

Abstract
Non-protective immune responses to highly virulent Cryptococcus neoformans strains, such as H99, are associated with Th2-type cytokine production, alternatively activated macrophages and inability of the host to clear the fungus. In contrast, experimental studies show that protective immune responses against cryptococcosis are associated with Th1-type cytokine production and classical macrophage activation. The protective response induced during C. neoformans strain H99γ (C. neoformans strain H99 engineered to produce murine interferon-γ) infection correlates with enhanced phosphorylation of the transcription factor STAT1 in macrophages; however, the role of STAT1 in protective immunity to C. neoformans is unknown. The current studies examined the effect of STAT1-deletion in murine models of protective immunity to C. neoformans. Survival and fungal burden were evaluated in WT and STAT1 KO mice infected with either strain H99γ or C. neoformans strain 52D (unmodified clinical isolate). Both strains H99γ and 52D were rapidly cleared from the lungs, did not disseminate to the CNS, or cause mortality in the WT mice. Conversely, STAT1 KO mice infected with H99γ or 52D had significantly increased pulmonary fungal burden, CNS dissemination, and 90-100% mortality. STAT1-deletion resulted in a shift from Th1 to Th2 cytokine bias, pronounced lung inflammation and defective classical macrophage activation. Pulmonary macrophages from STAT1 KO mice exhibited defects in nitric oxide production correlating with inefficient inhibition of fungal proliferation. These studies demonstrate that STAT1 signaling is essential not only for regulation of immune polarization but for the classical activation of macrophages that occurs during protective anti-cryptococcal immune responses.
Introduction
Cryptococcus neoformans, the predominant etiological agent of cryptococcosis, is an opportunistic fungal pathogen and frequent cause of life-threatening infection in individuals with impaired T cell function (i.e., persons with AIDS, lymphoid malignancies, and recipients of immunosuppressive therapies) (1-13). Cryptococcosis is considered an AIDS-defining illness and the most common mycological cause of morbidity and mortality among AIDS patients (14). Global estimates show that one million cases of cryptococcal meningitis occur in AIDS patients each year resulting in approximately 625,000 deaths (14). C. neoformans also remains the third most common invasive fungal infection among organ transplant recipients (13, 15, 16). Current anti-fungal drug therapies are oftentimes rendered ineffective due to the development of drug resistance or an inability of the host’s immune system to participate in eradicating the pathogen (17-19), necessitating new therapeutic strategies that consider the immune status of the host population commonly impacted by cryptococcal disease.
C. neoformans is ubiquitous in the environment and exposure via inhalation of desiccated basidiospores or yeast into the lung alveoli often occurs in early childhood (20-22). The overwhelming majority of exposures result in asymptomatic disease and possible latency in immunocompetent individuals. However, in immunocompromised individuals, C. neoformans can spread from the lungs to the central nervous system (CNS) resulting in life threatening meningoencephalitis (23). Since inhalation is the principle route of entry, the host is dependent on resident pulmonary phagocytes, such as macrophages, to contain the pathogen and prevent dissemination from the lungs. Studies using various experimental models of pulmonary cryptococcosis indicate that protection against C. neoformans infection is associated with the induction of Th1-type cytokine responses (IL-2, IL-12, IFN-γ, TNF-α), increased lymphocyte infiltration, and classical macrophage (M1) activation (24-30). In contrast, Th2-type cytokine responses (IL-4, IL-5, and IL-13) are detrimental to the host and are associated with alternative macrophage (M2) activation (31-33). M1 macrophages are known for their capacity to efficiently kill phagocytized organisms through the production of reactive oxygen and nitrogen species (25, 31, 34-36). M2 macrophages, however, are associated with wound healing and are not efficiently microbicidal against C. neoformans (25, 31, 34-36). M1 macrophages produce nitric oxide (NO) and L-citrulline via inducible NO synthase (iNOS) by acting on the substrate L-arginine. Arginase-1 (Arg1) in M2 macrophages competes with iNOS for L-arginine to produce L-ornithine and urea (37), which is not fungicidal against C. neoformans (38, 39). Therefore, macrophage polarization towards an M2 phenotype, which occurs during infection with wild type (WT) C. neoformans strain H99, results in uncontrolled fungal growth, dissemination, and exacerbation of disease (31, 35, 40, 41).
Experimental pulmonary infection in mice with a C. neoformans strain engineered to express interferon (IFN)-γ, designated H99γ, results in Th1-type and IL-17A cytokine responses, M1 macrophage activation, and resolution of the acute infection (26, 42). Additionally, mice protectively immunized with C. neoformans strain H99γ developed an M1 macrophage activation phenotype during secondary challenge with a non-IFN-γ producing WT C. neoformans strain (H99) and displayed enhanced fungistasis and NO production compared to macrophages from non-protectively immunized mice following secondary challenge (24). Specifically, resolution of pulmonary C. neoformans infection in protectively immunized mice was associated with M1 macrophage activation that coincided with robust signal transducer and activator of transcription 1 (STAT1) phosphorylation (24). STAT1 signaling via the IFN-γ-JAK1/2-STAT1 pathway leads to the formation of phospho-STAT1 homodimers that translocate to the nucleus, bind to gamma-activated sequences (GAS), and promote the transcription of STAT1 target genes (reviewed in (43)). STAT1 signaling and its downstream products have been tied to the generation of Th1-type immune responses (44-46) that are protective against cryptococcosis and associated with M1 macrophage activation (24-30, 32). Little is known about the role of STAT1 in antifungal immunity. Specific mutations that alter STAT1 function (loss-of-function or gain-of-function mutations) have been shown to increase susceptibility/interfere with host defenses to fungal infections such as chronic mucocutaneous candidiasis (47, 48), coccidioidomycosis and histoplasmosis (49). The role of STAT1 signaling during Cryptococcus infections has not previously been studied.
The objective of the studies presented herein was to determine the role of STAT1 in the generation of protective immunity against C. neoformans, including its role in mediating M1 macrophage activation and anti-fungal activity in mice. Utilizing STAT1 KO mice, we demonstrated that STAT1 signaling promotes Th1-type immune responses, but most importantly, is required for M1 macrophage activation and subsequent protection against experimental pulmonary infection with C. neoformans strain H99γ. These data provide the first evidence of an effector cell population and the intracellular signaling mechanism by which the host mounts a protective immune response to C. neoformans.
Methods
Mice
Female STAT1 KO (129S6/SvEv-Stat1tm1Rds) and WT controls mice (129S6/SvEvTac) from Taconic Farms (Germantown, NY) and female IFN-γ KO (C.129S7(B6)-Ifngtm1Ts/J) and WT control mice (BALB/cJ) from The Jackson Laboratory (Bar Harbor, ME) were used throughout these studies. Mice were housed at The University of Texas at San Antonio Small Animal Laboratory Vivarium and handled according to guidelines approved by the Institutional Animal Care and Use Committee.
Strains and media
C. neoformans strain H99γ (derived from H99 serotype A, mating type α) (50) and C. neoformans strain 52D (serotype D) (a kind gift from Dr. Brian Wickes, University of Texas Health Science Center at San Antonio, San Antonio, TX) was recovered from a 15% glycerol stock stored at −80°C prior to use in the experiments described in this study. The strain was maintained on yeast extract/peptone/dextrose (YPD) medium agar plates (Becton Dickinson, Sparks, MD). Yeast cells were grown for 16-18 h at 30°C with shaking in liquid YPD broth, collected by centrifugation, washed three times with sterile phosphate buffered saline (PBS), and viable yeasts were quantified using trypan blue dye exclusion on a hemacytometer.
Pulmonary Cryptococcal Infections
Mice were anesthetized with 2% isoflurane using a rodent anesthesia device (Eagle Eye Anesthesia, Jacksonville, FL) then given an intranasal inoculation with 1 × 104 CFU of C. neoformans strains H99γ or 52D in 50 μl of sterile PBS. The inocula used for nasal inhalation were verified by quantitative culture on YPD agar. Mice were euthanized on predetermined days by CO2 inhalation followed by cervical dislocation, and lung tissues were excised using aseptic technique. For survival analyses, mice were inoculated as stated above and monitored twice daily for up to 60 days post-inoculation.
Fungal Burden
For fungal burden analysis, the left lobe of the lung was removed and homogenized in 1ml sterile PBS. A 50 μl aliquot was removed, serially diluted in sterile PBS, and plated on YPD agar supplemented with chloramphenicol. Plates were incubated for 48 hours at 30°C and fungal colonies recorded.
Cytokine Analysis
Cytokine production in lung tissues was analyzed using the Bio-Plex protein array system (Luminex-based technology; Bio-Rad Laboratories, Hercules, CA). Briefly, the left lobe of the lung was excised and homogenized in ice-cold sterile PBS (1 ml). An aliquot (50 μl) was taken to quantify the pulmonary fungal burden and an anti-protease buffer solution containing PBS, protease inhibitors (inhibiting cysteine, serine, and other metalloproteinases), and 0.05% Triton X-100 was added to the homogenate and then clarified by centrifugation (800 × g) for 10 min. Supernatants from pulmonary homogenates were assayed for the presence of IL-1α, IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-9, IL-10, IL-12(p40), IL-12(p70), IL-13, IL-17A, CCL5/RANTES, Eotaxin, CXCL1/KC, CCL3/MIP-1α, CCL4/MIP-1β, CCL2/MCP-1, G-CSF, GM-CSF, TNF-α, and IFN-γ.
Pulmonary leukocyte isolation
Lungs were excised on days 7 and 10 post-inoculation and digested enzymatically at 37°C for 30 min in 10 ml digestion buffer (RPMI 1640 and 1 mg/ml collagenase type IV (Sigma-Aldrich, St. Louis, MO) with intermittent (every 10 min) stomacher homogenizations. The digested tissues were then successively filtered through sterile 70- and 40-μm nylon filters (BD Biosciences, San Diego, CA) to enrich for leukocytes, and then cells were washed with sterile HBSS. Erythrocytes were lysed by incubation in NH4Cl buffer (0.859% NH4Cl, 0.1% KHCO3, 0.0372% Na2EDTA [pH 7.4]; Sigma-Aldrich) for 3 min on ice followed by a 2-fold excess of PBS. The resulting leukocyte population was then enriched for macrophages by positive selection using magnetic beads labeled with F4/80 antibody (Miltenyi Biotec, Auburn, CA) according to the manufacturer’s recommendations.
