

Abstract
Therapeutic monoclonal antibodies (mAbs) have been successful for therapy of a number of diseases mostly cancer and immune disorders. However, the vast majority of mAbs approved for clinical use are full size, typically in IgG1 format. These mAbs may exhibit relatively poor tissue penetration and restricted epitope access due to their large size. A promising solution to this fundamental limitation is the engineering of smaller scaffolds based on the IgG1 Fc region. These scaffolds can be used for generation of libraries of mutants from which high-affinity binders can be selected. Comprised of the CH2 and CH3 domains, the Fc region is important not only for the antibody effector function but also for its long half-life. This review focuses on engineered Fc based antibody fragments and domains including native (dimeric) Fc and monomeric Fc as well as CH2 and monomeric CH3, and their use as novel scaffolds and binders. The Fc based binders are promising candidate therapeutics with optimized half-life, enhanced tissue penetration and access to sterically restricted binding sites resulting in an increased therapeutic efficacy.
Keywords: monoclonal antibody, domain antibody, antibody engineering, Fc, monomeric Fc, CH3 domain, CH2 domain
1. Introduction
The vast majority of the more than 40 monoclonal antibodies (mAbs) approved for clinical use are full-size antibodies in IgG1 format [1, 2]. Although these mAbs have significant impact on clinical benefits in several diseases, they still have limitations due to their relatively large size which causes poor penetration into tissues (e.g., solid tumors) and a lack of binding to epitopes on the surface of some targets that are accessible only by molecules of smaller size [3–5]. A variety of antibody fragments of smaller size such as Fab, Fv, scFv, and domains such as heavy chain variable domain (VH) and light chain variable domain (VL) have been previously developed [6–8]. However, these antibody fragments and domains have been of limited therapeutic applications because they display greatly reduced half-lives compared to that of the full-size IgG. To increase the serum half-life, various approaches including fusion with Fc, albumin, additional peptides to bind with the neonatal Fc receptor (FcRn) or albumin, as well as pegylation have been used [9]. But, the advantage of smaller size is essentially lost as additional modifications to improve the half-life significantly increase the molecule’s size.
The IgG Fc is a homodimer consisting of two heavy chain constant domains and has various effector functions. Moreover, the Fc region contributes to the long half-life of IgG through its pH-dependent association with FcRn [10, 11]. The IgG Fc can bind to FcRn in the acidic environment of the endosome after internalization and then be recycled into the cell surface and released into circulation. This protects IgG from degradation and increases its serum half-life [12]. Therefore, to overcome the problem of short half-life in smaller antibody domains and fragments, the IgG Fc itself and its constant domains were proposed as scaffolds that could be engineered for binding to antigens while retaining its binding to human FcRn [13–17].
From a structural point of view the constant domains share the topology and three-dimensional structure with the variable domains but lack the C′ and C″ strands and the C′C″ loop [18]. Hence, structural components of isolated constant domains, namely, beta strands A through G and exposed loop regions between these strands could provide scaffold functionality including some intrinsic stability and exposed regions tolerant to amino acid mutations as well as grafting of complementarity-determining regions (CDRs) into the scaffolds [15, 16, 19]. Previously, other approaches through chemically programmed antibodies (cpAbs) including the modification of Fc domains with antigen binding capability were also described [20–22], and was recently reviewed elsewhere [23]. These chemical programing with antigen-binding small molecules in the Fc based scaffolds could also be applied to the engineered Fc based antibody domains and fragments. Here, we review the strategies and technologies that have been adopted to develop novel antigen binding scaffolds derived from different Fc based antibody fragments and domains, including Fc, monomeric Fc (mFc), CH2 and monomeric CH3 (mCH3) domains (Fig. 1). We also discuss some of the engineered scaffolds as the potential candidates with better tissue penetration and reduced steric hindrance resulting in increased therapeutic efficacy. Further development of these Fc based antibody scaffolds would offer the next-generation of binders of smaller size with potentially enhanced half-life, which could make them promising candidate therapeutics and diagnostics.
Fig. 1.

Schematic diagram of Fc based scaffolds.
