

Abstract
Background
Bipolar disorder (BD) and major depressive disorder (MDD) are highly heritable and genetically overlapping conditions characterised by episodic elevation and/or depression of mood. Both demonstrate abnormalities in white matter integrity, measured using diffusion tensor magnetic resonance imaging (MRI), that are also heritable. However it is unclear how these abnormalities relate to the underlying genetic architecture of each disorder. Genome-wide association studies (GWAS) have demonstrated a significant polygenic contribution to BD and MDD, where risk is attributed to the summation of many alleles of small effect. Determining the effects of an overall polygenic risk profile score on neuroimaging abnormalities may help to identify proxy measures of genetic susceptibility and thereby inform models of risk prediction.
Methods
In the current study we determined the extent to which common genetic variation underlying risk to mood disorders (BD and MDD) was related to fractional anisotropy, an index of white matter integrity. This was conducted in unaffected individuals at familial risk of mood disorder (n=70) and comparison subjects (n=62). Polygenic risk scores were calculated separately for BD and MDD based on GWAS data from the Psychiatric Genome Consortia.
Results
We report that a higher polygenic risk allele load for MDD was significantly associated with decreased white matter integrity across both groups in a large cluster with a peak in the right-sided superior longitudinal fasciculus.
Conclusions
These findings suggest that the polygenic approach to examining brain imaging data may be a useful means of identifying traits linked to the genetic risk of mood disorders.
Keywords: polygenic, bipolar disorder, major depressive disorder, white matter, diffusion tensor MRI
Introduction
Mood disorders, comprising bipolar disorder (BD) and major depressive disorder (MDD), are common disabling psychiatric conditions characterised by periods of elevated or depressed mood. These disorders are known to be heritable and share complex overlapping genetic architecture(1-5). Neuroimaging studies indicate abnormalities in mood-related brain circuitry in both individuals affected with mood disorder, and in unaffected relatives, suggesting that these features are heritable and represent trait-related markers of illness(6-8). Improving the understanding of the relationship between genetic heritability and quantitative trait features of the disorder, such as neuroimaging measures, may help to identify biomarkers of genetic susceptibility and thereby inform models of risk prediction for these conditions.
Evidence from candidate gene and genome wide association studies (GWAS) have identified a number of single nucleotide polymorphisms (SNPs) that are significantly associated with mood disorders, particularly BD(9-15), with less success for MDD(16). Recent evidence however indicates that each individual risk variant makes only a small contribution to the overall heritability of these disorders(17,18). The modest number of associated variants, together with small effect sizes, suggests that a large number of causal variants contribute to overall risk, a hypothesis referred to as the polygenic model of inheritance(19). A recent study confirmed that a polygenic risk score could differentiate cases (schizophrenia and BD) from controls in an independent sample, concluding that the polygenic model could explain a small but significant portion of the total variation in liability to psychiatric disorders(17).
Similar studies have confirmed that additive genetic variance contributes significantly to MDD(20), and MDD polygene scores have been able to differentiate MDD cases versus controls in independent samples(21). We recently employed this approach to examine the relationship between BD polygene risk scores and functional imaging traits in individuals at familial risk for mood disorder(22). In the current study we examine the same cohort to explore relationships between these measures and white matter integrity. Since polygenic models capture a greater proportion of the overall genetic contribution to these disorders than individual SNPs, we considered this approach likely to identify brain regions linked to additive genetic risk. We previously reported that higher BD polygene scores were related to increased functional activation of limbic areas of the brain, the anterior cingulate and amygdala, regions which have consistently been linked to the aetiology of mood disorders(22). Other studies have successfully employed similar approaches, for example using symptom dimension scales as the quantitative trait in relation to polygenic risk in psychotic disorders(23).
Here we examine associations between polygene risk profile scores for mood disorder (BD and MDD, separately) and structural white matter integrity in a cohort of unaffected young individuals at high familial risk of mood disorder as previously described(6,7,22). These individuals were at high familial risk because they had first and/or second degree relatives with BD. The control subjects had no immediate family history of mood disorder. Since there is consistent evidence of an increased frequency of both BD and MDD in first degree relatives of affected bipolar patients, the familial group were considered at high-risk of both unipolar and bipolar mood disorders, (i.e. BD and MDD)(24), (25).
We examined the relationship between polygene scores for BD and MDD in relation to white matter integrity as measured using diffusion tensor MRI (DTI). DTI measures the random motion of water molecule protons within white matter fibre tracts in the brain. The parallel organisation of white matter axons and the surrounding myelin sheaths in major tracts causes water molecules to diffuse predominantly along, rather than across, the principal fibre direction. The magnitude of this diffusion anisotropy is commonly quantified as the fractional anisotropy (FA), and is thought to reflect white matter integrity(26). Differences in FA between different groups can be identified using voxel-based methods, in particular Tract-based Spatial Statistics (TBSS)(27,28), a technique designed to maximise inter-subject registration. TBSS projects each individual subject’s FA data onto a common FA skeleton before applying between-subject voxel statistics. In this way it improves the probability that a given voxel contains information from the same region of the same white matter tract within each individual(27,29,30).
