

Abstract
Prostate cancer (PCa) is the most commonly diagnosed malignancy among western men and accounts for the second leading cause of cancer-related deaths. PCa tends to grow slowly and recent studies suggest that it relies on lipid fuel more than on aerobic glycolysis. However, the biochemical mechanisms governing the relationships between lipid synthesis, lipid utilization, and cancer growth remain unknown. To address the role of lipid metabolism in PCa we have used Etomoxir and Orlistat, clinically safe drugs that block lipid oxidation and lipid synthesis/lipolysis, respectively. Etomoxir is an irreversible inhibitor of the carnitine palmitoyltransferase (CPT1) enzyme that decreases beta oxidation in the mitochondria. Combinatorial treatments using Etomoxir and Orlistat resulted in synergistic decreased viability in LNCaP, VCaP and patient-derived benign and PCa cells. These effects were associated with decreased androgen receptor (AR) expression, decreased mammalian target of Rapamycin (mTOR) signaling and increased caspase-3 activation. Knockdown of CPT1A enzyme in LNCaP cells resulted in decreased palmitate oxidation but increased sensitivity to Etomoxir, with inactivation of AKT kinase and activation of caspase-3. Systemic treatment with Etomoxir in nude nice resulted in decreased xenograft growth over 21 days, underscoring the therapeutic potential of blocking lipid catabolism to decrease PCa tumor growth.
Keywords: Lipid, CPT1, Etomoxir, Orlistat, Prostate, androgen receptor
Introduction
Prostate cancer (PCa) is the most commonly diagnosed malignancy and the second highest contributor to cancer deaths in men in the United States (1). Currently, the standard systemic treatment for advanced PCa is based on androgen deprivation with initial positive responses, but PCa tumors eventually become resistant and restore AR signaling (2). After PCa becomes castration-resistant no curative treatments exist, making the identification of novel therapies imperative.
The mechanisms by which PCa cells use lipids to their benefit are poorly understood. De novo fatty acid synthesis can occur in cancer cells from glucose, in a pathway largely controlled by the enzyme fatty acid synthase (FASN), and is associated with cell growth, survival and drug resistance (3, 4). However, the biochemical mechanisms governing the relationships between lipid synthesis, lipid utilization, and cancer growth are still largely unknown.
Overexpression of key enzymes in lipid synthesis in prostate cancer is characteristic of both primary and advanced disease (5), suggesting that targeting lipid metabolism enzymes in PCa may offer new avenues for therapeutic approaches. Recent research has focused on the development of small FASN inhibitors for PCa therapeutics (6). The lipase and FASN inhibitor Orlistat has been used in several preclinical studies to decrease tumor growth (7–9). However, much less attention is being focused on the oxidation of newly synthesized lipid in PCa cells. The lipid utilization pathways in these cells are inferred from indirect evidence, but they are not well studied or understood (10, 11).
Several lines of evidence indicate that intracellular lipid turnover (not just lipid synthesis) is important in cancer cell survival: monoacylglycerol lipase, which catalyzes the release of fatty acids from intracellular lipid stores, promotes tumor growth and survival (12); blocking fat oxidation results in significant death of leukemia cells exposed to pro-apoptotic agents (13); fatty acid oxidation is associated with increased resistance to radiation and chemotherapeutic agents (14); finally, fatty acid oxidation fuels the production of metabolites needed to synthesize lipids and to protect cells from oxidative stress (15). Altogether, lipid oxidation is an important component of cancer metabolism together with aerobic glycolysis and lipogenesis, but remains ill-defined in PCa metabolism.
One way to study the role of lipid oxidation in a translatable manner is through the use of safe metabolic inhibitors that can be used both in the laboratory and the clinic. Etomoxir is a safe irreversible inhibitor of the long-chain fatty acid transporter and has been used in the treatment of heart failure (16). Etomoxir works by inhibiting carnitine palmitoyltransferase 1 (CPT1) and blocking the entry of long-chain fatty acids into mitochondria for oxidation, forcing cells to use the oxidation of glucose for energy (17). Only a few studies describe the effect of Etomoxir on cancer survival (13, 18), but there are no studies of its effects on PCa tumor metabolism.
In this report we examined the effects of pharmacologically blocking lipid synthesis and oxidation in PCa cell viability, AR content, molecular signaling and tumor growth. Our results suggest that PCa cells are dependent on lipid oxidation for their survival and this may represent a novel avenue to investigate new non-toxic therapeutic approaches to PCa treatment.
Materials and Methods
Cell culture and drug treatment
Cell lines were obtained from the University of Colorado Cancer Center Tissue Culture Core (year 2011) and were authenticated by Single Tandem Repeat analysis. Cells were used at low passage number and grown in RPMI or DMEM (for VCaP cells) containing 5% FBS supplemented with amino acids and Insulin (Hyclone). Charcoal stripped serum (CSS) was used for androgen-deprived conditions. Human prostate derived cells were isolated from de-identified surgical specimen at Wake Forest University using our previously described protocol (19). The histological origin of the sample was determined by analysis of the tissue surrounding the plug used for culture. Etomoxir-HCl (Sigma) was dissolved as a 15 mM stock solution; Orlistat (Sigma) was dissolved as a 50 mM stock in DMSO.
Cell viability and proliferation analysis
Cell proliferation was analyzed using the Beckman Coulter Vi-Cell Automated Cell Viability Analyzer. MTS proliferation assays were carried out using CellTiter 96® AQueous One solution from Promega according to manufacturer’s protocols.
Quantitative RT-PCR
Total RNA was isolated from cells (RNeasy, Qiagen), and cDNA was synthesized (high-capacity cDNA reverse transcription kit, Applied Biosystems) and quantified by real-time PCR using SYBR green (Applied Biosystems) detection. Results were normalized to the housekeeping gene Beta-2-microglobulin for mRNA and expressed as arbitrary units of 2−ΔΔCT relative to the control group. X-box binding protein-1 was amplified with Taq polymerase and products were resolved on 2% Tris-acetate-EDTA agarose gels and imaged on AlphaImager HP. Supplementary Table 1 contains the primer sequences used for the qPCR studies.