2. Engineered IgG1 Fc as a scaffold
Although the Fc domain lacks the antigen binding capability of full-size IgG, it governs their cytotoxic effector functions and long serum half-life. Therefore, extensive efforts have been made to engineer the Fc domain to fulfill a variety of therapeutic demands. The Fc region mediates cellular cytotoxic effector functions such as antibody-dependent cellular cytotoxicity (ADCC), antibody-dependent cell phagocytosis (ADCP) and complement-dependent cytotoxicity (CDC) through its interactions with Fcγ receptors (activating receptors: FcγRI, FcγRIIa and FcγRIIIa; inhibitory receptor FcγRIIb) and complement factor C1q [24–27]. The cytotoxicity of IgGs is correlated with the affinity of interactions between Fc and the Fcγ receptors and C1q [28, 29]. In knockout mice, it has also been shown that the presence of activating Fcγ receptors is necessary for the cytotoxicity of IgGs, while a deficiency of the inhibitory Fcγ receptor, FcγRIIb, further elevates ADCC [30]. In one example of effector function optimization, Lazar et al. engineered Fc variants with enhanced affinity for activating receptors and reduced affinity for the inhibitory receptor FcγRIIb, which resulted in enhanced effector functions in vitro and improved in vivo cytotoxicity in macaques [31]. The inhibitory effects of FcγRIIb have also been capitalized on to suppress the immune response by increasing affinity to FcγRIIb, which confers anti-inflammatory effects [32]. Groups have also worked to eliminate C1q binding [33] and to silence effector functions entirely [34] to reduce side effects such as injection site reactions and cross-targeting, respectively.
The interactions of the Fc region with FcRn significantly contribute to the exceptionally long serum half-life of IgG1 (about three weeks) compared to that of small-molecule drugs (minutes to hours) [35]. However, due to the unique pH-dependent association of the Fc with FcRn, in which Fc binds with FcRn at the endosomal pH but is released back to circulation at a physiological pH, enhanced affinity does not immediately equate to improved half-life in vivo [36]. Only the selectively improved binding of Fc to FcRn at pH 6.0 but not at pH 7.4 enhances half-life; for instance, the engineered Fc variants of bevacizumab (Avastin) that exhibit increased affinity with FcRn at pH 6.0 have been shown to not only possess longer half-lives but also improved antitumor activity in vivo [37]. However, in some cases, such as that of antibodies which serve as targeted carriers of radioisotopes and other toxic therapies, antibodies may benefit from greatly reduced half-lives. The Fc region has therefore also been engineered for reduced FcRn binding to FcRn [38].
It has recently been shown that the Fc region itself can serve as an antibody scaffold by engineering the loop regions at the C-terminal of the CH3 domains of Fc to form new antigen-binding sites [15]. To identify Fc binders (Fcab; Fc antigen binding) specific to HER2/neu, Wozniak-Knopp et al. generated a large yeast display library of human IgG1 Fc regions in which these loop sequences were randomized. FACS sorting against the HER2/neu resulted in the identification of H242-9 and, through subsequent affinity maturation of the clone, H10-03-6. The latter Fcab exhibited specific and selective binding to HER2-positive cells and elicited ADCC in vitro, as well as an in vivo half-life comparable to that of wild-type Fc in mice. However, although the Fc domain is considered a fairly stable molecule, the mutations in these loop structures have been shown to result in a loss of stability. Therefore, this same group engineered additional intradomain disulfide bonds to connect the N-terminus of the CH3 domain to the F-strand and the BC loop of the CH3 domain with the D-strand, which not only enhanced thermal stability in wild-type Fc but also in Fcabs [39]. These results demonstrate that, even in its small-size format of 60 kDa, Fcabs can possess the antigen specificity, effector functions and long serum half-life of full-size IgG antibodies.
3. Engineered monomeric IgG1 Fc as a scaffold
Recently several engineered monomeric IgG1 Fc have been developed in our laboratory [17]. A large phage library was generated displaying clones that have extensive mutations in the CH3 dimerization interface of IgG1 Fc. This library was used to select desired clones using a novel multiple panning/screening strategy (Fig. 2). It was first panned against protein G to enrich soluble and well-folded clones. After depletion of the poor behavior clones, the phage pool was further panned against FcRn to enrich clones that bind FcRn in a pH-dependent manner, and dominant clones were further screened to select purely monomeric mutants. Three Fc monomers were successfully selected using this strategy. They are purely monomeric, highly soluble, bind to FcRn and can be high efficiently expressed in E. coli.
Fig. 2.

Illustration of a phage-display based multiple panning/screening strategy to generate monomeric IgG1 Fc. A phage display library containing 109 different rational-designed IgG1 Fc mutants was panned against protein G, FcRn and screened by expression levels. The three IgG1 Fc monomers selected using this strategy were highly soluble, efficiently expressed in E. Coli and possessed pH-dependent FcRn binding capability.