Although there is some inconsistency, in general, DTI studies in BD patients have reported reductions in white matter integrity in a number of brain regions including the superior longitudinal fasciculus (SLF), inferior fronto-occipital fasciculus (IFOF), inferior longitudinal fasciculus (ILF), thalamic radiations, uncinate fasciculus (UF), and corpus callosum (CC)(31,32), with some replication in individuals at familial risk(6,33). DTI studies of MDD have yielded more heterogenous results, however a recent meta-analysis implicated the SLF, and IFOF, where there was also a correlation between low FA and increasing severity of illness and with increased duration of illness(34). Studies on individuals at familial risk of depression have also reported FA reductions in the SLF(35). In the current study we therefore hypothesised that there would be a negative relationship between FA values in regions previously linked to BD and MDD and the polygenic risk score for these mood disorders.
Methods and materials
Study population
The two groups examined in this study comprised a group of individuals at familial risk of mood disorder and comparison group of healthy comparison subjects(7). In brief, individuals at high genetic risk of mood disorder because of a close family history of bipolar disorder (BD I) and control subjects were recruited as part of the Scottish Bipolar Family Study as described previously(6,7). Since there is an increased risk of both BD and MDD in unaffected first degree relatives of BD patients, we considered these individuals at enhanced familial risk of both MDD and BD and we refer to them throughout as being at high risk of mood disorder. To identify high-risk participants, caseloads of psychiatrists across Scotland were searched for individuals diagnosed with BDI. Diagnoses were confirmed with the Structural Clinical Interview for DSM-IV-TR Axis I Disorders (SCID-I)(36) or the symptom checklist of the Operational Criteria (OPCRIT)(37). Subsequently, subjects affected by BD were asked to identify a first or second-degree relative aged 16-25 years not suffering from the disorder. These unaffected individuals were invited to participate in this study providing that they had at least one first degree, or two second-degree relatives suffering from BDI. Control subjects with no personal history of BD or family history of a mood disorder in first-degree relatives were identified from the personal contacts of the bipolar high-risk subjects. Only unrelated individuals were included in the current analysis. Exclusion criteria for all groups included a personal history of major depression, mania or hypomania, generalised anxiety disorder, panic disorder, eating disorder, psychosis, substance dependence, an IQ < 70 or clinical diagnosis of learning disability, any major neurological disorder or history of head injury that included loss of consciousness, and any contraindications to MRI. A total of 70 high risk and 62 control subjects provided genetic and DTI data. All participants provided written informed consent and the study was approved by the multicentre research ethics committee for Scotland. We would also like to note that, to the best of our knowledge, none of the participants in the current study were related to individuals within the GWAS discovery sample.
Genotyping and derivation of polygenic scores
Genomic DNA was extracted from venous blood. Genotyping was conducted at the Wellcome Trust Clinical Research Facility, Edinburgh, United Kingdom (www.wtcrf.ed.ac.uk) and used the Illumina OmniExpress 730K SNP array. SNPs were excluded where the minor allele frequency was less than 1%, or if the call rate was less than 95% or if the X2 test for Hardy Weinberg Equilibrium was less than 1×10−3. Strand ambiguous SNPs were also removed. The resulting SNP set was then used to calculate four multidimensional scaling (MDS) components to assess and adjust for population stratification in later analyses. The data was then imputed to HapMap Version 3 (HM3; http://hapmap.ncbi.nlm.nih.gov) in Mach software(38) and then converted back to Plink (http://pngu.mgh.harvard.edu/~purcell/plink; map/ped) format for later analysis(39).
Summary results from the most recent international GWAS of BD and MDD were obtained from the publicly available data published by the Psychiatric GWAS Consortia (PGC)(17,21). Polygenic scores were calculated according to the methods by Purcell et al(17), and as reported in our previous study(22). Four lists of significant SNPs were generated from the PGC-BD and PGCMDD association data at significance thresholds of p=0.5, p=0.1, p=0.05 and p=0.01. These were then used to select common SNPs from our imaging-GWAS datasets resulting in four separate files (for BD and MDD) that contained the genotype of each individual. Our primary analyses concerned those SNPs from PGC-BD/MDD that met a significance level of p=0.5 or less, as this was the level that most efficiently discriminated individuals with and without BD in an independent test set(17). Full details of findings for the other thresholds are presented in supplementary information (Supplementary Table 1). In order to identify polygenic effects due to independent SNPs in linkage disequilibrium (LD) with one another, each SNP set was then pruned using a published method based on the variance inflation factor (VIF)(39). LD based SNP pruning was conducted in a sliding window of 50 SNPs with each calculation performed iteratively by moving the window by 5 SNPs. SNPs were conservatively selected on the basis of a VIF of 2 or less. Finally, these 4 SNP sets were then scored using the sum of the number of reference alleles multiplied by the logarithm of the odds ratio across the whole genome. All analyses were performed in Plink(39) with the exception of imputation to HM3 and data manipulation which were performed in Mach and R respectively(38,40).