Immunoblots
Protein extracts (20 μg) were separated on a 7.5% or 4–20% SDS-PAGE gels and transferred to PVDF (General Electric) as described (20). All antibodies were from Cell Signaling (Supplementary Table 2). Band signals were visualized with ECL substrate (Pierce). Cell lysates were run on different blots to avoid stripping and re-probing except for phospho-antibody blots.
Glucose uptake
Basal glucose uptakes were determined as previously reported for human cells in vitro (20). Briefly, cells (105/well) in 6-well plates were incubated with 2-deoxyglucose (0.5 mM; Sigma-Aldrich), and [1,2-3H]2-deoxy-D-glucose (GE). Counts were converted to moles of glucose taken-up and normalized to the protein concentration of the lysates.
Lipid oxidation
Cells were plated in 12-well plates and grown to 70% confluence in their respective growth media conditions and with 150 μM Etomoxir at the indicated times. At the time of the assay, 1 mM carnitine, 100 μM BSA-conjugated fatty acids (Sigma) and C14-labeled fatty acids (1 uCi/mL; PerkinElmer) and fresh medium were added to the cells for 3 h. Entrapment of the generated 14CO2 was done by injecting perchloric acid as described (21). Radioactivity was measured by scintillation (Beckman) and normalized to protein.
CPT1A shRNA transductions
TRCN0000036279 (CPTsh1) and TRCN0000036281(CPTsh2) CPT1A shRNAs and the non-targeting control SHC002 were purchased from Functional Genomics Core, Boulder, Colorado. Lentiviral transduction and selection were performed according to Sigma’s MISSION protocol but using lentiviral packing plasmids pCMV-R8.74psPAX2 and VSV-G/pMD2.G and transfection reagent TransIT-LT1 (Mirius).
Lipid fractionation and analysis by Gas Chromatography coupled to Mass Spectrometry (GC/MS)
LNCaP cells were grown in 60 mm plates and treated with Etomoxir or vehicle for 24 h. Incubation was stopped by adding 1 vol. of methanol. To an aliquot representing 25% of the total sample, a mixture of internal standards was added: 20 μL of a solution containing 0.5 nmol each of 12:0-ceramide, 12:0-sphingomyelin, glucosyl(β)-C12-ceramide and Lactosyl(β)-C12-ceramide (Cer/Sph Mixture I; Avanti Polar Lipids, Alabaster, AL). After extracting lipids using Bligh & Dyer method (22), sphingolipids were analyzed by liquid chromatography/tandem mass spectrometry essentially as described (23). Data were analyzed using MultiQuant software from AB Sciex (Framingham, MA), and are presented as the ratios between the integrated area of the intensity peak of each analyte and the intensity peak of the corresponding internal standard.
Xenograft production in nude mice
Male athymic nude mice, 4–6 weeks old, were purchased from Harlan Laboratories. Tumor xenografts were generated by injecting human VCaP cells in the flank of nude male mice as described (24). Approximately 2×106 cells were used for each injection. When tumors were palpable the mice were randomized into 2 groups: vehicle or Etomoxir. Treatment was carried out with intraperitoneal injection of Etomoxir (40 mg/kg) or vehicle (water) every other day for 3 weeks. All procedures were carried out under a protocol approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Colorado. After treatment, xenografts were collected and processed for immunohistochemical studies using standard protocols at the UC Denver Pathology core.
Statistics
ANOVA tests were used to compare between groups followed by post hoc Tukey tests when appropriate. Analysis of in vivo tumor growth was done with ANOVA followed by t-tests with SPSS v20 software. All tests were 2-sided. p<0.05 was considered significant. Data represent mean ± sd except for qPCR that is mean ± sem. Synergism was analyzed using CalcuSyn 2.0 (Biosoft) as described (25).
Results
Lipid metabolic inhibitors reduce the viability of prostate cancer cell lines
Benign (BPH-1, epithelial and WPMY-1, stroma) and PCa (VCaP, LNCaP and PC3) cell lines were treated with Etomoxir (75 μM) for 48 h and subjected to viability analysis using trypan blue exclusion. Figure 1A shows that PCa cells havedecreased viability in response to Etomoxir when compared to normal BPH-1 (epithelial) and WPMY-1 (stroma) prostate-derived cell lines. VCaP cells showed the highest sensitivity to Etomoxir treatment, with a 60% reduction in viability (p<0.01), followed by LNCaP (50% reduction, p<0.01) and PC3 (40% reduction, p<0.01).
Figure 1. Lipid metabolic inhibitors reduce the viability of prostate cancer cell lines.

A. Relative cell viability of prostate-derived cell lines exposed to Etomoxir (75μM) for 48 hours, *p<0.001 compared to vehicle. B, Viability of LNCaP cells exposed to inhibitors Etomoxir (75 μM), Orlistat (20 μM): ^p≤0.001, compared to vehicle, # p≤0.016, compared to single drug. C, Viability of VCaP cells: *p≤0.001, compared to vehicle, # p≤0.001, compared to single drug. D, MTS proliferation assay of LNCaP cells in FBS media. *p<0.02, **p=0.001 combination vs. single drug. E, CSS media *p<0.001 combination vs. single drug. F, MTS assay of VCaP cells in FBS: *p<0.001 combination vs. single drug. G, CSS media *p≤0.003, ^p= 0.026 combination vs. single drug. H–I, MTS assay of patient-matched prostate-derived benign (H) and cancer (I) cells exposed to inhibitors for 48 hours. Two-tailed t-tests: a, p<0.01 compared to Orlistat treatment in benign cells. b, p<0.05 compared to Etomoxir treatment in benign cells. c, p<0.05 compared to combinatorial treatment in benign cells. Combinatorial index is shown at bottom of the graph, where CI<1.0 indicates synergy.
Since LNCaP cells were sensitive to Etomoxir and they are also known to be sensitive to Orlistat (7), we used Etomoxir (75 μM) and Orlistat (20 μM) to study the viability and proliferation of LNCaP and VCaP cells exposed to both inhibitors. Treatments were done in FBS and androgen-deprived CSS conditions. Figure 1B and 1C show the effects of the drug combination on cell viability for LNCaP and VCaP cells respectively, indicating a strong effect (p<0.001) of both inhibitors compared to control.