Real-time PCR Analysis
Total RNA was isolated from purified F4/80+ cells using TRIzol reagent (Invitrogen, Carlsbad, CA) and then DNase (Qiagen, Germantown, MD) treated to remove possible traces of contaminating DNA according to the manufacturer’s instructions. Total RNA was subsequently recovered using the Qiagen RNeasy kit. cDNA was synthesized from 1 μg total RNA using the oligo(dT) primer and reagents supplied in the SuperScript III RT kit (Invitrogen) according to the manufacturer’s instructions. The cDNA was used as a template for real-time PCR analysis using the TaqMan gene expression assay (Applied Biosystems, Foster City, CA) according to the manufacturer’s instructions. All real-time PCR reactions were performed using the 7300 real-time PCR system (Applied Biosystems). For each real-time PCR reaction, a master mix was prepared on ice with TaqMan gene expression assays specific for iNOS, IFN-γ, TNF-α, CXCL9, CXCL10, IRF-1, SOCS-1, IL-17A, Ym1, FIZZ1, Arg1, IL-4, IL-13, and CD206 (Applied Biosystems). TaqMan rodent GAPDH (Applied Biosystems) was used as an internal control. The thermal cycling parameters contained an initial denaturing cycle of 95°C for 10 min followed by 40 cycles of 95°C for 15s and 60°C for 60s. Results of the real-time PCR data were derived using the comparative Ct method to detect relative gene expression as described previously (26).
Immunohistochemistry and Histology
Mice were sacrificed according to approved protocols, and the lungs were immediately perfused with sterile PBS by transcardial perfusion through the right ventricle. The pericardium and trachea were exposed by dissection, and an incision was made in the trachea for the insertion of a sterile flexible cannula attached to a 3-ml syringe. The lungs were slowly inflated with 0.5-0.7 ml 10% ultrapure formaldehyde (Polysciences, Inc., Warrington, PA), excised and immediately fixed in 10% ultrapure formaldehyde for 24-48 hours. The lungs were then transferred to 70% ethanol and subsequently mounted into cassettes and paraffin embedded by personnel at The University of Texas Health Science Center at San Antonio Histology and Immunohistochemistry Laboratory. After paraffin embedding, 5-mm sections were cut and stained using hematoxylin-eosin (H&E) with mucicarmine at McClinchey Histology Labs, Stockbridge, MI. Sections were analyzed with light microscopy using a Nikon microscope and microphotographs taken using Digital Microphotography system DFX1200 with ACT-1 software (Nikon Co, Tokyo, Japan) at the Ann Arbor VA Health System.
Macrophage Anti-cryptococcal Assay, ROS Detection Assay and NO Production Assay
Cell viability and quantity of F4/80+-enriched macrophage populations were assessed using trypan blue exclusion in a hemacytometer. Macrophages were cultured at a density of 5 × 105 cells per well in a 96-well tissue culture plate in RPMI 1640 (without phenol red) (Life Technologies, Grand Island, NY) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 2mM L-glutamine, and 100 μg/ml penicillin-streptomycin (complete medium). Macrophages were incubated in complete medium at 37°C with 5% CO2. Initial fungal burden was determined by lysis of the macrophages using sterile deionized water followed by serial dilution and plating on YPD agar supplemented with chloramphenicol (Mediatech, Manassas, VA) for 48 h at 30°C. After 24 h incubation, cell supernatants were collected and an aliquot (50ul) was used to determine NO production with Griess reagent (Sigma-Aldrich) according to manufacturer’s instructions. Light absorbance values were measured at 540 nm using a BioTek Elx808 absorbance microplate reader with Gen5 v1.04.5 software. Alternatively, F4/80+ macrophages were isolated and adjusted to 5 × 105 cells per well as described above and ROS measured using CellROX® Deep Red Reagent (Life Technologies) according to manufacturer’s instructions and measured using BD FACSArray software™ on a BD FACSArray flow cytometer (BD Biosciences).
Flow cytometry
Standard methodology was employed for the direct immunofluorescence of pulmonary leukocytes. Briefly, in 96-well U-bottom plates, 100 μl containing 1 × 106 cells in PBS + 2% FBS (FACS buffer) were incubated with 50 μl of Fc Block™ (BD Biosciences) diluted in FACS buffer for 5 minutes to block non-specific binding of antibodies to cellular Fc receptors. Subsequently, an optimal concentration of fluorochrome-conjugated antibodies (between 0.06-0.5 μg/1 × 106 cells in 50 μl of FACS buffer) were added in various combinations to allow for dual or triple staining experiments, and cells were incubated for 30 minutes at 4°C. Following incubation, the cells were washed three times with FACS buffer and were fixed in 200 μl of 2% ultrapure formaldehyde (Polysciences, Inc., Warrington, PA) diluted in FACS buffer (fixation buffer). Cells incubated with either FACS buffer alone or single fluorochrome-conjugated antibodies were used to determine positive staining and spillover/compensation calculations and the flow cytometer determined background fluorescence. The samples were analyzed using BD FACSArray software™ on a BD FACSArray flow cytometer (BD Biosciences). Dead cells were excluded on the basis of forward angle and 90° light scatter. For data analyses, 30,000 events (cells) were evaluated from a predominantly leukocyte population identified by back-gating from CD45+-stained cells. The absolute number of total leukocytes was quantified by multiplying the total number of cells observed by hemacytometer counting by the percentage of CD45+ cells determined by flow cytometry. The absolute number of leukocytes (CD45+ cells), CD4+/CD3+ T cells, CD8+/CD3+ T cells, CD19+/B220+ B cells, PMNs (1A8+/CD45+), MΦs (F4/80+/CD45+), DCs (CD11bint/CD11c+/CD45+), NK cells (Nkp46+/DX5+), NKT cells (Nkp46+/DX5+/CD4+), and eosinophils (SiglecF+/CD11bint) was determined by multiplying the percentage of each gated population by the total number of CD45+ cells.
Statistical analysis
The unpaired Student’s t test was used to analyze fungal burden, cytokine/chemokine levels, pulmonary cell populations, ROS and NO Assays (two-tailed) using GraphPad Prism version 5.00 for Windows (GraphPad Software, San Diego, CA) to detect statistically significant differences. The One-Way ANOVA with Tukey’s Post-Test was used for real-time PCR analysis (GraphPad Software) to detect significant differences. Survival data was analyzed using the log-rank test (GraphPad Software). Significant differences were defined as *p < 0.01, **p < 0.001 or ***p < 0.0001.
Results
STAT1 is critical for the induction of protection against C. neoformans infection in mice
Previous studies in our lab demonstrated that experimental pulmonary infection with C. neoformans strain H99γ in BALB/c and A/Jcr mice (24, 26, 50-52) and C129 mice (unpublished results) results in clearance of the acute infection and the induction of protective immunity against a subsequent challenge with a WT C. neoformans strain that does not produce IFN-γ. Protection in H99γ-immunized mice against WT C. neoformans challenge occurred concurrent to the induction of STAT1 signaling within macrophages and M1 macrophage activation (24). To determine the requirement for STAT1 signaling in mediating protection against pulmonary cryptococcosis, STAT1 KO (129S6/SvEv-Stat1tm1Rds) and WT controls (129S6/SvEvTac) were intranasally inoculated with H99γ. Mice survival (morbidity) was monitored for 60 days post-inoculation in one subgroup, while pulmonary fungal burden was quantified in another subgroup of infected animals at selected time points. WT mice had a 90% survival rate at day 60 post-inoculation with H99γ (Figure 1A). In comparison, STAT1 KO mice began to succumb to H99γ infection on day 12 post-inoculation and had only a 10% survival rate by day 60 post-inoculation (p < 0.0001; Figure 1A) with a median survival time of 18 days. The fungal burden was evaluated on days 7 and 10 post-inoculation with H99γ, i.e. at the time points prior to the onset of mortality in STAT1 KO mice, which ensured the most accurate statistical analysis of the fungal burden. STAT1 KO mice showed significantly higher pulmonary fungal burden on day 10 post-inoculation compared to infected WT mice (p < 0.0001; Figure 1B). Fungal burden in the WT mice did not increase from day 7 to day 10, indicating that the host was able to control the growth of the yeast. However, fungal burden in STAT1 KO mice was significantly increased on day 10 compared to day 7 in these mice (p < 0.0001), revealing that they were unable to control proliferation of C. neoformans in the lungs. Therefore, STAT1 signaling is required for control of pulmonary infection with C. neoformans strain H99γ in mice.
Figure 1. STAT1 signaling is required for control of pulmonary fungal burden and survival following inoculation with C. neoformans strain H99γ.

129S6/SvEv (WT) and STAT1 KO mice were given an intranasal inoculation with 1 × 104 CFU of C. neoformans strain H99γ. Mice were observed for up to 60 days for survival analysis (A) or pulmonary fungal burden was analyzed at days 7 and 10 post inoculation (B). Experiments are cumulative of 2 experiments using 5 mice per group per time point (A) or 2 experiments using 4 mice per group per time point (B). (**p < 0.001; ***p < 0.0001)
To determine the requirement of STAT1 signaling for protection against infection with a WT strain of C. neoformans, WT and STAT1 KO mice were intranasally inoculated with an unmodified clinical isolate, C. neoformans strain 52D, and survival monitored for 45 days post-infection. Inoculation with this strain is generally controlled in BALB/c mice, however, it is capable of inducing a chronic infection in C57BL/6 mice (53, 54). All WT mice infected with C. neoformans strain 52D were alive and appeared healthy upon conclusion of the experiment (Figure 2A). In contrast, all STAT1 KO mice succumbed to the infection or were moribund by day 28 with a median survival time of 26.5 days. Post-mortem analysis showed that moribund STAT1 KO mice had significantly higher pulmonary fungal burden compared to WT mice (p < 0.01; Figure 2B). In addition, STAT1 KO mice showed evidence of massive CNS dissemination (Figure 2B) unlike the WT mice which were negative for CNS fungal cultures with the exception of 1 out of 10 WT mice that had detectable (1 colony) C. neoformans in the brain. These results indicate that STAT1 signaling is required for the induction of immune protection against C. neoformans and prevention of fungal dissemination into the CNS.
Figure 2. Control of pulmonary fungal burden, CNS dissemination, and survival during WT C. neoformans strain 52D inoculation are dependent on STAT1 signaling.