Compared to the dimeric Fc, the engineered Fc monomers could have several advantages when used as scaffolds. First, the size advantage could lead to better tissue penetration and the targeting of sterically restricted epitopes. The size of monomeric Fc (~27 kDa) is comparable to scFv but could have much longer serum half-life due to the contribution of pH-dependent FcRn binding. Importantly, compared to the wild-type Fc, a large surface area (>1000 Å2) is exposed in the Fc monomer due to the exposure of the CH3 dimerization interface, providing more accessibility for antibody engineering (Fig. 3). This area does not overlap with the binding sites of FcRn and Fcγ receptors, presenting a unique opportunity for the grafting of complementarity determining regions or the introduction of extensive mutations.
Fig. 3.
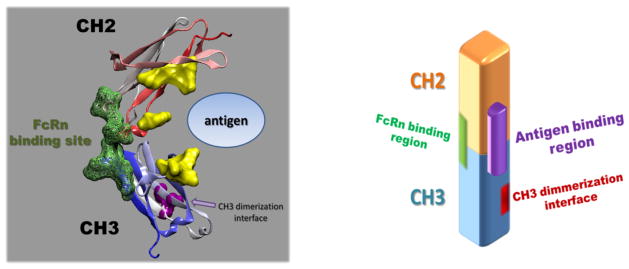
Molecular structure (left panel) and cartoon representation (right panel) for monomeric Fc serving as scaffold. The FcRn binding sites were colored in green. The yellow ball and sticks in the left panel show the residues that were mutated in a library to generate the antigen-binding sites. Compared to dimeric Fc, a large surface was exposed in the monomeric Fc due to the breaking of CH3 dimerization interface, providing more accessibility for antibody engineering (colored in purple in right panel).
4. Ig CH2 as a scaffold
CH2 in IgG, IgA, and IgD, and CH3 in IgE and IgM, exhibit very weak carbohydrate-mediated interchain protein-protein interactions in contrast to the extensive interchain interactions that occur between the other Ig domains. This property of CH2 results in its ~14 kDa independently folded monomeric form when expressed in bacteria [40, 41]. The structure of isolated CH2 has been characterized [41, 42] and shown to contain seven β-strands connected by three loops (loop BC, DE and FG) and two helices (helix 1 and 2). The three loops in CH2 are relatively flexible and similar to that of the three CDRs in VH. After replacement of the original residues in these loops by other sequences for construction of the library, specific binders based on CH2 that target certain antigens could be selected by panning (Fig. 4A). In addition, as part of Fc, CH2 contains binding sites or portions of binding sites for Fc receptors (e.g., FcRn) and complement C1q [43], which could naturally extend half-life and mediate stability and effector functions in vivo. Therefore, CH2 was proposed as a new scaffold for the development of novel therapeutic candidates, termed nanoantibodies (nAbs), that can not only bind to specific antigens but also confer Fc functions (e.g. bind to FcRn) [13, 44].
Fig. 4.

Schematic diagram of CH2 (A) and mCH3 (B) scaffolds. The green circles represent FcRn binding sites of CH2 and mCH3 scaffolds. The purple circles represent the loop regions that are suitable for introducing antigen binding sites.
5. Engineered IgG1 CH2 as a scaffold
Although isolated native CH2 exists as a monomer independently, it has significantly lower thermal stability compared with other scaffolds in a similar size range, such as the 10th type III domain of human fibronectin (FN3) [40, 41, 45]. It also has a high probability of instability when engineered for binding to antigens and enhanced effector functions. In native IgG and Fc molecules, the N-terminal residues of CH2 from the two heavy chains interact with each other and form hinge regions. By contrast, the N-terminal residues are highly disordered in isolated CH2. We have hypothesized that the removal of the CH2 N-terminal residues may not only increase its stability but also its aggregation resistance. Therefore, we generated a shortened human IgG1 CH2 variant, CH2s, by removing seven residues from the N-terminus. As expected, CH2s showed increased stability and aggregation resistance [46]. We also tried to further increase the stability of CH2s by introducing an additional disulfide bond between strands A (the first strand) and G (the last strand). We engineered a new mutant m01s that is extremely stable with high Tm (> 80°C) [14]. The half-life of m01s is about 10 hours as measured in normal B6 mice, human FcRn transgenic mice and cynomolgus macaques [47], which is much longer than that of other scaffolds of similar size such as human VH (range in minutes). This extended half-life may be due to its binding to FcRn. The binders based on m01s scaffold may exhibit very unique advantages when administered in vivo.