Figures 1D and 1E show the proliferation of LNCaP cells treated with drugs in the presence of FBS or CSS media, respectively. A dose-dependent effect and a strong inhibition of proliferation were observed with the combination of drugs (p<0.001 compared to either drug alone). The combinatorial index (CI) for the drugs is indicated at the bottom of figures. A number less than 1 indicates synergism between both drugs, while 1 or greater reflects additive or antagonistic effects, respectively. These results indicate that lower doses were needed to obtain a synergistic effect on proliferation. CSS media increased the sensitivity of LNCaP cells to the drugs, especially Etomoxir. Figures 1F and 1G show the same paradigm for VCaP cells. Treatments in the presence of CSS media resulted in increased sensitivity to the combination of drugs compared to treatments with FBS-containing media. This was reflected in the strong synergistic CI scores.
We also examined the effect of Etomoxir and Orlistat on the proliferation of patient-derived primary human prostate epithelial cells. Purity of the epithelial cultures was assessed by E-cadherin and Vimentin expression (Supplemetary Figure 1). Figures 1H–I shows the effect of inhibitors on benign and cancer primary cells, respectively. Increasing drug concentrations decreased proliferation (p<0.001) for both cell types. However, the cancer cells were more sensitive to each drug than the benign cells at the lower drug concentrations, and more sensitive to the combination of drugs at the higher concentrations (p<0.05). Strong synergism was observed in the cancer cell lines (CI=0.5) compared to the benign cells (CI = 0.75), suggesting a higher sensitivity of the cancer cells to the inhibitors, especially Etomoxir. Additional patient-derived primary prostate cell lines are shown in Supplementary Figure 2.
Etomoxir and Orlistat decrease AR isoform expression and modify lipid oxidation and glucose uptake
Since AR activity is associated with lipid metabolism, we examined the expression of AR mRNA as well as its downstream targets PSA and NKX3.1. Figures 2A–2B show a significant decrease of transcripts in the presence of inhibitors, regardless of the presence of androgens (p≤0.05). Similar results were obtained for VCaP cells but to a lesser extent (Figures 2C–2D). Examination of the effect of inhibitors on lipid oxidation in PCa and BPH-1 cells was done by trapping the CO2 produced by the cells after treatment. Figure 2E shows increased lipid oxidation in PCa cells compared to the benign BPH-1 cells (~5-fold, p<0.01). Orlistat incubation did not affect oxidation rate significantly, except for VCaP cells. However, addition of Etomoxir decreased the oxidation rate by 50% in LNCaP and VCaP cells (p≤0.05). These results demonstrate that PCa cells are lipolytic, and suggest that their survival strongly depends on AR action and lipid utilization.
Figure 2. Etomoxir and Orlistat decrease AR isoform expression and modify lipid oxidation and glucose uptake.

Expression of full length AR (ARfl), variant 7 (ARv7), Total AR, PSA and NKX3.1 genes in LNCaP (A–B) and VCaP (C–D) cells treated with Etomoxir (75 μM) and/or Orlistat (20 μM). Post hoc tests compared to vehicle: A *p≤0.004, B *p≤0.05, C *p≤0.03, D *p≤0.05. E, Rate of 14C-palmitate oxidation in LNCaP (ANOVA, p=0.008), VCaP (ANOVA p=0.02) and BPH-1 cells exposed to inhibitors for 6 hours. *p<0.01, ^p≤0.02 compared to vehicle. $p<0.02 PCa cells compared to BPH-1. F, Glucose uptake of LNCaP (ANOVA, p<0.0001), VCaP (ANOVA p<0.001) and BPH-1(ANOVA, p=0.002) cells exposed to inhibitors for 6 hours. Post hoc tests: Comparisons to vehicle treatment: #p<0.05, *p<0.05, ^p≤0.007. VCaP and BPH-1 compared to LNCaP treated with etomoxir,ap<0.05.
Since Etomoxir is known to increase glucose uptake in heart cells (26) we also examined the effect of inhibitors on glucose uptake, Figure 2F. Interestingly, the drug combination produced a significant increase in glucose uptake in all the cells but was greater in BPH-1 cells, suggesting that different metabolic pathways operate in benign and cancer cells.
Lipid catabolism blockade results in decreased mTOR signaling and increased apoptosis
In order to study the molecular mechanisms of Etomoxir and Orlistat on PCa cells we examined the phosphorylation status of the pro-apoptotic BAD protein, which has been shown to be associated with the metabolic status of the cell and is necessary to protect PCa cells from apoptosis, likely mediated by AKT activation (27). Figures 3A–3B show blots of Etomoxir and/or Orlistat-treated LNCaP and VCaP lysates respectively. Decreased BAD S112 phosphorylation was observed in both cells lines with etomoxir treatment at 6 hours.
Figure 3. Lipid catabolism blockade results in decreased mTOR signaling and increased apoptosis.

AKT and BAD phosphorylation of LNCaP (A) and VCaP (B) lysates treated with Etomoxir (75 μM) and/or Orlistat (20 μM) for 6 hours. C, Expression of mTOR-S6K-BAD-Caspase3 axis after metabolic treatments for 16 hours in LNCaP cells. D, Blot of AMPK activation and ACC2 inactivation of LNCaP lysates E, Diagram of molecular pathway likely involved in the LNCaP cells. F, Expression of mTOR-S6K-BAD-Caspase3 axis after 16-hour treatments in VCaP cells. G, AMPK and phospho-ACC2 in VCaP lysates.
mTOR and AMPK are also involved in nutrient sensing and integrate fuel homeostasis and cell survival. Etomoxir treatment resulted in decreased activation of mTORC1 and its downstream substrates S6K and 4EBP1, which are involved in protein synthesis and survival, especially the S6K-BAD signaling axis (28) (Figure 3C). Interestingly, less caspase-3 activation was observed in the BPH-1 cells (Supplementary Figure 3). Additionally, strong suppression of ACC, an enzyme involved in fat synthesis, was also observed with treatment, indicating AMPK activation. Figure 3D shows AMPK activation after 16 h of treatment in LNCaP cells. Figure 3E shows a putative diagram of these molecular players. Figures 3F–3G show blots for VCaP lysates. Decreased mTOR signaling axis and increased caspase-3 activation was observed for the drug combination. Orlistat also increased caspase-3, suggesting increased sensitivity to orlistat in VCaP cells.