129S6/SvEv (WT) and STAT1 KO mice were given an intranasal inoculation with 1 × 104 CFU of C. neoformans strain 52D. Mice were observed for up to 45 days for survival analysis (A) and pulmonary and brain fungal burden was analyzed post-mortem (B). Experiments are cumulative of 2 experiments using 5 mice per group per time point. (**p < 0.001; ***p < 0.0001)
STAT1 deletion results in the development of severe pathology in C. neoformans strain H99γ infected lungs
To further characterize the defect in pulmonary control of cryptococcal growth in STAT1 KO compared to WT mice, we examined histological sections of the infected and uninfected mouse lungs at day 10 post-inoculation. While the uninfected lungs of WT and STAT1 KO mice showed normal morphology (Figure 3A-B), histological examination of infected lungs at day 10 post-inoculation revealed the development of severe lung pathology in the absence of STAT1 expression (Figure 3D,F). The inflamed areas in the lungs of the WT mice at day 10 show that both infection and the inflammatory response are well contained within granulomatous foci, while the majority of the lung tissue remains clear of the infection/inflammation (Figure 3C). In contrast, virtually all lung tissue from STAT1 KO mice is severely laden with cryptococcal organisms and infiltrated with inflammatory cells (Figure 3D), consistent with uncontrolled growth of the microbe and a non-protective immune/inflammatory response. Analysis of histological sections under high power magnification demonstrates that in addition to major differences in fungal burden, cellular composition of inflammatory infiltrates varies between the WT and STAT1 KO mice. Minimal presence of cryptococcal organisms and predominance of lymphoid cells and macrophage-type cells within the infiltrates of WT mice is consistent with the development of a protective immune response (Figure 3E). In contrast, the abundance of cryptococcal organisms accompanied by mixed inflammatory infiltrates with enhanced presence of granulocytes (eosinophils and PMNs) and lower frequency of lymphoid cells characterize the lesions within the infected lungs of STAT1 KO mice (Figure 3F). Such severe pneumonia and lung consolidation could explain the mortality observed in STAT1 KO mice, and indicate that the immune response in the lungs of STAT1 KO mice, albeit robust, is non-protective.
Figure 3. Uncontrolled C. neoformans expansion and severe lung pathology develop in H99γ-infected STAT1 KO mice by day 10 post-infection.

129S6/SvEv (WT) and STAT1 KO mice were given an intranasal inoculation with 1 × 104 CFU of C. neoformans strain H99γ or left uninfected. Lungs of uninfected (A-B) and H99γ-infected (C-F), WT (upper row) and STAT1 KO mice (lower row) were collected on day 10 post-inoculation, processed and analyzed using light microscope. Deletion of STAT1 that did not affect morphology of uninfected lungs has resulted in the development of severe lung pathology and massive expansion of fungus by day 10 post-inoculation. Note that the infection and inflammatory response are contained within portions of the lungs in the WT mice (C), while virtually all lungs are infected and consolidated in STAT1 KO mice (D). High power images show minimal presence of cryptococcal organisms and predominantly mononuclear cell infiltrates with lymphoid and macrophage-type morphologies within infected lungs areas in the WT mice (E), contrasting with granulocyte-enriched mixed cellular infiltrate surrounding clusters of proliferating cryptococci (green arrows) and in the lungs of STAT1 KO mice (F). Histological slides were stained with H&E and mucicarmine, and images were taken at 10X (A-D) and 40X (E-F) objective power. Images are representative of images derived from 2 experiments using 3 mice per group.
STAT1 deletion results in altered inflammatory cell accumulation in C. neoformans strain H99γ infected lungs
Our histology data suggest that STAT1 deletion results in a significantly altered immune/inflammatory response in H99γ infected lungs. To further characterize and quantify these differences, flow cytometry analysis of isolated pulmonary leukocytes from enzymatically dispersed lungs of WT and STAT1 KO mice was performed at days 7 and 10 post-inoculation. A trend towards increased leukocyte infiltration to the lungs of the STAT1 KO mice has been noted at day 10. However, we observed no statistically significant differences in total CD45+ leukocyte numbers (Figure 4A), suggesting that the differences in histological appearance of the inflammatory response were caused predominantly by differential distribution/density of the inflammatory infiltrates, rather than the major changes in total leukocyte number. Additionally, there were no significant differences in the numbers of infiltrating leukocyte subsets including macrophages, dendritic cells, CD4+ and CD8+ T cells, B cells, and neutrophils (Figures 4B-G) at days 7 and 10 post-inoculation with H99γ in STAT1 KO mice compared to WT mice. However, the absolute number of NK cells in STAT1 KO mice were significantly lower (p < 0.01; Figure 4I) and the absolute number of CD4+ T cells in STAT1 KO mice trended lower (p < 0.08; Figure 4D) at day 10 post-inoculation compared to WT mice. In contrast, eosinophils, which are associated with a Th2-type immune response and a hallmark of disease progression, were significantly increased in STAT1 KO mice at days 7 and 10 post-inoculation compared to WT mice (p < 0.01; Figure 4H). Neutrophils also showed an increasing trend (p < 0.146; Figure 4G) in STAT1 KO mice at day 10 post-inoculation. Collectively, these data suggest that STAT1 is not required for induction of the inflammatory response/recruitment of leukocytes into the lungs; however, STAT1 ablation does impact the phenotype of the cellular response to C. neoformans strain H99γ.
Figure 4. STAT1 is not required for pulmonary infiltration by immune cells in response to C. neoformans strain H99γ infection.

129S6/SvEv (WT) and STAT1 KO mice were given a pulmonary inoculation with 1 × 104 C. neoformans strain H99γ yeast cells. At days 7 and 10 post-inoculation, lungs were excised, tissue digested, and pulmonary infiltrates were analyzed by flow cytometry. Leukocytes were labeled with anti-CD45 antibodies for total leukocytes (A) or dual labeled with anti-CD45 and antibodies for specific cell types (B-I) and analyzed by flow cytometry. Data shown are the mean ± SEM of absolute cell numbers from 3 independent experiments performed using 4 mice per group per experiment. (*p < 0.01)
STAT1 signaling does not affect the induction of a cell-mediated immune response, but does contribute to an increased pro-inflammatory cytokine response and a shift in Th1-type/Th2-type cytokine bias
The changes in fungal control and cellular composition of the inflammatory infiltrates suggested that STAT1 expression affected the type of immune response/cytokine response during pulmonary inoculation with C. neoformans strain H99γ. We next determined how STAT1 deletion impacted cytokine expression in the infected lungs by analyzing lung homogenates from STAT1 KO and WT mice on days 7 and 10 post-inoculation with C. neoformans strain H99γ for cytokine protein content. The levels of Th1-type cytokines IL-2, IL-12p40 and IL-12p70 in STAT1 KO mice were similar relative to WT mice on day 7 post-inoculation (Table 1). IL-12p70 was significantly increased in the STAT1 KO mice compared to WT mice at day 10 post-inoculation (p < 0.01), however, IL-12p40 levels were significantly decreased at day 10 post-inoculation in STAT1 KO mice compared to WT mice (p < 0.001). IFN-γ levels were significantly decreased at days 7 (p < 0.01) and 10 (p < 0.001) post-inoculation in STAT1 KO mice compared to WT mice, despite the increase in pulmonary fungal burden of the IFN-γ producing C. neoformans strain in the lungs of STAT1 KO mice (Fig 1B). The available methodology does not allow us to directly distinguish what portion of the IFN-γ measured in vivo is derived from the IFN-γ producing C. neoformans strain versus the host leukocytes. To evaluate whether differences in IFN-γ levels are due to changes in production by the host or the IFN-γ producing C. neoformans strain, IFN-γ KO (C.129S7(B6)-Ifngtm1Ts/J) and WT controls (BALB/cJ) were inoculated with C. neoformans strain H99γ as described above and IFN-γ protein levels quantified from lung homogenates at day 7 post-inoculation (4-6 mice per group). There was no significant difference in pulmonary IFN-γ production in WT mice (17.6 ± 1.947 pg/ml) compared to IFN-γ KO mice (11.00 ± 2.203 pg/ml) and overall amounts of IFN-γ were low. This comparison suggests that the majority of IFN-γ detected in the STAT1 KO mice was produced by the microbe. In contrast, H99γ infected WT mice have greater levels of IFN-γ in the lungs, indicating that the host's cells produce a detectable amount of IFN-γ (p < 0.05) above what is produced by the microbe alone and that this amount contributes to the global level of IFN-γ in the infected lungs of the WT mice. Thus, the data acquired from IFN-γ KO mice suggests that the pulmonary leukocytes in STAT1 KO mice have diminished IFN-γ production when infected with the IFN-γ producing C. neoformans strain.
Table 1.
Cytokine analysis of lung homogenates in WT mice compared to STAT1 KO mice
| Cytokine or Chemokine | Level (pg/ml) in pulmonary homogenate tissue at day: | |||
|---|---|---|---|---|
|
| ||||
| 7 | 10 | |||
|
| ||||
| WT | STAT 1 KO | WT | STAT 1 KO | |
| Th1-type cytokine | ||||
| IFN-γ | 21.1 ± 3.89 | 12.48 ± 0.74* | 20.48 ± 1.96 | 13.74 ± 0.79** |
| IL-2 | 32.57 ± 3.19 | 46.33 ± 3.47 | 23.34 ± 1.47 | 20.12 ± 1.39 |
| IL-12 p40 | 81.27 ± 9.62 | 53.26 ± 12.15 | 227.46 ± 18.73 | 147.26 ± 18.48** |
| IL-12 p70 | 78.98 ± 12.20 | 88.52 ± 13.28 | 106.59 ± 15.52 | 153.50 ± 10.94* |
| Th2-type cytokine | ||||
| IL-4 | 18.37 ± 5.98 | 41.31 ± 5.00* | 10.01 ± 2.08 | 12.67 ± 1.58 |
| IL-5 | 7.35 ± 1.02 | 18.02 ± 3.75 | 7.06 ± 0.85 | 8.14 ± 0.68 |
| IL-10 | 31.75 ± 2.94 | 31.95 ± 1.74 | 48.52 ± 4.23 | 47.80 ± 2.81 |
| IL-13 | 101.54 ± 14.09 | 106.21 ± 5.39 | 96.84 ± 8.14 | 109.80 ± 4.60 |
| Pro-inflammatory cytokine | ||||
| IL-1α | 461.78 ± 114.37 | 310.38 ± 95.16 | 842.92 ± 185.14 | 1340.21 ± 153.47 |
| IL-1β | 2114.39 ± 549.41 | 2759.83 ± 845.82 | 3403.94 ± 654.74 | 5891.87 ± 611.89* |
| IL-6 | 112.10 ± 23.18 | 155.52 ± 59.42 | 72.76 ± 15.66 | 453.48 ± 86.54*** |
| IL-17A | 26.32 ± 3.63 | 240.85 ± 59.96*** | 20.15 ± 1.96 | 209.17 ± 33.27*** |
| TNF-α | 130.3 ± 21.03 | 157.33 ± 17.90 | 239.29 ± 59.03 | 458.83 ± 37.63* |
| G-CSF | 348.32 ± 43.58 | 909.00 ± 281.48 | 294.15 ± 54.97 | 2709.22 ± 214.13** |
| GM-CSF | 102.30 ± 8.10 | 103.72 ± 10.42 | 100.22 ± 3.59 | 129.84 ± 2.87 |
| Chemokine | ||||
| Eotaxin | 719.54 ± 171.16 | 606.13 ± 87.17 | 647.81 ± 35.75 | 705.59 ± 32.28 |
| CXCL1/KC | 765.54 ± 109.51 | 955.89 ± 213.18 | 729.03 ± 141.35 | 1838.48 ± 178.00*** |
| CCL5/RANTES | 715.31 ± 160.53 | 212.17 ± 42.91*** | 1058.68 ± 110.91 | 132.78 ± 24.12*** |
| CCL3/MIP-lα | 770.42 ± 160.53 | 1114.23 ± 301.66 | 1215.11 ± 314.48 | 2080.93 ± 254.31 |
| CCL4/MIP-1β | 175.68 ± 41.64 | 157.79 ± 38.92 | 255.24 ± 43.82 | 305.41 ± 39.42 |
| CCL2/MCP-1 | 924.17 ± 98.47 | 497.23 ± 98.31** | 883.19 ± 146.02 | 1874.04 ± 192.94** |
Data shown are expressed as mean ± SEM and are cumulative of 2 experiments utilizing 4 mice per group per time point.