A previous study reported the construction of a large phage-displayed library by using the human CH2 as a scaffold [19]. Several binders were identified after panning against an HIV-1 gp120-soluble CD4 complex. The highest-affinity binder, m1a1, specifically binds to a highly conserved CD4i epitope that is also recognized by other broadly neutralizing antibodies [43]. m1a1 can neutralize seven of nine HIV-1 isolates in a pseudovirus model from different clades. Recently, a newly designed library based on m01s was constructed by novel strategies: rational mutagenesis on loop BC according to the frequency of the occurrence of the amino acids in its partner VH CDR1; limited mutagenesis on loop DE without significantly changing the properties of each mutated residue; precise replacement of loop FG (no changes in its flanking sequences) by VH CDR3 from VH libraries by multi-step PCR [48]. Panning of this library against a peptide from HIV-1 membrane proximal external regions resulted in the identification of a bivalent binder m2a1, which interacts non-competitively with an HIV-1 neutralizing epitope and FcRn. Similar to m1a1, m2a1 could also neutralize a panel of HIV-1 isolates based on a pseudovirus assay. Therefore, m2a1 could be considered the first prototype of a nanoantibody. However, both m1a1 and m2a1 can only bind to the target with low affinity, which results in only modest neutralizing activity. They should therefore be further improved for sufficient efficacy as therapeutic candidates.
Although we developed binders based on CH2-based scaffolds further studies are needed to develop them as therapeutics of potential clinical use. Firstly, as we found, the stability and aggregation resistance of CH2 could be increased through proper mutations. Therefore, a possible direction is to continue resolving these issues to prevent the molecule from incorrectly folding after the introduction of foreign sequences. Secondly, the CH2 scaffold can naturally possess Fc or part of Fc functions because it is derived from the Fc fragment, but the molecule may not be flexible enough to present foreign sequencing for perfect docking to the antigen, which leads to poor affinity. Thus, appropriate mutations should be carried out in the CH2 molecule to increase its flexibility or a new strategy for library construction should be adopted for better, high affinity interactions between the antibody and antigen. Thirdly, for treatment in vivo, more modifications of CH2 may be considered to further extend the half-life, by enhancing its binding to FcRn, or attain other necessary Fc functions.
6. Engineered monomeric IgG1 CH3 as a scaffold
By using a similar strategy for generating monomeric IgG1 Fc [17], we also developed a monomeric IgG1 CH3 domain [12] as a potential scaffold. mCH3 has four mutations to the wild-type CH3 and exists in pure monomer. Its thermal stability was low but was largely enhanced by introducing a disulfide bond connecting its N-terminal A strand and C-terminal G strand. It contains a part of the FcRn binding site of Fc, and can bind FcRn in a pH-dependent manner, although this binding is weaker than that of the wild-type Fc and Fc monomers.
Similar to that of the Fc monomers, the monomeric IgG1 CH3 domain would possess exposed surface areas that were inaccessible in the dimeric Fc and CH3 domains. These areas would enable the engineering of antigen binding sites and the use of this novel antibody domain as a potential scaffold (Fig. 4B). Their size is even smaller than mFc-based binders, leading to improved penetration, better targeting of cavities and sterically restricted regions resulting in increased potency per dose and reduction in overall manufacturing cost. Moreover, the mCH3-based binders could be advantageous in certain circumstances. For instance, the lack of CH2 domain in mCH3 would lead to a loss of binding to Fcγ receptors and C1q, which are critical for ADCC and CDC induced by antibodies. This particular advantage of engineered Fc based domains and scaffolds (e.g., mCH3) with a lack of Fcγ receptor binding could be their lower immunogenicity as these potential scaffolds with non-self amino acid sequences in their engineered paratopes may not be directly delivered and processed in Fcγ receptor-expressing antigen binding cells that trigger T-cell dependent antibody responses. Thus, they may be used under some circumstances where Fc-induced cytotoxic effects are disadvantageous.
7. Conclusions and prospects
Engineered Fc based antibody fragments and domains including the dimeric Fc, mFc, CH2 and mCH3 have been shown to be promising novel scaffolds. Table 1 summarizes the Fc based scaffolds with engineering strategies and characteristics of the binders developed. Particularly, novel panning/screening strategies were generated for the development of monomeric Fc based scaffolds. These monomers could be expressed in E. coli with high efficiency and were found to be relatively stable while retaining the ability to bind FcRn in a pH-dependent manner. Another scaffold, CH2, was derived from the native sequence without any mutations and formed a stable monomer, which is in contrast to all other constant domains and most of the variable domains. The mCH3 scaffold, which has only four mutations compared to dimeric CH3 and exists in pure monomer, exhibited higher FcRn binding compared to CH2, and may possess more accessibility for antibody engineering due to the exposure of a large surface area that was buried in dimeric Fc and CH3 domains. However, protein design strategies through disulphide bond engineering and random or site-directed mutagenesis combined with structural analysis may be further required to enhance their drugability properties [49, 50]. Because of their smaller size and the FcRn binding capability, the Fc based engineered fragments and domains have potential as novel scaffolds for construction of libraries. Several large libraries were already constructed and antigen-specific binders were successfully identified [19, 48, 51]. Thus, the Fc based scaffolds and binders could be used for the development of candidate therapeutics with optimized half-life, enhanced tissue penetration and access to sterically restricted binding sites resulting in increased therapeutic efficacy.