ER stress and apoptotic ceramides are increased after lipid metabolism blockade in LNCaP cells
Since mTOR inactivation is associated with survival signals like endoplasmic reticulum (ER) stress and autophagy, we examined the expression of canonical markers in LNCaP and VCaP lysates. Figure 4A shows activation of the ER stress transcription factor XBP-1 by splicing in response to Etomoxir. Interestingly, addition of palmitate to the treatments resulted in less XBP-1 splicing, underscoring the role of lipogenesis/lipolysis cycle in cell homeostasis (Supplementary Figure 4). ER stress-related factors ATF4, CHOP, GADD34 and GRP78 were also increased after Etomoxir treatment (Figure 4B). The most dramatic changes were observed in the expression of CHOP (C/EBP homologous protein) and GADD34 (Growth arrest and DNA damage 34) expression, both of which have been associated with the pro-apoptotic side of the ER stress response (29). No changes in the mRNA expression of ER stress-related factors were observed in VCaP cells (not shown).
Figure 4. ER stress and apoptotic ceramides are increased after lipid metabolism blockade in LNCaP cells.

A, Orlistat and Etomoxir treatments induce strong activation of the XBP-1 transcription factor after 16 hours of treatment. B, Relative qPCR analysis of the ER stress related factors after 16 hours. For each gene examined, ANOVA<0.001 across treatments. Post hoc tests: *p<0.01, a p=0.002 compared to vehicle. C–D, Western blots for phospho-eIF2a and LC3 fragments of LNCaP and VCaP lysates, respectively, treated with inhibitors. Total eIF2a bands were used as loading controls. E, Ceramide species in LNCaP cells were treated with Etomoxir for 24 hours and harvested for lipid extraction and ceramide analysis. Two-sided t test: *p≤0.05. Fatty acid composition of the ceramide molecules are indicated in the x-axis.
Since ER stress also leads to a block in protein translation, we examined phospho-eIF2α in the same LNCaP samples that had XBP1 activation (Figure 4C). A slight increase in p-eIF2a was observed with Orlistat as shown before (30), but stronger signals were observed in the Etomoxir-treated samples. The weaker p-eIF2a signal in the combination treatment was parallel to the increased expression of the GADD34 phosphatase regulator (Figure 4B), potentially leading to suppression of the unfolded protein response and induction of apoptosis (31). Lastly, since unresolved ER stress activates autophagy (32), we examined the conversion of LC3-II (17 KDa), which was evident in the Etomoxir-treated samples (Figure 4C). VCaP cells did not show increased phospho-eIF2a with etomoxir, but changes in autophagy were noticeable with Orlistat (Figure 4D).
Since ceramides containing 16- and 18-carbon fatty acids are also associated with decreased mTOR activity and autophagy (33), we examined the levels of different ceramide species present in Etomoxir-treated LNCaP cells after 24 hours. Figure 4E shows a significant increase in ceramide species containing palmitic (16:0) and stearic (18:0) acyl chains. Interestingly, 16C and 18C containing-ceramides appear to be most important for intrinsic apoptosis induction (34).
Downregulation of CPT1A decreases fat oxidation and leads to apoptosis
To verify the role of CPT1A as the target of Etomoxir action we used two different shRNAs to knock down (KD) CPT1A expression in LNCaP cells. Control cells were transduced with a non-targeting shRNA construct. Clones were treated with vehicle (V, H2O/DMSO), Orlistat (O, 20 μM), Orlistat/Etomoxir (OE, 20 μM/75 μM) or Etomoxir alone (E, 75 μM) for 24 h. Figure 5A shows the decrease in CPT1A expression in the KD clones compared to control cells (V lanes). An unexpected increase in CPT1A expression with Etomoxir was observed in all clones, suggesting a compensatory feedback effect. A lack of S112 and S155 phosphorylation of BAD was observed in the combinatorial treatment, suggesting an activation of BAD pro-apoptotic activity (27, 28). Decreases in pAKT and mTOR action (via pS6K) were also observed, concomitant with cleaved caspase-3 signal, suggesting apoptosis induction. VCaP cells were not viable after CPT1A KD selection but they also showed a slight increase of CPT1A protein expression with Etomoxir (Figure 5B).
Figure 5. Downregulation of CPT1A decreases fat oxidation and leads to apoptosis.
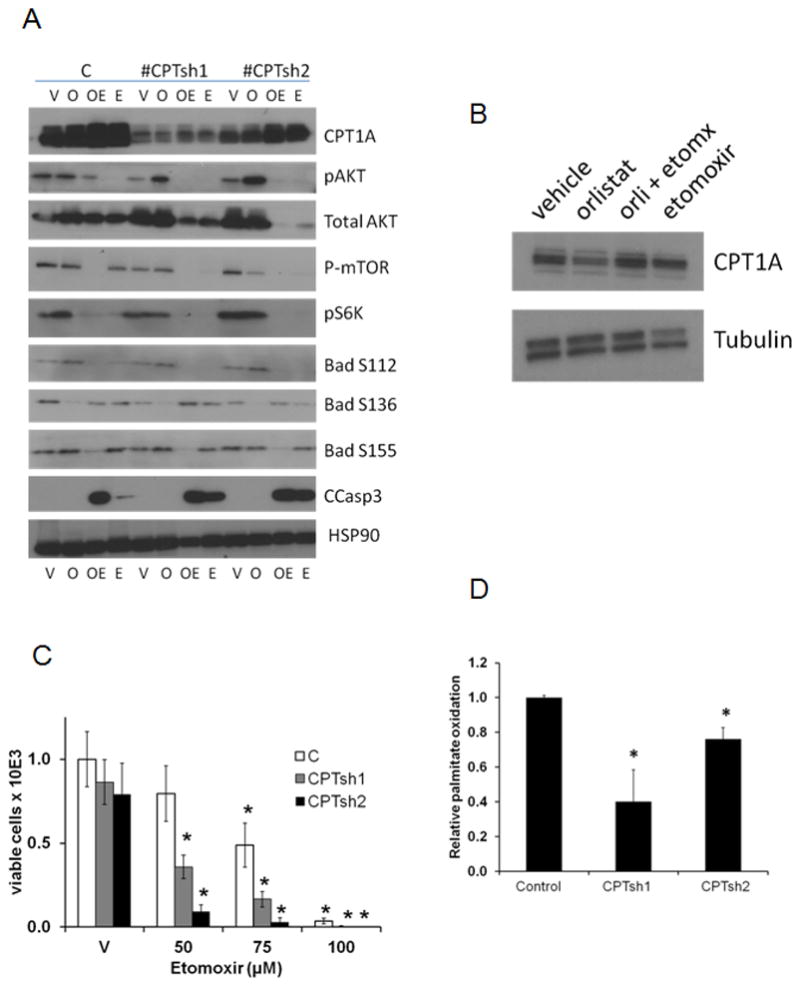
A, Western blots of CPT1A KD cells treated with vehicle (V), Orlistat (O), Orlistat+Etomoxir (OE) or Etomoxir alone (E) for 24 hours. B CPT1A expression in VCaP cells treated with inhibitors C, Trypan Blue viability assay of shRNA clones treated with 3 doses of Etomoxir for 2 days; *P<0.01 compared to control cells treated with vehicle. D, Palmitate oxidation rate in KD clones compared to control, *p≤0.01compared to control shRNA clone.