p < 0.01,
p < 0.001,
p < 0.0001 compared to the infected counterpart on the same day postinoculation
The STAT1 KO mice demonstrated significantly higher levels of Th2-type cytokine IL-4 in the lungs at day 7 post-inoculation compared to WT mice (p < 0.01). However, IL-4 levels on day 10 and levels of IL-5 and immune-regulatory cytokine IL-10 in STAT1 KO and WT mice were not significantly different during the time course evaluated. Overall, the cytokine milieu indicates that a drift in Th1/Th2 cytokine balance away from Th1 and towards Th2 occurred early in C. neoformans strain H99γ inoculated STAT1 deficient mice. However, the subsequent increases in pro-inflammatory cytokine and chemokine production in the lungs of STAT1 KO mice compared to WT type mice during H99γ infection were much more dramatic, an indication of aberrant immune responses.
We observed significant increases in IL-17A at days 7 and 10 post-inoculation (p < 0.0001) and of IL-1β, IL-6, G-CSF, and TNF-α at day 10 post-inoculation (p < 0.01) within lung homogenates of STAT1 KO mice compared to WT mice during C. neoformans strain H99γ infection (Table 1). IL-1α levels were also increased, though not significantly, at day 10 post-inoculation in the STAT1 KO mice compared to WT mice. Additionally, we observed significantly decreased CCL5 production (p < 0.0001) at day 7 and 10 post-inoculation in STAT1 KO mice compared to WT mice but significantly increased CXCL1 and CCL2 production (p < 0.001) at day 10 post-inoculation in STAT1 KO mice compared to WT mice. These data show that more robust pro-inflammatory and altered chemokine responses occur in STAT1 KO mice compared to the WT mice during infection with C. neoformans strain H99γ. This increase in inflammatory cytokines was highly pronounced at day 10, consistent with an increasingly dysregulated inflammatory response promoting inflammatory pathology in response to the expansion of C. neoformans in the lungs.
STAT1 signaling is required for the induction of classically activated macrophages
While STAT1 signaling was required to mediate protection against pulmonary cryptococcosis, the changes in inflammatory responses and cytokine profiles could only partially explain the severe defect in fungal clearance of STAT1 KO mice. We sought to establish a causal link between STAT1 signaling and M1 macrophage activation, which we have previously shown correlates with disease resolution (24). Therefore, to examine macrophage polarization, we quantified the expression of genes associated with M1 and M2 macrophage activation in macrophages isolated from H99γ infected lungs of STAT1 KO compared to WT mice. Pulmonary F4/80+ macrophages were isolated at days 7 and 10 post-inoculation with H99γ and total RNA was evaluated for the expression of genes commonly associated with macrophage activation using real-time PCR (Figure 5). Gene expression of the M1 activation marker iNOS was significantly decreased in STAT1 KO mice compared to WT mice at day 7 by 200-fold (p < 0.0001; Figure 5A) and at day 10 by 10-fold (p < 0.001). Conversely, mRNA expression for M2 macrophage activation markers Arg1 and CD206 were significantly increased at days 7 (p < 0.01 and p < 0.001, respectively; Figure 5B) and 10 (p < 0.01) post-inoculation in STAT1 KO mice compared to WT mice. M2 activation marker FIZZ1 expression was also increased in macrophages from STAT1 KO mice, though not significantly, and Ym1 expression was significantly increased in macrophages from STAT1 KO mice at day 7 post-inoculation (p < 0.001; Figure 5B) and increased, though not significantly, at day 10 post-inoculation compared to expression in macrophages from WT mice. Gene expression of the pro-inflammatory cytokine IFN-γ was increased in macrophages from WT mice, though not significantly, compared to macrophages from STAT1 KO mice at days 7 and 10, however no difference was observed in the expression of TNF-α or IL-17A. Gene expression for cytokines IL-4 and IL-13, which are associated with a non-protective Th2-type response and M2 macrophages, was not different in macrophages from STAT1 KO mice compared to WT mice. Also, gene expression of CXCL9 and CXCL10, downstream chemokines in the STAT1 signaling pathway, was significantly decreased in macrophages from STAT1 KO mice compared to WT mice (p < 0.0001; Figure 5A). Gene expression of CXCL9 was increased by approximately 13,000-fold and CXCL10 was increased by 300-fold at day 7 post-inoculation, suggesting an increase in chemokine-induced infiltration of leukocytes to the site of infection. Furthermore, gene expression for transcription factor IRF-1, which is induced by STAT1, was significantly decreased in macrophages from STAT1 KO mice compared to macrophages WT from mice (p < 0.0001; Figure 5A). Altogether, the gene expression profile demonstrates that STAT1 signaling is required for M1 macrophage polarization during pulmonary C. neoformans H99γ infection.
Figure 5. STAT1 signaling is required for polarization of M1 macrophages in response to Cryptococcus strain H99γ infection.
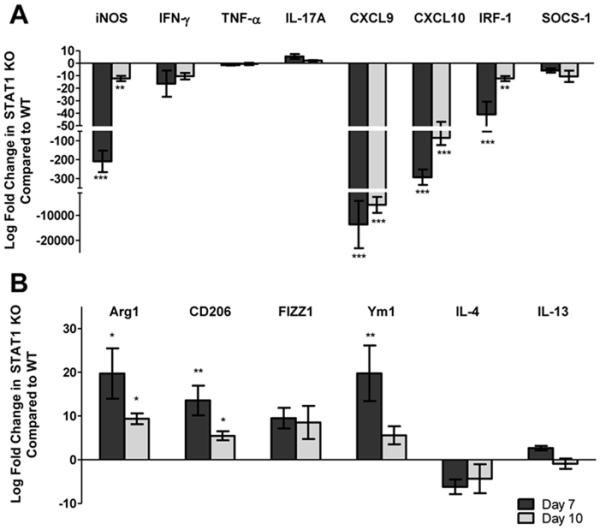
129S6/SvEv (WT) and STAT1 KO mice were inoculated with 1 × 104 CFU of C. neoformans strain H99γ. Pulmonary F4/80+ macrophages were isolated from the lungs of WT and STAT1 KO mice at days 7 and 10 post-inoculation with C. neoformans strain H99γ. Total RNA was extracted and transcripts were analyzed for markers and cytokines associated with M1 (A) and M2 (B) macrophage activation. The indicated mRNA levels were normalized to GAPDH. Results are expressed as mean ± SEM and are cumulative of 3 experiments utilizing 5 mice per group per time point. (*p < 0.01, **p < 0.001, ***p < 0.0001)
STAT1 signaling is important for optimal macrophage nitric oxide production and anti-fungal activity against C. neoformans
NO and reactive oxygen species (ROS) are by-products of M1 activated macrophages and these molecules together account for the oxidative microbicidal arsenal of phagocytes. Both NO and ROS are thought to be important for C. neoformans killing by host cells (24, 55-60). To assess the anti-fungal activity of STAT1 deficient macrophages in response to C. neoformans strain H99γ, we isolated pulmonary macrophages from STAT1 KO and WT mice at days 7 and 10 post-inoculation. The macrophages were lysed to release any intracellular cryptococci and the fungal burden was determined in order to quantify anti-fungal efficiency of macrophages. Macrophages were also examined for ROS production or cultured for 24 hours in vitro and nitrite production measured in the culture supernatants. Additional C. neoformans cells were not added to the cell culture medium as the macrophages were already infected in vivo.
We observed no significant difference in the number of cryptococci found within macrophages obtained from STAT1 KO and WT mice at day 7 post-inoculation; however, significantly more yeast were found within macrophages isolated from STAT1 deficient mice compared to macrophages from WT mice at day 10 post-inoculation (p < 0.001; Figure 6A). Furthermore, the morphology of intracellular organisms was different between WT and STAT1 KO mice. By day 10, macrophages from WT mice contained predominantly degraded organisms as documented by the presence of vacuoles that contained contracted and/or fragmented yeasts, or the capsular material alone (Figure 6B). In contrast, macrophages from STAT1 KO mice contained large and heavily capsulated yeast cells that grouped in multi-cellular clusters and robustly stained with mucicarmine (Figure 6B), consistent with viable and intracellular proliferating yeasts and illustrating the diminished capacity of the macrophages for control of intracellular fungal proliferation.
Figure 6. Anti-cryptococcal activity of pulmonary macrophages requires STAT1-dependent NO production.

129S6/SvEv (WT) and STAT1 KO mice were intranasally inoculated with 1 × 104 CFU of C. neoformans strain H99γ. Macrophages were isolated from the lungs of WT and STAT1 KO mice at days 7 and 10 post-inoculation and intracellular cryptococci enumerated (A). Lungs of H99γ -infected WT and STAT1 KO mice at day 10 post-inoculation were processed and analyzed using a light microscope (B). Note that pulmonary macrophages in the WT mice contain small vacuoles/inclusions consistent with degraded cryptococci and no evidence of intracellular growth. Macrophages in STAT1 KO mice harbor large cryptococci with well-developed capsules grouped in clusters of newly divided cells. Isolated pulmonary macrophages were examined for ROS production (C) or cultured ex vivo for 24 h and supernatants analyzed for the presence of NO (D). Data are expressed as the mean ± SEM and are cumulative of 3 experiments utilizing 3-5 mice per group per time point (A, C, D) or are representative images derived from 2 experiments utilizing 3 mice per group and photographed at 100X objective power (B). (*p < 0.01, **p < 0.001, ***p < 0.0001)
To further demonstrate a molecular basis for the diminished control of intracellular cryptococci by STAT1 KO mice, we assessed ROS and NO production by macrophages isolated from WT and STAT1 KO mice at days 7 and 10 post-inoculation. At day 7 post-inoculation, a small but significant reduction of ROS was detected in macrophages from STAT1 KO mice compared to WT mice (p < 0.0001; Figure 6C). However, at day 10 post-inoculation, there was no difference in the ROS production by macrophages between the two groups. This suggests that STAT1 plays only a minor role in induction of ROS production by macrophages and that ROS generation is insufficient for control of C. neoformans proliferation within macrophages (Figure 6A). NO production was assessed by measuring nitrite accumulation in the supernatants of the 24h macrophage cell cultures. Macrophages isolated from STAT1 KO mice inoculated with C. neoformans strain H99γ at days 7 and 10 post-inoculation had a profound decrease in nitrite accumulation following 24h of culture compared to macrophages from H99γ infected WT mice (p < 0.01, p < 0.0001, respectively; Figure 6D). These data correlate with the significant decrease in gene expression for iNOS, the enzyme that catalyzes NO production and hallmark marker of M1 macrophage activation, in macrophages from STAT1 KO mice compared to WT mice (Figure 5A). Thus, loss of STAT1 signaling results in profound defect in NO production by macrophages in response to C. neoformans H99γ infection which correlates with decreased anti-fungal efficiency and increased intra-macrophage residence of cryptococci.