Table 1.
Different Fc based antibody domain scaffolds that are used to develop novel binders along with strategies applied and characteristics
| Scaffold | Size | Source | Strategy | Characteristics |
|---|---|---|---|---|
| Fc | 60 kDa | Wozniak- Knopp et al, PEDS, 2010 | Generated yeast display library of Fc regions with randomly mutated loop regions at the CH3 C-terminal, which could serve as new antigen-binding sites; Performed FACS sorting against antigen. | Specific and selective antigen binding; In vitro ADCC elicitation; In vivo half-life similar to wild-type Fc; Loss of stability(Improved with additional engineered intradomain disulfide bonds) |
| Wozniak- Knopp et al, PloS One, 2012 | ||||
| mFc | 27 kDa | Ying et al, JBC, 2012 | Generated large phage library of mFc with mutations at the CH3 dimerization interface of Fc; Panned against protein G followed by FcRn. | Highly soluble; High affinity binding to FcRn (59–204 nM); Large exposed surface area (>1000 Å2 ) - potential for engineering antigen binding sites |
| CH2 | 14 kDa | Xiao et al, BBRC, 2009 | m1a1: Generated large phage library of CH2 with its two loops (BC and FG) mutated to four residues (Y, A, D or S); Panned against antigen. | m1a1 & m2a1: Specifically binds to epitope recognized by other broadly neutralizing mAbs; Low affinity to target antigen; Modest neutralization of pseudoty ped HIV-1 from multiple clades. |
| Gong et al, PLoS One, 2012 | ||||
| m2a1: Generated large phage library based on m01s (shortened CH2 variant) with rational mutagenesis on loop BC, limited mutagenesis on loop DE and precise replacement of loop FG; Panned against antigen. | m2a1 (based on m01s): Increased stability; Aggregation resistance; Modest pH-dependent binding to FcRn (~4 μM); Long in vivo half-life; Non-competitive interactions with both HIV-1 neutralizing epitope and FcRn. | |||
| mCH3 | 14 kDa | Ying et al, JBC, 2013 | Similar to mFc strategy: Generated large phage library with mutations at the CH3 dimerization interface; Panned against protein G followed by FcRn. | Moderate pH- dependent binding to FcRn (940 nM); Low thermal stability - enhanced by additional disulfide bond; Greater exposed surface area - potential for new antigen binding sites; Lack of Fc-induced cytotoxic effects. |
Highlights.
The full-size monoclonal antibodies may exhibit poor tissue penetration due to their large size.
This review focuses on engineered Fc based antibody fragments and domains as novel scaffolds.
The Fc based binders are promising candidate therapeutics with small size and long half-life.
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research, and by Federal funds from the NIH, National Cancer Institute, under contract numbers NO1-CO-12400 and HHSN261200800001E and CRADA with RCT. We thank our RCT collaborators Kurt Gehlsen and David Bramhill for useful discussions and support. We thank the Wuhan Key Laboratory on Emerging Infectious Diseases and Biosafety for its support.