Analysis of cell viability and sensitivity of clones to Etomoxir was also examined (Figure 5C). CPT1A KD clones were sensitive to 50 μM Etomoxir (reduced by 60%, p<0.001) while the control cells showed a 20% decrease in viability. This effect was dose-dependent as the 75 and 100 μM doses decreased viability in all the clones. Since the total AKT expression was strongly reduced in the CPT1A KD clones with the Etomoxir treatments, this suggests a lack of compensatory survival pathway leading to decreased viability. Concomitant with decreased CPT1A expression, we also observed lower palmitate oxidation in the KD clones (Figure 5D). Clone CPTsh1 showed a stronger decrease in fat oxidation (60% decrease, p<0.01) followed by CPTsh2 (25% decrease, p=0.01).
Systemic treatment with Etomoxir decreases xenograft tumor growth in nude mice
Male nude mice were grafted with VCaP cancer cells subcutaneously, randomized to four groups (2 vehicles and 2 treatment doses) when the tumors were palpable, and treated with Etomoxir systemically for 21 days. VCaP cells were used instead of LNCaP because they grow well as xenografts in nude mice (35) and are also sensitive to Etomoxir, Figure 1A. To account for possible toxicity we used two different doses of Etomoxir. Figure 6A shows the progressive growth of tumors using a 40 mg/kg dose every other day. Significant differences were observed in the last week of treatment (ANOVA p<0.001, post-hoc p<0.05) suggesting that systemic inhibition of fat oxidation impairs growth of implanted tumors. Animals were healthy and their body weight remained stable over the 3 weeks of treatments, Figure 6B. CPT1A content in the tumors was still strong after 21 days, albeit weaker in the Etomoxir group, Figure 6C. Phospho-S6 (marker of mTOR action), did not show any changes. The study using 20mg/kg did not produce significant results over 3 weeks (data not shown).
Figure 6. Systemic treatment with Etomoxir decreases xenograft tumor growth in nude mice.
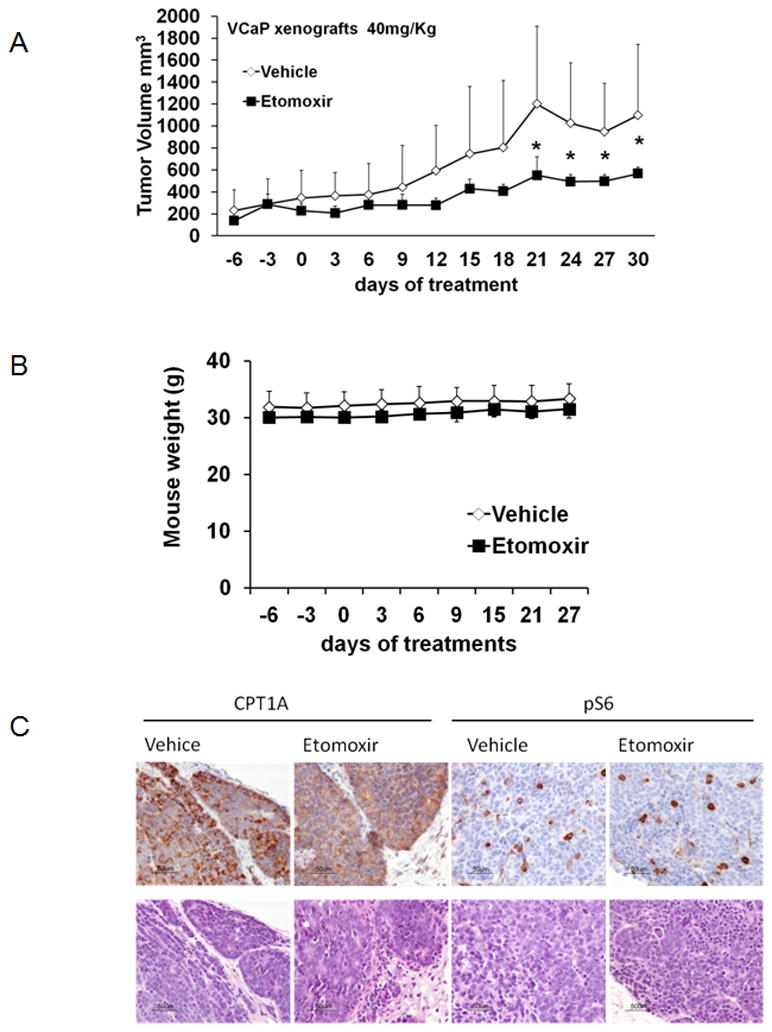
A, Tumor growth progression (mean ±SD) in mice treated with 40mg/Kg Etomoxir injections for 3 weeks. (*P<0.05 Tukey, compared to vehicle-treated tumors). B, Mouse body weight over the course of the experimental treatments. C, Representative CPT1A and phospho-S6 stains of tumor xenografts at the end of study.