Discussion
The identification of specific mechanisms that mediate the induction of protective immune responses following C. neoformans infection are likely to aid in the development of novel immunotherapies for treatment of cryptococcosis. We have developed a murine infection model with C. neoformans strain H99γ, a strain that secretes murine IFN-γ, which rapidly induces: 1) a protective Th1-type immune response in mice, 2) classical, M1, macrophage activation, and 3) resolution of infection with a protective memory (24, 26, 42, 50-52). This immune response to C. neoformans strain H99γ results in up-regulation of the STAT1 pathway in macrophages. STAT1 has been shown to play a major role in mediating immune and pro-inflammatory actions of IFN-γ (61), a cytokine important for regulation of the immune response (reviewed in (62)). Signaling via the IFN-γ-JAK1/2-STAT1 pathway is imperative for resistance to various intracellular pathogens including Listeria monocytogenes and Toxoplasma gondii (61, 63-65). Previous studies have shown that loss of STAT1 signaling in mice results in a complete lack of responsiveness to IFN-γ and IFN-α, correlating with high sensitivity to infection by some microbial pathogens (61, 65, 66). However, the role of STAT1 in the development of protective immunity to C. neoformans has not been investigated. Here we present for the first time that STAT1 is required for the generation of immune protection against C. neoformans. Our data further highlight that STAT1 is not absolutely required for the development of T cell mediated immune responses; however, it contributes to Th1 development and is critical for M1 polarization of macrophages and their fungicidal function.
The present studies were designed to evaluate a putative role of STAT1 signaling in mediating protection against pulmonary infection with C. neoformans. We previously examined the JAK/STAT pathway and TLR pathways in which we observed increased gene expression of members of the IFN-γR pathway, including STAT1, IRF-1, CXCL9, and CXCL10 (24). However, we detected no changes in expression of other STAT genes, MAPK genes, or genes within the TLR pathways (24). Our results demonstrate that STAT1 deficiency in mice results in increased fungal burden and decreased survival when inoculated with C. neoformans strain 52D and C. neoformans strain H99γ, while WT mice were able to clear infection with both of these strains. Furthermore, inoculation with C. neoformans strain 52D resulted in increased dissemination to the brain in STAT1 KO mice compared to WT mice. A similar trend occurred during inoculation with C. neoformans strain H99γ where lack of STAT1 signaling also resulted in increased trafficking of the yeast to the brain (data not shown). The increased trafficking to the CNS in the STAT1 deficient mice may be due to the loss of control of the pulmonary infection which then leads to dissemination. Altogether, these data indicate that STAT1 signaling has a role in prevention of dissemination to the CNS. Collectively, these results indicate that STAT1 signaling is essential for the generation of the protective immune response to C. neoformans infection in mice.
The data further show that loss of STAT1 signaling did not result in deficient leukocyte accumulation in the lungs. A trend towards increased leukocyte recruitment and an increase in the production of some but not all pro-inflammatory cytokines and chemokines occurred within the lungs of STAT1 KO mice. These data imply that the deficient cryptococcal clearance from the lungs of inoculated STAT1 KO mice is a result of a dysregulated immune response. Consistent with this view, the overall magnitude of the T cell response did not appear to be defective in the STAT1 KO mice. IFN-γ and STAT1 induce the transcription factor T-bet, which in turn promotes Th-1 type T cell development (67, 68). Additionally, T-bet suppresses GATA3, a transcription factor that aids in regulation of Th2-type immune responses (69-72). Interestingly, while some decrease in Th1 cytokines was observed, the levels of Th2-associated cytokines IL-4, IL-5, and IL-13 were not dramatically different between WT and STAT1 deficient mice, except for IL-4 at day 7 post-inoculation. The lack of major changes in expression of these cytokines that are transcriptionally enhanced by GATA3, suggest that STAT1 deletion did not significantly affect GATA3 levels. These findings suggest that STAT1 is more important for execution of downstream effector mechanisms, such as M1 macrophage activation, rather than the upstream regulation of T cell polarizing responses during cryptococcal infection. However, further studies including T-bet and GATA3 expression levels by T cells during cryptococcal infection in STAT1 KO mice are needed to definitively address this point.
Apart from the increased cryptococcal antigen load, the uncontrolled inflammation may be a result of the loss of regulatory proteins that are induced down-stream of STAT1 signaling. We observed that transcripts for IRF-1, a regulatory protein induced by STAT1 (62, 73, 74), were decreased in macrophages from STAT1 KO mice compared to WT mice. While IRF-1 participates in regulation of the IFN-γ/STAT1 pathway, this protein also plays a role in other pathways to restrain the pro-inflammatory immune response, thus limiting damage to the host. Macrophages from STAT1 KO mice also had a decrease in transcripts for CXCL9 and CXCL10, IFN-γ induced chemokines that play a role in attracting other immune cells including antigen-specific Th1 cells and NK cells (46, 75). This may partially explain the decreasing trend in CD4+ T cells and the significant decrease in NK cells trafficking into the lungs of the STAT1 deficient mice at day 10 post-inoculation. The necessity of NK cell activity during cryptococcal infection is unclear. While it has been demonstrated that the absence of NK cells results in a reduction of fungal clearance from the lungs and spleens (76, 77), studies conducted in our laboratory show that mice depleted of NK cells maintain a protective immune response to C. neoformans strain H99γ (unpublished results). Additionally, CXCL9 has been reported to inhibit eosinophil infiltration (78, 79), therefore, the decrease in CXCL9 gene expression by macrophages may be, to some degree, responsible for the increased eosinophilia observed in the STAT1 KO mice.
It is very important for the host to be able to respond to an invading pathogen with a balance between pro- and anti-inflammatory factors as excessive inflammation can result in severe lung pathology and ultimately death (80, 81). Cytokine analysis of pulmonary tissue from STAT1 KO mice showed increased levels of pro-inflammatory cytokines and a substantial increase in pulmonary leukocyte infiltrates compared to WT mice, however, the STAT1 deficient mice were unable to eradicate the yeast and permitted rapid fungal proliferation. This increase in pulmonary fungal burden could contribute to the increased production of pro-inflammatory factors leading to increased inflammation. Taken together, these studies demonstrate that though STAT1 is not required for leukocyte recruitment in response to C. neoformans strain H99γ infection, they strongly suggest that STAT1 signaling may be directly (through the regulatory negative feedback loops) and/or indirectly (increased fungal burden) responsible for limiting damage-inducing inflammation.
It is accepted that macrophage activation phenotype is important for host defense against Cryptococcus infection (24-26, 35, 36, 42, 82). Our data show that when STAT1 signaling is ablated in the host, there is an increase in Arg1 gene expression and a decrease in iNOS gene expression compared to WT mice. Overall, this indicates a shift in the Arg1: iNOS ratio favoring Arg1 and M2 macrophage activation, suggesting that the loss of STAT1 signaling leads to alternative activation of macrophages which are associated with non-protective responses to C. neoformans. Though loss of STAT1 signaling does not significantly alter the total numbers of macrophages trafficking into the lungs during inoculation with C. neoformans strain H99γ, it appears that it is the quality and not quantity of macrophages that aids in eradication of the yeast. Assays measuring macrophage production of ROS and NO, molecules associated with anti-cryptococcal activity, reveal that STAT1 has minimal effect in promoting ROS generation but has a profound effect on NO production. While sufficient in ROS generation, STAT1 deficient macrophages produced significantly less NO compared to WT macrophages, which correlated with a defect in cryptococcal killing. These effects are attributed to dramatically decreased iNOS expression but also significantly increased Arg1. Arg1 competes with iNOS for the substrate L-arginine (37), therefore, the observed decrease in NO production in STAT1 deficient macrophages could be linked to the changes in both of these enzymes. In support of this theory, previous studies have shown that the ratio Arg1/iNOS is predictive of the fungicidal activity of macrophages during C. neoformans infection (31, 54, 83). Our results showing that STAT1 deficiency result in an over one hundred-fold change in Arg1/iNOS ratio at day 7 and day 10 post-infection is consistent with the diminished fungicidal capacity observed by macrophages from STAT1 KO mice infected with C. neoformans strain H99γ. Collectively, our data indicate that the most important function of STAT1 in anti-cryptococcal immunity is to mediate M1 polarization of macrophages, which is then crucial for protection against pulmonary cryptococcosis by the production of NO with possible contribution of other factors such as activation of lysosome enzymes, which was not investigated at this time. Our new data combined with our previous observation of increased STAT1 phosphorylation in macrophages from protectively immunized mice strongly suggests that this is an intrinsic effect of STAT1 determining the fate of macrophage polarization down-stream of IFN-γ signaling. One caveat to this conclusion is that the general STAT1 deletion also resulted in diminished IFN-γ production in the infected lungs. Since IFN-γ is the major factor driving M1 polarization, some of the in vivo effects of the STAT1 deletion could be associated with an indirect effect of STAT1 on IFN-γ production. Future studies using transgenic mice with cell-restricted STAT1 abrogation will be useful tools to specifically address this point.
The applications of these findings have the potential to spread beyond the scope of cryptococcosis. IFN-γ activated macrophages have been shown to have fungicidal activity against Histoplasma capsulatum (84-86) while presence of IL-4 and IL-10 results in inhibition of apoptosis and increase in disease severity (87). M1 activated macrophages have also been associated with antifungal effects against Aspergillus fumigatus (88), Blastomyces dermatitidis (89-91), and Paracoccidioides brasiliensis (90, 92, 93). Therefore, the development of therapeutics that induce M1 macrophage activation via an IFN-γ/STAT1 mechanism may provide protection against a myriad of microbial pathogens.
