg3305. [DOI] [PubMed] [Google Scholar]
- 6.Chan J. Genome sequencing in clinical microbiology. Nature Biotechnology. 2012;30:1068–1071. doi: 10.1038/nbt.2410. [DOI] [PubMed] [Google Scholar]
- 7.Johnson P, et al. Assuring the quality of next-generation sequencing in clinical laboratory practice. Nature Biotechnology. 2012;30:1033–1036. doi: 10.1038/nbt.2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Couger MB, Pipes L, Blood PD, Mason CE. Proceedings of the Conference on Extreme Science and Engineering Discovery Environment: Gateway to Discovery (XSEDE '13)) ACM; New York, NY, USA: 2013. Enabling large-scale next-generation sequence assembly with Black-light. Article 27, 6 pages. [DOI] [Google Scholar]
- 9.Brownlee AG. Remarkably AT-rich genomic DNA from the anaerobic fungus Neocallimastix. Nucleic Acids Research. 1989;17(4):1327–1335. doi: 10.1093/nar/17.4.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Youssef NH, Couger MB, Struchtemeyer CG, Liggenstoffer AS, Prade RA, Najar FZ, Atiyeh HK, Wilkins MR, Elshahed MS. The genome of the anaerobic fungus Orpinomyces sp. strain C1A reveals the unique evolutionary history of a remarkable plant biomass degrader. Applied and Environmental Microbiology. 2013;79(15):4620–4634. doi: 10.1128/AEM.00821-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berger ED, McKinley KS, Blumofe RD, Wilson PR. Proceedings of the ninth international conference on Architectural support for programming languages and operating systems (ASPLOS IX) ACM; New York, NY, USA: 2000. Hoard: a scalable memory allocator for multithreaded applications; pp. 117–128. to appear in print. [DOI] [Google Scholar]
- 12.Meyer F, Paarmann D, D'Souza M, Olson R, Glass EM, Kubal M, Paczian T, Rodriguez A, Stevens R, Wilke A, Wilkening J, Edwards RA. The metagenomics RAST server - a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics. 2008;9:386. doi: 10.1186/1471-2105-9-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sanderson K. Lignocellulose: a chewy problem. Nature. 2011;474:S12–S14. doi: 10.1038/474S0. [DOI] [PubMed] [Google Scholar]
- 14.Zhu W, Lomsadze A, Borodovsky M. Ab initio gene identification in metagenomic sequences. Nucleic Acids Research. 2010;38(12):e132. doi: 10.1093/nar/gkq27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eddy SR. Profile hidden Markov models. Bioinformatics. 1998;14(9):755–63. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- 16.Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Research. 2009;37(Database issue):D233–8. doi: 10.1093/nar/gkn663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yin Y, Mao X, Yang J, Chen X, Mao F, Xu Y. dbCAN: a web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Research. 2012;40(W1):W445–W451. doi: 10.1093/nar/gks479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hess M, et al. Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science. 2011;331(6016):463–7. doi: 10.1126/science.1200387. [DOI] [PubMed] [Google Scholar]
- 19.Pipes L, et al. The non-human primate reference transcriptome resource (NHPRTR) for comparative functional genomics. Nucleic Acids Research. 2013;41(D1):D906–14. doi: 10.1093/nar/gks1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Henschel R, Lieber M, Wu LS, Nista PM, Haas BJ, LeDuc RD. Proceedings of the 1st Conference of the Extreme Science and Engineering Discovery Environment: Bridging from the eXtreme to the campus and beyond (XSEDE '12) ACM; New York, NY, USA: 2012. Trinity RNA-Seq assembler performance optimization. Article 45, 8 pages. [DOI] [Google Scholar]
- 21.Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nature Reviews Genetics. 2009;10:155–159. doi: 10.1038/nrg2521. [DOI] [PubMed] [Google Scholar]
- 22.Illumina's Body Map 2.0 transcriptome. http://www.ebi.ac.uk/arrayexpress/browse.html?keywords=E-MTAB-513.