Discussion
The results of our study point to an important role of beta-oxidation of fatty acids in PCa. We have observed a significant decrease in viability when PCa cells are grown in the presence of the CPT1 inhibitor Etomoxir, but this effect was not replicated in non-tumor forming cells, suggesting a possible therapeutic window for PCa. Furthermore, since lipogenesis from sugar carbons is a well documented observation in PCa (36), the combinatorial approach of inhibiting fat oxidation and fat synthesis simultaneously, with Etomoxir and Orlistat respectively, generated synergistic results in PCa cell growth assays. Unfortunately, the main problem with targeting FASN is the low solubility and bioavailability of currently approved drugs (like Orlistat), mainly due to the hydrophobicity of the FASN active site (37), making it difficult to advance to clinical trials.
Androgen receptors (AR) are involved in the activation of lipid metabolism (38) as well as the growth of PCa cells, even in castration-resistant or recurrent PCa (39). Very little is known about how lipids regulate the AR and its variants in PCa. Our results blocking lipid catabolism and significantly decreasing AR expression in both LNCaP and VCaP cells suggest that thwarting the ability of PCa cells to utilize lipids, regardless of the PTEN status, may synergize with current anti-androgen therapies for a more effective AR blockade. Expression of PSA and NKX3.1 was also decreased, suggesting a reduced AR-signaling axis. Furthermore, benign BPH-1 cells showed higher glucose uptake than PCa cells when treated with both Etomoxir and Orlistat, suggesting that they were able to compensate for the lipid blockade. This lack of metabolic flexibility in PCa cells may contribute to their decreased viability when challenged with metabolic stress, opening doors for combinatorial treatments to be explored clinically, like Etomoxir and Enzalutamide.
The mammalian kinase mTOR is deregulated in nearly 100% of advanced human prostate cancers. However there are not clinically effective drugs that target mTOR activity (40). We have observed decreased mTOR activation when cells were challenged with metabolic inhibitors, leading to increased 4EBP1 inhibitor activity (less phosphorylated) that likely reduces protein translation and growth (41). Interestingly, activation of caspase-3 was different between LNCaP and VCaP cells. Etomoxir was the driver for apoptosis in LNCaP cells, while VCaP apoptosis appeared dependent on Orlistat treatment. These cell line differences in response to the inhibitors may rest in their genetic differences: LNCaP cells have activation of survival AKT pathways while VCaP likely rely on other pathways. The observation that pAKT was increased with Orlistat in the CPT1A KD clones was unexpected, but could reflect a compensatory mechanism to increase FASN activity, as was previously observed in PCa (42).
The role of BAD proteins in sensing mitochondrial metabolism is well described (43), linking glucose use with apoptosis. Our studies provide evidence that lipid use by PCa cells is also connected to the apoptotic machinery, since phospho-BAD S112 was decreased in both cell lines leading to caspase activation with the drug combination. The possibility that the observed decreased mTOR-S6K axis is responsible for this effect needs to be further validated.
Additional consequences of blocking lipid turnover in LNCaP cells are ER stress, autophagy and ceramide production. It is unknown which phenomenon occurs first but it is possible that accumulated palmitate in the ER (that is not oxidized) leads to activation of the unfolded protein response and a survival autophagic response, an effect that has been previously reported in yeast (44) and more recently in leukemia cells (45), where phosphorylation of eIF2a by PERK leads to a survival autophagy response. Ceramide synthesis in PCa is also another potential target for therapeutic intervention. We have observed significant increases in ceramide species in LNCaP cells treated with Etomoxir, suggesting that the excess fatty acids (mainly C16:0 and C18:0) that could not get oxidized in the mitochondria due to CPT1 inhibition, were used to generate pro-apoptotic ceramides. Indeed, ceramidases are becoming therapeutic targets for advanced PCa, since these degrading enzymes are abundant and contribute to chemoresistance (34).
The most significant preclinical extension of our work is the result from the PCa xenografts in mice. The effect of fat oxidation inhibition in leukemia cells is well documented (13, 46) but there are no reports of its effects on PCa cells, which depend on fat metabolism for survival. Results from injections with Etomoxir revealed a dose-dependent effect on tumor growth without affecting the body weight or health of the mice. These results emphasize the dependence of PCa cells on fatty acid availability for oxidation and ATP production. Since the use of Orlistat in our in vitro studies further decreased the viability of the PCa cells, this suggests that de novo lipogenesis and/or lipase activity are likely the sources of fatty acids for beta-oxidation in PCa cells. The possibility of using 2-DG (non-usable glucose) and Etomoxir in combination is a promising therapeutic avenue for PCa that needs to be explored.
Several studies have indicated that fat availability to tumors (via high-fat diets or obesity) leads to PCa growth (47, 48). However, lipid markers like FASN and IGF-1 levels do not fully explain the association between obesity and poor PCa outcome (48), indicating that the availability of lipids to tumors may be an important factor worth exploring in depth. This is relevant in the setting of androgen deprivation therapy, where the metabolic syndrome with altered blood lipid profile favors increased fatty acid availability to the growing PCa tumors. Additionally our data suggests that lipid catabolism also modulates AR content, likely creating a feed-forward cycle that sustains PCa growth. In conclusion, systemically targeting lipid use by tumors offers possibilities to reduce PCa tumor burden.