Collectively, our data show that STAT1 plays an essential role in the development and execution of crucial effector mechanisms in protective immunity to pulmonary cryptococcosis: Th1 and M1 polarizations of T cells and macrophages, respectively. Activation of M2 macrophages, as demonstrated in the H99γ-inoculated STAT1 KO mice, does not appear to aid in clearance of Cryptococcus from the lungs, but instead hinders anti-fungal mechanisms. Furthermore, NO but not ROS production appears to be critical for control of C. neoformans strain H99γ proliferation within macrophages. This is consistent with the hypothesis that STAT1-induced M1 macrophages are essential to mount a protective immune response against C. neoformans. These data represent the first evidence of an intracellular signaling mechanism in an effector cell population by which the host executes a protective immune response to C. neoformans.
Acknowledgments
Supported by research grant 2RO1 AI071752 from the National Institute of Allergy and Infectious Diseases (NIAID) of the National Institutes of Health (NIH) and W911NF-11-1-0136 from the Army Research Office of the Department of Defense (F.L.W. Jr.) and VA Merit Review Grant 1I01BX000656 (M. A. O.).
Footnotes
The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious diseases, the National Institutes of Health, or The University of Texas at San Antonio. The authors declare no conflict of interest.
- Arg1
- Arginase-1
- IRF
- IFN regulatory factor
- iNOS
- inducible nitric oxide synthase
- H99γ
- IFN-γ producing C. neoformans strain
- KO
- knock out
- SOCS
- suppressor of cytokine signaling
- YPD
- yeast extract/peptone/dextrose
References
- 1.Chuck SL, Sande MA. Infections with Cryptococcus neoformans in the acquired immunodeficiency syndrome. N Engl J Med. 1989;321:794–799. doi: 10.1056/NEJM198909213211205. [DOI] [PubMed] [Google Scholar]
- 2.Dismukes WE. Cryptococcal meningitis in patients with AIDS. J Infect Dis. 1988;157:624–628. doi: 10.1093/infdis/157.4.624. [DOI] [PubMed] [Google Scholar]
- 3.Eng RH, Bishburg E, Smith SM, Kapila R. Cryptococcal infections in patients with acquired immune deficiency syndrome. Am J Med. 1986;81:19–23. doi: 10.1016/0002-9343(86)90176-2. [DOI] [PubMed] [Google Scholar]
- 4.Gal AA, Koss MN, Hawkins J, Evans S, Einstein H. The pathology of pulmonary cryptococcal infections in the acquired immunodeficiency syndrome. Arch Pathol Lab Med. 1986;110:502–507. [PubMed] [Google Scholar]
- 5.Kovacs JA, Kovacs AA, Polis M, Wright WC, Gill VJ, Tuazon CU, Gelmann EP, Lane HC, Longfield R, Overturf G, et al. Cryptococcosis in the acquired immunodeficiency syndrome. Ann Intern Med. 1985;103:533–538. doi: 10.7326/0003-4819-103-4-533. [DOI] [PubMed] [Google Scholar]
- 6.Diamond RD, Bennett JE. Prognostic factors in cryptococcal meningitis. A study in 111 cases. Ann Intern Med. 1974;80:176–181. doi: 10.7326/0003-4819-80-2-176. [DOI] [PubMed] [Google Scholar]
- 7.Duperval R, Hermans PE, Brewer NS, Roberts GD. Cryptococcosis, with emphasis on the significance of isolation of Cryptococcus neoformans from the respiratory tract. Chest. 1977;72:13–19. doi: 10.1378/chest.72.1.13. [DOI] [PubMed] [Google Scholar]
- 8.Goldstein E, Rambo ON. Cryptococcal infection following steroid therapy. Ann Intern Med. 1962;56:114–120. doi: 10.7326/0003-4819-56-1-114. [DOI] [PubMed] [Google Scholar]
- 9.Collins VP, Gellhorn A, Trimble JR. The coincidence of cryptococcosis and disease of the reticulo-endothelial and lymphatic systems. Cancer. 1951;4:883–889. doi: 10.1002/1097-0142(195107)4:4<883::aid-cncr2820040426>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 10.Gendel BR, Ende M, Norman SL. Cryptococcosis; a review with special reference to apparent association with Hodgkin's disease. Am J Med. 1950;9:343–355. doi: 10.1016/0002-9343(50)90430-x. [DOI] [PubMed] [Google Scholar]
- 11.Keye JD, Jr., Magee WE. Fungal diseases in a general hospital; a study of 88 patients. Am J Clin Pathol. 1956;26:1235–1253. doi: 10.1093/ajcp/26.11.1235. [DOI] [PubMed] [Google Scholar]
- 12.Lewis JL, Rabinovich S. The wide spectrum of cryptococcal infections. Am J Med. 1972;53:315–322. doi: 10.1016/0002-9343(72)90174-x. [DOI] [PubMed] [Google Scholar]
- 13.Husain S, Wagener MM, Singh N. Cryptococcus neoformans infection in organ transplant recipients: variables influencing clinical characteristics and outcome. Emerg Infect Dis. 2001;7:375–381. doi: 10.3201/eid0703.010302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Park BJ, Wannemuehler KA, Marston BJ, Govender N, Pappas PG, Chiller TM. Estimation of the current global burden of cryptococcal meningitis among persons living with HIV/AIDS. Aids. 2009;23:525–530. doi: 10.1097/QAD.0b013e328322ffac. [DOI] [PubMed] [Google Scholar]
- 15.Singh N, Lortholary O, Alexander BD, Gupta KL, John GT, Pursell K, Munoz P, Klintmalm GB, Stosor V, del Busto R, Limaye AP, Somani J, Lyon M, Houston S, House AA, Pruett TL, Orloff S, Humar A, Dowdy L, Garcia-Diaz J, Kalil AC, Fisher RA, Husain S, G. Cryptococcal Collaborative Transplant Study An immune reconstitution syndrome-like illness associated with Cryptococcus neoformans infection in organ transplant recipients. Clin Infect Dis. 2005;40:1756–1761. doi: 10.1086/430606. [DOI] [PubMed] [Google Scholar]
- 16.Pappas PG, Alexander BD, Andes DR, Hadley S, Kauffman CA, Freifeld A, Anaissie EJ, Brumble LM, Herwaldt L, Ito J, Kontoyiannis DP, Lyon GM, Marr KA, Morrison VA, Park BJ, Patterson TF, Perl TM, Oster RA, Schuster MG, Walker R, Walsh TJ, Wannemuehler KA, Chiller TM. Invasive fungal infections among organ transplant recipients: results of the Transplant-Associated Infection Surveillance Network (TRANSNET) Clin Infect Dis. 2010;50:1101–1111. doi: 10.1086/651262. [DOI] [PubMed] [Google Scholar]
- 17.Coker RJ, Viviani M, Gazzard BG, Du Pont B, Pohle HD, Murphy SM, Atouguia J, Champalimaud JL, Harris JR. Treatment of cryptococcosis with liposomal amphotericin B (AmBisome) in 23 patients with AIDS. AIDS (London, England) 1993;7:829–835. doi: 10.1097/00002030-199306000-00011. [DOI] [PubMed] [Google Scholar]
- 18.Perfect JR, Cox GM. Drug resistance in Cryptococcus neoformans. Drug Resistance Updates. 1999;2:259–269. doi: 10.1054/drup.1999.0090. [DOI] [PubMed] [Google Scholar]
- 19.Roh TY, Cuddapah S, Cui K, Zhao K. The genomic landscape of histone modifications in human T cells. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:15782–15787. doi: 10.1073/pnas.0607617103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mitchell TG, Perfect JR. Cryptococcosis in the era of AIDS--100 years after the discovery of Cryptococcus neoformans. Clin Microbiol Rev. 1995;8:515–548. doi: 10.1128/cmr.8.4.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goldman DL, Khine H, Abadi J, Lindenberg DJ, Pirofski L, Niang R, Casadevall A. Serologic evidence for Cryptococcus neoformans infection in early childhood. Pediatrics. 2001;107:E66. doi: 10.1542/peds.107.5.e66. [DOI] [PubMed] [Google Scholar]
- 22.Abadi J, Nachman S, Kressel AB, Pirofski L. Cryptococcosis in children with AIDS. Clin Infect Dis. 1999;28:309–313. doi: 10.1086/515130. [DOI] [PubMed] [Google Scholar]
- 23.Chayakulkeeree M, Perfect JR. Cryptococcosis. Infect Dis Clin North Am. 2006;20:507–544. doi: 10.1016/j.idc.2006.07.001. v-vi. [DOI] [PubMed] [Google Scholar]
- 24.Hardison SE, Herrera G, Young ML, Hole CR, Wozniak KL, Wormley FL., Jr. Protective immunity against pulmonary cryptococcosis is associated with STAT1-mediated classical macrophage activation. Journal of immunology. 2012;189:4060–4068. doi: 10.4049/jimmunol.1103455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Y, Wang F, Tompkins KC, McNamara A, Jain AV, Moore BB, Toews GB, Huffnagle GB, Olszewski MA. Robust Th1 and Th17 immunity supports pulmonary clearance but cannot prevent systemic dissemination of highly virulent Cryptococcus neoformans H99. Am J Pathol. 2009;175:2489–2500. doi: 10.2353/ajpath.2009.090530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hardison SE, Ravi S, Wozniak KL, Young ML, Olszewski MA, Wormley FL., Jr. Pulmonary infection with an interferon-gamma-producing Cryptococcus neoformans strain results in classical macrophage activation and protection. Am J Pathol. 2010;176:774–785. doi: 10.2353/ajpath.2010.090634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jain AV, Zhang Y, Fields WB, McNamara DA, Choe MY, Chen GH, Erb-Downward J, Osterholzer JJ, Toews GB, Huffnagle GB, Olszewski MA. Th2 but not Th1 immune bias results in altered lung functions in a murine model of pulmonary Cryptococcus neoformans infection. Infect Immun. 2009;77:5389–5399. doi: 10.1128/IAI.00809-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Herring AC, Lee J, McDonald RA, Toews GB, Huffnagle GB. Induction of interleukin-12 and gamma interferon requires tumor necrosis factor alpha for protective T1-cell-mediated immunity to pulmonary Cryptococcus neoformans infection. Infect Immun. 2002;70:2959–2964. doi: 10.1128/IAI.70.6.2959-2964.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huffnagle GB, Lipscomb MF, Lovchik JA, Hoag KA, Street NE. The role of CD4+ and CD8+ T cells in the protective inflammatory response to a pulmonary cryptococcal infection. J Leukoc Biol. 1994;55:35–42. doi: 10.1002/jlb.55.1.35. [DOI] [PubMed] [Google Scholar]