Footnotes
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does the mention of trade names, commercial products, or organizations imply endorsement by the U. S. Government.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Reichert JM. Marketed therapeutic antibodies compendium. mAbs. 2012;4:413–415. doi: 10.4161/mabs.19931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reichert JM. Antibodies to watch in 2014, mAbs. 2013;6 doi: 10.4161/mabs.27333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jain RK. Physiological barriers to delivery of monoclonal antibodies and other macromolecules in tumors. Cancer research. 1990;50:814s–819s. [PubMed] [Google Scholar]
- 4.Labrijn AF, Poignard P, Raja A, Zwick MB, Delgado K, Franti M, Binley J, Vivona V, Grundner C, Huang CC, Venturi M, Petropoulos CJ, Wrin T, Dimitrov DS, Robinson J, Kwong PD, Wyatt RT, Sodroski J, Burton DR. Access of antibody molecules to the conserved coreceptor binding site on glycoprotein gp120 is sterically restricted on primary human immunodeficiency virus type 1. Journal of virology. 2003;77:10557–10565. doi: 10.1128/JVI.77.19.10557-10565.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen W, Zhu Z, Feng Y, Dimitrov DS. Human domain antibodies to conserved sterically restricted regions on gp120 as exceptionally potent cross-reactive HIV-1 neutralizers. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:17121–17126. doi: 10.1073/pnas.0805297105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Holt LJ, Herring C, Jespers LS, Woolven BP, Tomlinson IM. Domain antibodies: proteins for therapy. Trends in biotechnology. 2003;21:484–490. doi: 10.1016/j.tibtech.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 7.Saerens D, Ghassabeh GH, Muyldermans S. Single-domain antibodies as building blocks for novel therapeutics. Curr Opin Pharmacol. 2008;8:600–608. doi: 10.1016/j.coph.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 8.Nelson AL, Reichert JM. Development trends for therapeutic antibody fragments. Nature biotechnology. 2009;27:331–337. doi: 10.1038/nbt0409-331. [DOI] [PubMed] [Google Scholar]
- 9.Kontermann RE. Strategies to extend plasma half-lives of recombinant antibodies. BioDrugs : clinical immunotherapeutics, biopharmaceuticals and gene therapy. 2009;23:93–109. doi: 10.2165/00063030-200923020-00003. [DOI] [PubMed] [Google Scholar]
- 10.Simister NE, Mostov KE. An Fc receptor structurally related to MHC class I antigens. Nature. 1989;337:184–187. doi: 10.1038/337184a0. [DOI] [PubMed] [Google Scholar]
- 11.Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nature reviews Immunology. 2007;7:715–725. doi: 10.1038/nri2155. [DOI] [PubMed] [Google Scholar]
- 12.Ying T, Chen W, Feng Y, Wang Y, Gong R, Dimitrov DS. Engineered soluble monomeric IgG1 CH3 domain: generation, mechanisms of function, and implications for design of biological therapeutics. The Journal of biological chemistry. 2013;288:25154–25164. doi: 10.1074/jbc.M113.484154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dimitrov DS. Engineered CH2 domains (nanoantibodies) mAbs. 2009;1:26–28. doi: 10.4161/mabs.1.1.7480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gong R, Wang Y, Feng Y, Zhao Q, Dimitrov DS. Shortened engineered human antibody CH2 domains: increased stability and binding to the human neonatal Fc receptor. The Journal of biological chemistry. 2011;286:27288–27293. doi: 10.1074/jbc.M111.254219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wozniak-Knopp G, Bartl S, Bauer A, Mostageer M, Woisetschlager M, Antes B, Ettl K, Kainer M, Weberhofer G, Wiederkum S, Himmler G, Mudde GC, Ruker F. Introducing antigen-binding sites in structural loops of immunoglobulin constant domains: Fc fragments with engineered HER2/neu-binding sites and antibody properties. Protein engineering, design & selection : PEDS. 2010;23:289–297. doi: 10.1093/protein/gzq005. [DOI] [PubMed] [Google Scholar]
- 16.Traxlmayr MW, Lobner E, Antes B, Kainer M, Wiederkum S, Hasenhindl C, Stadlmayr G, Ruker F, Woisetschlager M, Moulder K, Obinger C. Directed evolution of Her2/neu-binding IgG1-Fc for improved stability and resistance to aggregation by using yeast surface display. Protein engineering, design & selection : PEDS. 2013;26:255–265. doi: 10.1093/protein/gzs102. [DOI] [PubMed] [Google Scholar]