Supplementary Material
Acknowledgments
Financial support: NIH (K01CA168934 to I.R. Schlaepfer), ACS (PF-117219 and IRG-57-001-50 to I.R. Schlaepfer), the GMaP Cancer Research Network (Grant No. 3 U54 CA153511), and contributions from Herbert Crane Endowment, William R. Meyn Foundation, and Robert Rifkin Endowment (to L.M. Glodé)
Footnotes
Authors declare no conflict of interest
References
- 1.Carlsson S, Vickers AJ, Roobol M, Eastham J, Scardino P, Lilja H, et al. Prostate cancer screening: facts, statistics, and interpretation in response to the US Preventive Services Task Force Review. J Clin Oncol. 2012;30:2581–4. doi: 10.1200/JCO.2011.40.4327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Knudsen KE, Scher HI. Starving the addiction: new opportunities for durable suppression of AR signaling in prostate cancer. Clin Cancer Res. 2009;15:4792–8. doi: 10.1158/1078-0432.CCR-08-2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Suburu J, Chen YQ. Lipids and prostate cancer. Prostaglandins Other Lipid Mediat. 2012;98:1–10. doi: 10.1016/j.prostaglandins.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Swinnen JV, Brusselmans K, Verhoeven G. Increased lipogenesis in cancer cells: new players, novel targets. Curr Opin Clin Nutr Metab Care. 2006;9:358–65. doi: 10.1097/01.mco.0000232894.28674.30. [DOI] [PubMed] [Google Scholar]
- 5.Vavere AL, Kridel SJ, Wheeler FB, Lewis JS. 1-11C-acetate as a PET radiopharmaceutical for imaging fatty acid synthase expression in prostate cancer. J Nucl Med. 2008;49:327–34. doi: 10.2967/jnumed.107.046672. [DOI] [PubMed] [Google Scholar]
- 6.Flavin R, Zadra G, Loda M. Metabolic alterations and targeted therapies in prostate cancer. J Pathol. 2011;223:283–94. doi: 10.1002/path.2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kridel SJ, Axelrod F, Rozenkrantz N, Smith JW. Orlistat is a novel inhibitor of fatty acid synthase with antitumor activity. Cancer Res. 2004;64:2070–5. doi: 10.1158/0008-5472.can-03-3645. [DOI] [PubMed] [Google Scholar]
- 8.Kuhajda FP. Fatty acid synthase and cancer: new application of an old pathway. Cancer Res. 2006;66:5977–80. doi: 10.1158/0008-5472.CAN-05-4673. [DOI] [PubMed] [Google Scholar]
- 9.Pemble CW, Johnson LC, Kridel SJ, Lowther WT. Crystal structure of the thioesterase domain of human fatty acid synthase inhibited by Orlistat. Nat Struct Mol Biol. 2007;14:704–9. doi: 10.1038/nsmb1265. [DOI] [PubMed] [Google Scholar]
- 10.Carracedo A, Cantley LC, Pandolfi PP. Cancer metabolism: fatty acid oxidation in the limelight. Nat Rev Cancer. 2013;13:227–32. doi: 10.1038/nrc3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu Y. Fatty acid oxidation is a dominant bioenergetic pathway in prostate cancer. Prostate Cancer Prostatic Dis. 2006;9:230–4. doi: 10.1038/sj.pcan.4500879. [DOI] [PubMed] [Google Scholar]
- 12.Nomura DK, Long JZ, Niessen S, Hoover HS, Ng SW, Cravatt BF. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell. 2010;140:49–61. doi: 10.1016/j.cell.2009.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Samudio I, Harmancey R, Fiegl M, Kantarjian H, Konopleva M, Korchin B, et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J Clin Invest. 2010;120:142–56. doi: 10.1172/JCI38942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harper ME, Antoniou A, Villalobos-Menuey E, Russo A, Trauger R, Vendemelio M, et al. Characterization of a novel metabolic strategy used by drug-resistant tumor cells. FASEB J. 2002;16:1550–7. doi: 10.1096/fj.02-0541com. [DOI] [PubMed] [Google Scholar]
- 15.Pike LS, Smift AL, Croteau NJ, Ferrick DA, Wu M. Inhibition of fatty acid oxidation by etomoxir impairs NADPH production and increases reactive oxygen species resulting in ATP depletion and cell death in human glioblastoma cells. Biochim Biophys Acta. 2011;1807:726–34. doi: 10.1016/j.bbabio.2010.10.022. [DOI] [PubMed] [Google Scholar]
- 16.Abozguia K, Clarke K, Lee L, Frenneaux M. Modification of myocardial substrate use as a therapy for heart failure. Nat Clin Pract Cardiovasc Med. 2006;3:490–8. doi: 10.1038/ncpcardio0583. [DOI] [PubMed] [Google Scholar]
- 17.Schmidt-Schweda S, Holubarsch C. First clinical trial with etomoxir in patients with chronic congestive heart failure. Clin Sci (Lond) 2000;99:27–35. [PubMed] [Google Scholar]
- 18.Hernlund E, Ihrlund LS, Khan O, Ates YO, Linder S, Panaretakis T, et al. Potentiation of chemotherapeutic drugs by energy metabolism inhibitors 2-deoxyglucose and etomoxir. Int J Cancer. 2008;123:476–83. doi: 10.1002/ijc.23525. [DOI] [PubMed] [Google Scholar]
- 19.Barclay WW, Woodruff RD, Hall MC, Cramer SD. A system for studying epithelial-stromal interactions reveals distinct inductive abilities of stromal cells from benign prostatic hyperplasia and prostate cancer. Endocrinology. 2005;146:13–8. doi: 10.1210/en.2004-1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schlaepfer IR, Hitz CA, Gijon MA, Bergman BC, Eckel RH, Jacobsen BM. Progestin modulates the lipid profile and sensitivity of breast cancer cells to docetaxel. Mol Cell Endocrinol. 2012;363:111–21. doi: 10.1016/j.mce.2012.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Consitt LA, Bell JA, Koves TR, Muoio DM, Hulver MW, Haynie KR, et al. Peroxisome proliferator-activated receptor-gamma coactivator-1alpha overexpression increases lipid oxidation in myocytes from extremely obese individuals. Diabetes. 2010;59:1407–15. doi: 10.2337/db09-1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.BLIGH EG, DYER WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–7. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 23.Merrill AH, Jr, Sullards MC, Allegood JC, Kelly S, Wang E. Sphingolipidomics: high-throughput, structure-specific, and quantitative analysis of sphingolipids by liquid chromatography tandem mass spectrometry. Methods. 2005;36:207–24. doi: 10.1016/j.ymeth.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 24.Cai C, Wang H, Xu Y, Chen S, Balk SP. Reactivation of androgen receptor-regulated TMPRSS2:ERG gene expression in castration-resistant prostate cancer. Cancer Res. 2009;69:6027–32. doi: 10.1158/0008-5472.CAN-09-0395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rao A, Woodruff RD, Wade WN, Kute TE, Cramer SD. Genistein and vitamin D synergistically inhibit human prostatic epithelial cell growth. J Nutr. 2002;132:3191–4. doi: 10.1093/jn/131.10.3191. [DOI] [PubMed] [Google Scholar]