- 30.Huffnagle GB, Toews GB, Burdick MD, Boyd MB, McAllister KS, McDonald RA, Kunkel SL, Strieter RM. Afferent phase production of TNF-alpha is required for the development of protective T cell immunity to Cryptococcus neoformans. Journal of immunology. 1996;157:4529–4536. [PubMed] [Google Scholar]
- 31.Arora S, Hernandez Y, Erb-Downward JR, McDonald RA, Toews GB, Huffnagle GB. Role of IFN-gamma in regulating T2 immunity and the development of alternatively activated macrophages during allergic bronchopulmonary mycosis. Journal of immunology. 2005;174:6346–6356. doi: 10.4049/jimmunol.174.10.6346. [DOI] [PubMed] [Google Scholar]
- 32.Chen GH, McNamara DA, Hernandez Y, Huffnagle GB, Toews GB, Olszewski MA. Inheritance of immune polarization patterns is linked to resistance versus susceptibility to Cryptococcus neoformans in a mouse model. Infect Immun. 2008;76:2379–2391. doi: 10.1128/IAI.01143-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hernandez Y, Arora S, Erb-Downward JR, McDonald RA, Toews GB, Huffnagle GB. Distinct roles for IL-4 and IL-10 in regulating T2 immunity during allergic bronchopulmonary mycosis. Journal of immunology. 2005;174:1027–1036. doi: 10.4049/jimmunol.174.2.1027. [DOI] [PubMed] [Google Scholar]
- 34.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nature reviews. Immunology. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Muller U, Stenzel W, Kohler G, Werner C, Polte T, Hansen G, Schutze N, Straubinger RK, Blessing M, McKenzie AN, Brombacher F, Alber G. IL-13 induces disease-promoting type 2 cytokines, alternatively activated macrophages and allergic inflammation during pulmonary infection of mice with Cryptococcus neoformans. Journal of immunology. 2007;179:5367–5377. doi: 10.4049/jimmunol.179.8.5367. [DOI] [PubMed] [Google Scholar]
- 36.Osterholzer JJ, Surana R, Milam JE, Montano GT, Chen GH, Sonstein J, Curtis JL, Huffnagle GB, Toews GB, Olszewski MA. Cryptococcal urease promotes the accumulation of immature dendritic cells and a non-protective T2 immune response within the lung. Am J Pathol. 2009;174:932–943. doi: 10.2353/ajpath.2009.080673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hesse M, Modolell M, La Flamme AC, Schito M, Fuentes JM, Cheever AW, Pearce EJ, Wynn TA. Differential regulation of nitric oxide synthase-2 and arginase-1 by type 1/type 2 cytokines in vivo: granulomatous pathology is shaped by the pattern of L-arginine metabolism. Journal of immunology. 2001;167:6533–6544. doi: 10.4049/jimmunol.167.11.6533. [DOI] [PubMed] [Google Scholar]
- 38.Granger DL, Hibbs JB, Jr., Perfect JR, Durack DT. Specific amino acid (L-arginine) requirement for the microbiostatic activity of murine macrophages. J Clin Invest. 1988;81:1129–1136. doi: 10.1172/JCI113427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Granger DL, Hibbs JB, Jr., Perfect JR, Durack DT. Metabolic fate of L-arginine in relation to microbiostatic capability of murine macrophages. J Clin Invest. 1990;85:264–273. doi: 10.1172/JCI114422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen GH, Olszewski MA, McDonald RA, Wells JC, Paine R, 3rd, Huffnagle GB, Toews GB. Role of granulocyte macrophage colony-stimulating factor in host defense against pulmonary Cryptococcus neoformans infection during murine allergic bronchopulmonary mycosis. Am J Pathol. 2007;170:1028–1040. doi: 10.2353/ajpath.2007.060595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guerrero A, Jain N, Wang X, Fries BC. Cryptococcus neoformans variants generated by phenotypic switching differ in virulence through effects on macrophage activation. Infect Immun. 2010;78:1049–1057. doi: 10.1128/IAI.01049-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hardison SE, Wozniak KL, Kolls JK, Wormley FL., Jr. Interleukin-17 is not required for classical macrophage activation in a pulmonary mouse model of Cryptococcus neoformans infection. Infect Immun. 2010;78:5341–5351. doi: 10.1128/IAI.00845-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stark GR. How cells respond to interferons revisited: From early history to current complexity. Cytokine Growth F R. 2007;18:419–423. doi: 10.1016/j.cytogfr.2007.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Szabo SJ, Sullivan BM, Peng SL, Glimcher LH. Molecular mechanisms regulating Th1 immune responses. Annual review of immunology. 2003;21:713–758. doi: 10.1146/annurev.immunol.21.120601.140942. [DOI] [PubMed] [Google Scholar]
- 45.Taki S, Sato T, Ogasawara K, Fukuda T, Sato M, Hida S, Suzuki G, Mitsuyama M, Shin EH, Kojima S, Taniguchi T, Asano Y. Multistage regulation of Th1-type immune responses by the transcription factor IRF-1. Immunity. 1997;6:673–679. doi: 10.1016/s1074-7613(00)80443-4. [DOI] [PubMed] [Google Scholar]
- 46.Mikhak Z, Fleming CM, Medoff BD, Thomas SY, Tager AM, Campanella GS, Luster AD. STAT1 in peripheral tissue differentially regulates homing of antigen-specific Th1 and Th2 cells. Journal of immunology. 2006;176:4959–4967. doi: 10.4049/jimmunol.176.8.4959. [DOI] [PubMed] [Google Scholar]
- 47.Smeekens SP, Plantinga TS, van de Veerdonk FL, Heinhuis B, Hoischen A, Joosten LA, Arkwright PD, Gennery A, Kullberg BJ, Veltman JA, Lilic D, van der Meer JW, Netea MG. STAT1 hyperphosphorylation and defective IL12R/IL23R signaling underlie defective immunity in autosomal dominant chronic mucocutaneous candidiasis. PLoS One. 2011;6:e29248. doi: 10.1371/journal.pone.0029248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van de Veerdonk FL, Plantinga TS, Hoischen A, Smeekens SP, Joosten LA, Gilissen C, Arts P, Rosentul DC, Carmichael AJ, Smits-van der Graaf CA, Kullberg BJ, van der Meer JW, Lilic D, Veltman JA, Netea MG. STAT1 mutations in autosomal dominant chronic mucocutaneous candidiasis. N Engl J Med. 2011;365:54–61. doi: 10.1056/NEJMoa1100102. [DOI] [PubMed] [Google Scholar]
- 49.Sampaio EP, Hsu AP, Pechacek J, Bax HI, Dias DL, Paulson ML, Chandrasekaran P, Rosen LB, Carvalho DS, Ding L, Vinh DC, Browne SK, Datta S, Milner JD, Kuhns DB, Long Priel DA, Sadat MA, Shiloh M, De Marco B, Alvares M, Gillman JW, Ramarathnam V, de la Morena M, Bezrodnik L, Moreira I, Uzel G, Johnson D, Spalding C, Zerbe CS, Wiley H, Greenberg DE, Hoover SE, Rosenzweig SD, Galgiani JN, Holland SM. Signal transducer and activator of transcription 1 (STAT1) gain-of-function mutations and disseminated coccidioidomycosis and histoplasmosis. J Allergy Clin Immunol. 2013;131:1624–1634. doi: 10.1016/j.jaci.2013.01.052. e1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wormley FL, Jr., Perfect JR, Steele C, Cox GM. Protection against cryptococcosis by using a murine gamma interferon-producing Cryptococcus neoformans strain. Infect Immun. 2007;75:1453–1462. doi: 10.1128/IAI.00274-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wozniak KL, Ravi S, Macias S, Young ML, Olszewski MA, Steele C, Wormley FL. Insights into the mechanisms of protective immunity against Cryptococcus neoformans infection using a mouse model of pulmonary cryptococcosis. PLoS One. 2009;4:e6854. doi: 10.1371/journal.pone.0006854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wozniak KL, Young ML, Wormley FL., Jr. Protective immunity against experimental pulmonary cryptococcosis in T cell-depleted mice. Clin Vaccine Immunol. 2011;18:717–723. doi: 10.1128/CVI.00036-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huffnagle GB, Yates JL, Lipscomb MF. Immunity to a Pulmonary Cryptococcus neoformans Infection Requires Both Cd4+ and Cd8+ T-Cells. Journal of Experimental Medicine. 1991;173:793–800. doi: 10.1084/jem.173.4.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Arora S, Olszewski MA, Tsang TM, McDonald RA, Toews GB, Huffnagle GB. Effect of cytokine interplay on macrophage polarization during chronic pulmonary infection with Cryptococcus neoformans. Infect Immun. 2011;79:1915–1926. doi: 10.1128/IAI.01270-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cox GM, Harrison TS, McDade HC, Taborda CP, Heinrich G, Casadevall A, Perfect JR. Superoxide dismutase influences the virulence of Cryptococcus neoformans by affecting growth within macrophages. Infect Immun. 2003;71:173–180. doi: 10.1128/IAI.71.1.173-180.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wormley FL, Jr., Heinrich G, Miller JL, Perfect JR, Cox GM. Identification and characterization of an SKN7 homologue in Cryptococcus neoformans. Infect Immun. 2005;73:5022–5030. doi: 10.1128/IAI.73.8.5022-5030.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Giles SS, Stajich JE, Nichols C, Gerrald QD, Alspaugh JA, Dietrich F, Perfect JR. The Cryptococcus neoformans catalase gene family and its role in antioxidant defense. Eukaryot Cell. 2006;5:1447–1459. doi: 10.1128/EC.00098-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.de Jesus-Berrios M, Liu L, Nussbaum JC, Cox GM, Stamler JS, Heitman J. Enzymes that counteract nitrosative stress promote fungal virulence. Curr Biol. 2003;13:1963–1968. doi: 10.1016/j.cub.2003.10.029. [DOI] [PubMed] [Google Scholar]
- 59.Aguirre KM, Gibson GW. Differing requirement for inducible nitric oxide synthase activity in clearance of primary and secondary Cryptococcus neoformans infection. Medical mycology. 2000;38:343–353. doi: 10.1080/mmy.38.5.343.353. [DOI] [PubMed] [Google Scholar]
- 60.Lovchik JA, Lyons CR, Lipscomb MF. A role for gamma interferon-induced nitric oxide in pulmonary clearance of Cryptococcus neoformans. Am J Respir Cell Mol Biol. 1995;13:116–124. doi: 10.1165/ajrcmb.13.1.7598935. [DOI] [PubMed] [Google Scholar]