- 17.Ying T, Chen W, Gong R, Feng Y, Dimitrov DS. Soluble monomeric IgG1 Fc. The Journal of biological chemistry. 2012;287:19399–19408. doi: 10.1074/jbc.M112.368647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alamyar E, Giudicelli V, Duroux P, Lefranc MP. Antibody V and C Domain Sequence, Structure, and Interaction Analysis with Special Reference to IMGT((R)) Methods Mol Biol. 2014;1131:337–381. doi: 10.1007/978-1-62703-992-5_21. [DOI] [PubMed] [Google Scholar]
- 19.Xiao X, Feng Y, Vu BK, Ishima R, Dimitrov DS. A large library based on a novel (CH2) scaffold: identification of HIV-1 inhibitors. Biochemical and biophysical research communications. 2009;387:387–392. doi: 10.1016/j.bbrc.2009.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hofer T, Thomas JD, Burke TR, Jr, Rader C. An engineered selenocysteine defines a unique class of antibody derivatives. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:12451–12456. doi: 10.1073/pnas.0800800105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xiao J, Chen R, Pawlicki MA, Tolbert TJ. Targeting a homogeneously glycosylated antibody Fc to bind cancer cells using a synthetic receptor ligand. Journal of the American Chemical Society. 2009;131:13616–13618. doi: 10.1021/ja9045179. [DOI] [PubMed] [Google Scholar]
- 22.Chiang MJ, Holbert MA, Kalin JH, Ahn YH, Giddens J, Amin MN, Taylor MS, Collins SL, Chan-Li Y, Waickman A, Hsiao PY, Bolduc D, Leahy DJ, Horton MR, Wang LX, Powell JD, Cole PA. An Fc domain protein-small molecule conjugate as an enhanced immunomodulator. Journal of the American Chemical Society. 2014;136:3370–3373. doi: 10.1021/ja5006674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rader C. Chemically programmed antibodies. Trends in biotechnology. 2014;32:186–197. doi: 10.1016/j.tibtech.2014.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carter PJ. Potent antibody therapeutics by design. Nature reviews Immunology. 2006;6:343–357. doi: 10.1038/nri1837. [DOI] [PubMed] [Google Scholar]
- 25.Presta LG. Molecular engineering and design of therapeutic antibodies. Current opinion in immunology. 2008;20:460–470. doi: 10.1016/j.coi.2008.06.012. [DOI] [PubMed] [Google Scholar]
- 26.Jefferis R. Antibody therapeutics: isotype and glycoform selection. Expert opinion on biological therapy. 2007;7:1401–1413. doi: 10.1517/14712598.7.9.1401. [DOI] [PubMed] [Google Scholar]
- 27.Bruhns P, Iannascoli B, England P, Mancardi DA, Fernandez N, Jorieux S, Daeron M. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood. 2009;113:3716–3725. doi: 10.1182/blood-2008-09-179754. [DOI] [PubMed] [Google Scholar]
- 28.Shields RL, Namenuk AK, Hong K, Meng YG, Rae J, Briggs J, Xie D, Lai J, Stadlen A, Li B, Fox JA, Presta LG. High resolution mapping of the binding site on human IgG1 for Fc gamma RI, Fc gamma RII, Fc gamma RIII, and FcRn and design of IgG1 variants with improved binding to the Fc gamma R. The Journal of biological chemistry. 2001;276:6591–6604. doi: 10.1074/jbc.M009483200. [DOI] [PubMed] [Google Scholar]
- 29.Shields RL, Lai J, Keck R, O’Connell LY, Hong K, Meng YG, Weikert SH, Presta LG. Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human Fcgamma RIII and antibody-dependent cellular toxicity. The Journal of biological chemistry. 2002;277:26733–26740. doi: 10.1074/jbc.M202069200. [DOI] [PubMed] [Google Scholar]
- 30.Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nature medicine. 2000;6:443–446. doi: 10.1038/74704. [DOI] [PubMed] [Google Scholar]
- 31.Lazar GA, Dang W, Karki S, Vafa O, Peng JS, Hyun L, Chan C, Chung HS, Eivazi A, Yoder SC, Vielmetter J, Carmichael DF, Hayes RJ, Dahiyat BI. Engineered antibody Fc variants with enhanced effector function. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:4005–4010. doi: 10.1073/pnas.0508123103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chu SY, Vostiar I, Karki S, Moore GL, Lazar GA, Pong E, Joyce PF, Szymkowski DE, Desjarlais JR. Inhibition of B cell receptor-mediated activation of primary human B cells by coengagement of CD19 and FcgammaRIIb with Fc-engineered antibodies. Molecular immunology. 2008;45:3926–3933. doi: 10.1016/j.molimm.2008.06.027. [DOI] [PubMed] [Google Scholar]
- 33.Hayden-Ledbetter MS, Cerveny CG, Espling E, Brady WA, Grosmaire LS, Tan P, Bader R, Slater S, Nilsson CA, Barone DS, Simon A, Bradley C, Thompson PA, Wahl AF, Ledbetter JA. CD20-directed small modular immunopharmaceutical, TRU-015, depletes normal and malignant B cells. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009;15:2739–2746. doi: 10.1158/1078-0432.CCR-08-1694. [DOI] [PubMed] [Google Scholar]