- 26.Lionetti V, Stanley WC, Recchia FA. Modulating fatty acid oxidation in heart failure. Cardiovasc Res. 2011;90:202–9. doi: 10.1093/cvr/cvr038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith AJ, Karpova Y, D’Agostino R, Jr, Willingham M, Kulik G. Expression of the Bcl-2 protein BAD promotes prostate cancer growth. PLoS ONE. 2009;4:e6224. doi: 10.1371/journal.pone.0006224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scholl C, Frohling S, Dunn IF, Schinzel AC, Barbie DA, Kim SY, et al. Synthetic lethal interaction between oncogenic KRAS dependency and STK33 suppression in human cancer cells. Cell. 2009;137:821–34. doi: 10.1016/j.cell.2009.03.017. [DOI] [PubMed] [Google Scholar]
- 29.Kraskiewicz H, FitzGerald U. InterfERing with endoplasmic reticulum stress. Trends Pharmacol Sci. 2012;33:53–63. doi: 10.1016/j.tips.2011.10.002. [DOI] [PubMed] [Google Scholar]
- 30.Little JL, Wheeler FB, Koumenis C, Kridel SJ. Disruption of crosstalk between the fatty acid synthesis and proteasome pathways enhances unfolded protein response signaling and cell death. Mol Cancer Ther. 2008;7:3816–24. doi: 10.1158/1535-7163.MCT-08-0558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Connor JH, Weiser DC, Li S, Hallenbeck JM, Shenolikar S. Growth arrest and DNA damage-inducible protein GADD34 assembles a novel signaling complex containing protein phosphatase 1 and inhibitor 1. Mol Cell Biol. 2001;21:6841–50. doi: 10.1128/MCB.21.20.6841-6850.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qin L, Wang Z, Tao L, Wang Y. ER stress negatively regulates AKT/TSC/mTOR pathway to enhance autophagy. Autophagy. 2010;6:239–47. doi: 10.4161/auto.6.2.11062. [DOI] [PubMed] [Google Scholar]
- 33.Taniguchi M, Kitatani K, Kondo T, Hashimoto-Nishimura M, Asano S, Hayashi A, et al. Regulation of autophagy and its associated cell death by “sphingolipid rheostat”: reciprocal role of ceramide and sphingosine 1-phosphate in the mammalian target of rapamycin pathway. J Biol Chem. 2012;287:39898–910. doi: 10.1074/jbc.M112.416552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grosch S, Schiffmann S, Geisslinger G. Chain length-specific properties of ceramides. Prog Lipid Res. 2012;51:50–62. doi: 10.1016/j.plipres.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 35.Korenchuk S, Lehr JE, MClean L, Lee YG, Whitney S, Vessella R, et al. VCaP, a cell-based model system of human prostate cancer. In Vivo. 2001;15:163–8. [PubMed] [Google Scholar]
- 36.Migita T, Ruiz S, Fornari A, Fiorentino M, Priolo C, Zadra G, et al. Fatty acid synthase: a metabolic enzyme and candidate oncogene in prostate cancer. J Natl Cancer Inst. 2009;101:519–32. doi: 10.1093/jnci/djp030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maier T, Leibundgut M, Ban N. The crystal structure of a mammalian fatty acid synthase. Science. 2008;321:1315–22. doi: 10.1126/science.1161269. [DOI] [PubMed] [Google Scholar]
- 38.Swinnen JV, Van Veldhoven PP, Esquenet M, Heyns W, Verhoeven G. Androgens markedly stimulate the accumulation of neutral lipids in the human prostatic adenocarcinoma cell line LNCaP. Endocrinology. 1996;137:4468–74. doi: 10.1210/endo.137.10.8828509. [DOI] [PubMed] [Google Scholar]
- 39.Li Y, Chan SC, Brand LJ, Hwang TH, Silverstein KA, Dehm SM. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res. 2013;73:483–9. doi: 10.1158/0008-5472.CAN-12-3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hsieh AC, Liu Y, Edlind MP, Ingolia NT, Janes MR, Sher A, et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature. 2012;485:55–61. doi: 10.1038/nature10912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pourdehnad M, Truitt ML, Siddiqi IN, Ducker GS, Shokat KM, Ruggero D. Myc and mTOR converge on a common node in protein synthesis control that confers synthetic lethality in Myc-driven cancers. Proc Natl Acad Sci U S A. 2013;110:11988–93. doi: 10.1073/pnas.1310230110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Van de Sande T, Roskams T, Lerut E, Joniau S, Van PH, Verhoeven G, et al. High-level expression of fatty acid synthase in human prostate cancer tissues is linked to activation and nuclear localization of Akt/PKB. J Pathol. 2005;206:214–9. doi: 10.1002/path.1760. [DOI] [PubMed] [Google Scholar]
- 43.Danial NN. BAD: undertaker by night, candyman by day. Oncogene. 2008;27 (Suppl 1):S53–S70. doi: 10.1038/onc.2009.44. [DOI] [PubMed] [Google Scholar]
- 44.Yorimitsu T, Nair U, Yang Z, Klionsky DJ. Endoplasmic reticulum stress triggers autophagy. J Biol Chem. 2006;281:30299–304. doi: 10.1074/jbc.M607007200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hart LS, Cunningham JT, Datta T, Dey S, Tameire F, Lehman SL, et al. ER stress-mediated autophagy promotes Myc-dependent transformation and tumor growth. J Clin Invest. 2012;122:4621–34. doi: 10.1172/JCI62973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tsunekawa-Imai N, Miwa H, Shikami M, Suganuma K, Goto M, Mizuno S, et al. Growth of xenotransplanted leukemia cells is influenced by diet nutrients and is attenuated with 2-deoxyglucose. Leuk Res. 2013;37:1132–6. doi: 10.1016/j.leukres.2013.05.017. [DOI] [PubMed] [Google Scholar]
- 47.Kobayashi N, Barnard RJ, Said J, Hong-Gonzalez J, Corman DM, Ku M, et al. Effect of low-fat diet on development of prostate cancer and Akt phosphorylation in the Hi-Myc transgenic mouse model. Cancer Res. 2008;68:3066–73. doi: 10.1158/0008-5472.CAN-07-5616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pettersson A, Lis RT, Meisner A, Flavin R, Stack EC, Fiorentino M, et al. Modification of the Association Between Obesity and Lethal Prostate Cancer by TMPRSS2:ERG. J Natl Cancer Inst. 2013;105:1881–90. doi: 10.1093/jnci/djt332. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.