- 61.Meraz MA, White JM, Sheehan KC, Bach EA, Rodig SJ, Dighe AS, Kaplan DH, Riley JK, Greenlund AC, Campbell D, Carver-Moore K, DuBois RN, Clark R, Aguet M, Schreiber RD. Targeted disruption of the STAT1 gene in mice reveals unexpected physiologic specificity in the JAK-STAT signaling pathway. Cell. 1996;84:431–442. doi: 10.1016/s0092-8674(00)81288-x. [DOI] [PubMed] [Google Scholar]
- 62.Saha B, Jyothi Prasanna S, Chandrasekar B, Nandi D. Gene modulation and immunoregulatory roles of interferon gamma. Cytokine. 2010;50:1–14. doi: 10.1016/j.cyto.2009.11.021. [DOI] [PubMed] [Google Scholar]
- 63.Kernbauer E, Maier V, Stoiber D, Strobl B, Schneckenleithner C, Sexl V, Reichart U, Reizis B, Kalinke U, Jamieson A, Muller M, Decker T. Conditional STAT1 ablation reveals the importance of interferon signaling for immunity to Listeria monocytogenes infection. PLoS Pathog. 2012;8:e1002763. doi: 10.1371/journal.ppat.1002763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lang C, Hildebrandt A, Brand F, Opitz L, Dihazi H, Luder CG. Impaired chromatin remodelling at STAT1-regulated promoters leads to global unresponsiveness of Toxoplasma gondii-Infected macrophages to IFN-gamma. PLoS Pathog. 2012;8:e1002483. doi: 10.1371/journal.ppat.1002483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gavrilescu LC, Butcher BA, Del Rio L, Taylor GA, Denkers EY. STAT1 is essential for antimicrobial effector function but dispensable for gamma interferon production during Toxoplasma gondii infection. Infect Immun. 2004;72:1257–1264. doi: 10.1128/IAI.72.3.1257-1264.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lieberman LA, Banica M, Reiner SL, Hunter CA. STAT1 plays a critical role in the regulation of antimicrobial effector mechanisms, but not in the development of Th1-type responses during toxoplasmosis. Journal of immunology. 2004;172:457–463. doi: 10.4049/jimmunol.172.1.457. [DOI] [PubMed] [Google Scholar]
- 67.Afkarian M, Sedy JR, Yang J, Jacobson NG, Cereb N, Yang SY, Murphy TL, Murphy KM. T-bet is a STAT1-induced regulator of IL-12R expression in naive CD4+ T cells. Nature immunology. 2002;3:549–557. doi: 10.1038/ni794. [DOI] [PubMed] [Google Scholar]
- 68.Lighvani AA, Frucht DM, Jankovic D, Yamane H, Aliberti J, Hissong BD, Nguyen BV, Gadina M, Sher A, Paul WE, O'Shea JJ. T-bet is rapidly induced by interferon-gamma in lymphoid and myeloid cells. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:15137–15142. doi: 10.1073/pnas.261570598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Usui T, Preiss JC, Kanno Y, Yao ZJ, Bream JH, O'Shea JJ, Strober W. T-bet regulates Th1 responses through essential effects on GATA-3 function rather than on IFNG gene acetylation and transcription. The Journal of experimental medicine. 2006;203:755–766. doi: 10.1084/jem.20052165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hwang ES, Szabo SJ, Schwartzberg PL, Glimcher LH. T helper cell fate specified by kinase-mediated interaction of T-bet with GATA-3. Science. 2005;307:430–433. doi: 10.1126/science.1103336. [DOI] [PubMed] [Google Scholar]
- 71.Zheng W, Flavell RA. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell. 1997;89:587–596. doi: 10.1016/s0092-8674(00)80240-8. [DOI] [PubMed] [Google Scholar]
- 72.Zhang DH, Cohn L, Ray P, Bottomly K, Ray A. Transcription factor GATA-3 is differentially expressed in murine Th1 and Th2 cells and controls Th2-specific expression of the interleukin-5 gene. J Biol Chem. 1997;272:21597–21603. doi: 10.1074/jbc.272.34.21597. [DOI] [PubMed] [Google Scholar]
- 73.Starr R, Willson TA, Viney EM, Murray LJ, Rayner JR, Jenkins BJ, Gonda TJ, Alexander WS, Metcalf D, Nicola NA, Hilton DJ. A family of cytokine-inducible inhibitors of signalling. Nature. 1997;387:917–921. doi: 10.1038/43206. [DOI] [PubMed] [Google Scholar]
- 74.Alexander WS, Starr R, Fenner JE, Scott CL, Handman E, Sprigg NS, Corbin JE, Cornish AL, Darwiche R, Owczarek CM, Kay TW, Nicola NA, Hertzog PJ, Metcalf D, Hilton DJ. SOCS1 is a critical inhibitor of interferon gamma signaling and prevents the potentially fatal neonatal actions of this cytokine. Cell. 1999;98:597–608. doi: 10.1016/s0092-8674(00)80047-1. [DOI] [PubMed] [Google Scholar]
- 75.Muller M, Carter S, Hofer MJ, Campbell IL. Review: The chemokine receptor CXCR3 and its ligands CXCL9, CXCL10 and CXCL11 in neuroimmunity--a tale of conflict and conundrum. Neuropathology and applied neurobiology. 2010;36:368–387. doi: 10.1111/j.1365-2990.2010.01089.x. [DOI] [PubMed] [Google Scholar]
- 76.Hidore MR, Murphy JW. Natural cellular resistance of beige mice against Cryptococcus neoformans. Journal of immunology. 1986;137:3624–3631. [PubMed] [Google Scholar]
- 77.Lipscomb MF, Alvarellos T, Toews GB, Tompkins R, Evans Z, Koo G, Kumar V. Role of natural killer cells in resistance to Cryptococcus neoformans infections in mice. Am J Pathol. 1987;128:354–361. [PMC free article] [PubMed] [Google Scholar]
- 78.Fulkerson PC, Zimmermann N, Brandt EB, Muntel EE, Doepker MP, Kavanaugh JL, Mishra A, Witte DP, Zhang H, Farber JM, Yang M, Foster PS, Rothenberg ME. Negative regulation of eosinophil recruitment to the lung by the chemokine monokine induced by IFN-gamma (Mig, CXCL9) Proceedings of the National Academy of Sciences of the United States of America. 2004;101:1987–1992. doi: 10.1073/pnas.0308544100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fulkerson PC, Zhu H, Williams DA, Zimmermann N, Rothenberg ME. CXCL9 inhibits eosinophil responses by a CCR3- and Rac2-dependent mechanism. Blood. 2005;106:436–443. doi: 10.1182/blood-2005-02-0489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Casadevall A, Pirofski LA. Virulence factors and their mechanisms of action: the view from a damage-response framework. J Water Health. 2009;7(Suppl 1):S2–S18. doi: 10.2166/wh.2009.036. [DOI] [PubMed] [Google Scholar]
- 81.Pirofski LA, Casadevall A. The damage-response framework of microbial pathogenesis and infectious diseases. Adv Exp Med Biol. 2008;635:135–146. doi: 10.1007/978-0-387-09550-9_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Stenzel W, Muller U, Kohler G, Heppner FL, Blessing M, McKenzie AN, Brombacher F, Alber G. IL-4/IL-13-dependent alternative activation of macrophages but not microglial cells is associated with uncontrolled cerebral cryptococcosis. Am J Pathol. 2009;174:486–496. doi: 10.2353/ajpath.2009.080598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Davis MJ, Tsang TM, Qiu Y, Dayrit JK, Freij JB, Huffnagle GB, Olszewski MA. Macrophage M1/M2 polarization dynamically adapts to changes in cytokine microenvironments in Cryptococcus neoformans infection. MBio. 2013;4:e00264–00213. doi: 10.1128/mBio.00264-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Youseff BH, Holbrook ED, Smolnycki KA, Rappleye CA. Extracellular superoxide dismutase protects Histoplasma yeast cells from host-derived oxidative stress. PLoS Pathog. 2012;8:e1002713. doi: 10.1371/journal.ppat.1002713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Newman SL, Gootee L, Hilty J, Morris RE. Human macrophages do not require phagosome acidification to mediate fungistatic/fungicidal activity against Histoplasma capsulatum. Journal of immunology. 2006;176:1806–1813. doi: 10.4049/jimmunol.176.3.1806. [DOI] [PubMed] [Google Scholar]
- 86.Newman SL. Macrophages in host defense against Histoplasma capsulatum. Trends Microbiol. 1999;7:67–71. doi: 10.1016/s0966-842x(98)01431-0. [DOI] [PubMed] [Google Scholar]
- 87.Allen HL, Deepe GS., Jr. Apoptosis modulates protective immunity to the pathogenic fungus Histoplasma capsulatum. J Clin Invest. 2005;115:2875–2885. doi: 10.1172/JCI25365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Moreira AP, Cavassani KA, Hullinger R, Rosada RS, Fong DJ, Murray L, Hesson DP, Hogaboam CM. Serum amyloid P attenuates M2 macrophage activation and protects against fungal spore-induced allergic airway disease. J Allergy Clin Immunol. 2010;126:712–721. doi: 10.1016/j.jaci.2010.06.010. e717. [DOI] [PubMed] [Google Scholar]
- 89.Brummer E, Kethineni N, Stevens DA. Immunological basis for susceptibility and resistance to pulmonary blastomycosis in mouse strains. Cytokine. 2005;32:12–19. doi: 10.1016/j.cyto.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 90.Brummer E, Hanson LH, Restrepo A, Stevens DA. In vivo and in vitro activation of pulmonary macrophages by IFN-gamma for enhanced killing of Paracoccidioides brasiliensis or Blastomyces dermatitidis. Journal of immunology. 1988;140:2786–2789. [PubMed] [Google Scholar]
- 91.Rocco NM, Carmen JC, Klein BS. Blastomyces dermatitidis yeast cells inhibit nitric oxide production by alveolar macrophage inducible nitric oxide synthase. Infect Immun. 2011;79:2385–2395. doi: 10.1128/IAI.01249-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Moreira AP, Dias-Melicio LA, Peracoli MT, Calvi SA, Victoriano de Campos Soares AM. Killing of Paracoccidioides brasiliensis yeast cells by IFN-gamma and TNF-alpha activated murine peritoneal macrophages: evidence of H(2)O (2) and NO effector mechanisms. Mycopathologia. 2008;166:17–23. doi: 10.1007/s11046-007-9046-3. [DOI] [PubMed] [Google Scholar]
- 93.Brummer E, Hanson LH, Stevens DA. Gamma-interferon activation of macrophages for killing of Paracoccidioides brasiliensis and evidence for nonoxidative mechanisms. Int J Immunopharmacol. 1988;10:945–952. doi: 10.1016/0192-0561(88)90041-0. [DOI] [PubMed] [Google Scholar]