- 34.Davis PM, Abraham R, Xu L, Nadler SG, Suchard SJ. Abatacept binds to the Fc receptor CD64 but does not mediate complement-dependent cytotoxicity or antibody-dependent cellular cytotoxicity. The Journal of rheumatology. 2007;34:2204–2210. [PubMed] [Google Scholar]
- 35.Wang Y, Tian Z, Thirumalai D, Zhang X. Neonatal Fc receptor (FcRn): a novel target for therapeutic antibodies and antibody engineering. Journal of drug targeting. 2014 doi: 10.3109/1061186X.2013.875030. [DOI] [PubMed] [Google Scholar]
- 36.Dall’Acqua WF, Woods RM, Ward ES, Palaszynski SR, Patel NK, Brewah YA, Wu H, Kiener PA, Langermann S. Increasing the affinity of a human IgG1 for the neonatal Fc receptor: biological consequences. Journal of immunology (Baltimore, Md : 1950) 2002;169:5171–5180. doi: 10.4049/jimmunol.169.9.5171. [DOI] [PubMed] [Google Scholar]
- 37.Zalevsky J, Chamberlain AK, Horton HM, Karki S, Leung IW, Sproule TJ, Lazar GA, Roopenian DC, Desjarlais JR. Enhanced antibody half-life improves in vivo activity. Nature biotechnology. 2010;28:157–159. doi: 10.1038/nbt.1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jain M, Kamal N, Batra SK. Engineering antibodies for clinical applications. Trends in biotechnology. 2007;25:307–316. doi: 10.1016/j.tibtech.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 39.Wozniak-Knopp G, Stadlmann J, Ruker F. Stabilisation of the Fc fragment of human IgG1 by engineered intradomain disulfide bonds. PloS one. 2012;7:e30083. doi: 10.1371/journal.pone.0030083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Feige MJ, Walter S, Buchner J. Folding mechanism of the CH2 antibody domain. Journal of molecular biology. 2004;344:107–118. doi: 10.1016/j.jmb.2004.09.033. [DOI] [PubMed] [Google Scholar]
- 41.Gong R, Vu BK, Feng Y, Prieto DA, Dyba MA, Walsh JD, Prabakaran P, Veenstra TD, Tarasov SG, Ishima R, Dimitrov DS. Engineered human antibody constant domains with increased stability. The Journal of biological chemistry. 2009;284:14203–14210. doi: 10.1074/jbc.M900769200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Prabakaran P, Vu BK, Gan J, Feng Y, Dimitrov DS, Ji X. Structure of an isolated unglycosylated antibody C(H)2 domain, Acta crystallographica. Section D Biological crystallography. 2008;64:1062–1067. doi: 10.1107/S0907444908025274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gong R, Chen W, Dimitrov DS. Candidate antibody-based therapeutics against HIV-1. BioDrugs : clinical immunotherapeutics, biopharmaceuticals and gene therapy. 2012;26:143–162. doi: 10.2165/11631400-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gong R, Xiao G. Engineered antibody variable and constant domains as therapeutic candidates. Pharmaceutical patent analyst. 2013;2:637–646. doi: 10.4155/ppa.13.44. [DOI] [PubMed] [Google Scholar]
- 45.Hackel BJ, Kapila A, Wittrup KD. Picomolar affinity fibronectin domains engineered utilizing loop length diversity, recursive mutagenesis, and loop shuffling. Journal of molecular biology. 2008;381:1238–1252. doi: 10.1016/j.jmb.2008.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gong R, Wang Y, Ying T, Feng Y, Streaker E, Prabakaran P, Dimitrov DS. N-Terminal Truncation of an Isolated Human IgG1 CH2 Domain Significantly Increases Its Stability and Aggregation Resistance. Molecular pharmaceutics. 2013 doi: 10.1021/mp400075f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gehlsen K, Gong R, Bramhill D, Wiersma D, Kirkpatrick S, Wang Y, Feng Y, Dimitrov DS. Pharmacokinetics of engineered human monomeric and dimeric CH2 domains. mAbs. 2012;4:466–474. doi: 10.4161/mabs.20652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gong R, Wang Y, Ying T, Dimitrov DS. Bispecific engineered antibody domains (nanoantibodies) that interact noncompetitively with an HIV-1 neutralizing epitope and FcRn. PloS one. 2012;7:e42288. doi: 10.1371/journal.pone.0042288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rouet R, Lowe D, Christ D. Stability engineering of the human antibody repertoire. FEBS letters. 2014;588:269–277. doi: 10.1016/j.febslet.2013.11.029. [DOI] [PubMed] [Google Scholar]
- 50.Perchiacca JM, Tessier PM. Engineering aggregation-resistant antibodies. Annual review of chemical and biomolecular engineering. 2012;3:263–286. doi: 10.1146/annurev-chembioeng-062011-081052. [DOI] [PubMed] [Google Scholar]
- 51.Chen W, Gong R, Ying T, Prabakaran P, Zhu Z, Feng Y, Dimitrov DS. Discovery of novel candidate therapeutics and diagnostics based on engineered human antibody domains. Current drug discovery technologies. 2014;11:28–40. doi: 10.2174/15701638113109990032. [DOI] [PMC free article] [PubMed] [Google Scholar]