Demographic and clinical assessments
All participants were interviewed by one of two experienced psychiatrists (AMM, JES) using the SCID(36) to confirm the absence of any lifetime axis I disorders. Current manic and depressive symptoms were rated using the Young Mania Rating Scale(41) and Hamilton Rating Scale for Depression (HAM-D)(42). Estimates of temperamental variations in minor affective symptoms were assessed using the Temperament Evaluation of Memphis, Pisa, Paris and San Diego Auto-questionnaire (TEMPS-A)(43), providing measures of cyclothymic, depressive, hyperthymic, irritable, and anxious temperament. Intelligence was measured using the National Adult Reading Test (NART) IQ. Statistical analysis of demographic characteristics was conducted using independent t-tests or chi-square (X2) tests. For the clinical assessments and measures of temperament, comparison of groups was conducted using Mann-Whitney U tests.
Scan Acquisition and Pre-processing
Scan acquisition and preprocessing details have been presented previously(6). Imaging was carried out at the Brain Research Imaging Centre (http://www.sbirc.ed.ac.uk) on a GE Signa 1.5 T clinical scanner (GE Medical, Milwaukee, USA). Whole brain DTI data were acquired using a single-shot pulsed gradient spin-echo echo-planar imaging (EPI) sequence with diffusion gradients (b = 1000 s/mm2) applied in 64 non-collinear directions, and 7 T2-weighted EPI baseline scans. Fifty-three 2.5 mm contiguous axial slices were acquired (field-of-view 240 × 240 mm, acquisition matrix 96 × 96, and zero-filled to 128 × 128), giving an isotropic acquisition voxel dimension of 2.5 mm. In addition, a T1-weighted volume was acquired (inversion time 500 ms, echo time 4 ms, flip angle 8°, acquisition matrix 192 × 192, 180 slices, giving voxel dimension 1.25 × 1.25 × 1.20 mm). The diffusion MRI data were converted to 4D NIfTI volumes and pre-processed using standard tools available from FSL (http://www.fmrib.ox.ac.uk/fsl). This included (i) correction for eddy current induced distortions and bulk subject motion by registering the diffusion weighed volumes to the first T2-weighted volume for each individual, (ii) brain extraction, and (iii) calculation of diffusion tensor characteristics including principal eigenvectors and FA values using DTIFIT.
Tract-Based Spatial Statistics
TBSS was carried out according to standard FSL procedures (http://www.fmrib.ox.ac.uk/fsl)(6,27,30). Initially, all subject’s FA volumes were linearly and non-linearly registered to a standard FA template. A mean of all registered FA volumes was then calculated and a white matter “skeleton” created. A threshold of FA > 0.25 was applied to the FA skeleton to exclude predominantly non-white matter voxels. For each subject’s FA volume, at each point on the skeleton the maximum voxel perpendicular to the local skeleton direction was projected onto the skeleton. This resulted in one FA skeleton map per subject, assumed to contain anatomically corresponding centres of white matter structure. Initial group comparison findings have been presented previously(6).
With the obtained TBSS skeletons we performed separate statistical analyses for each polygene score (BD and MDD). All analyses modelled the above four multidimensional scaling factors to control for population stratification effects. Our analysis plan consisted of firstly looking for evidence of polygene by group interaction effects. If these were not significant then we would assume that the relationship did not differ between groups and we would examine effects across all subject irrespective of group. Firstly, to test for group × polygene interaction effects we modelled the polygene scores as two separate regressors in the design matrix, i.e one per group. Secondly, we examined associations between the polygene scores and FA values across all subjects, irrespective of group, where each polygene score was modelled as a single regressor. To fully explore whether any effects seen across all subjects were influenced by group effects we performed an additional analysis modelling ‘group’ as a regressor in the statistical design.
All analyses were performing using the “randomise” functions in FSL. Threshold-free cluster enhancement (TFCE) was applied to obtain cluster-wise statistics corrected for multiple comparisons. The current analyses therefore present p-values corrected for whole brain using family-wise error via permutation testing with 20,000 permutations. The TFCE corrected p-maps were thresholded at pFWE < 0.05, and we report sizes of contiguous clusters of supra-threshold voxels (clusters > 10 voxels). Significant results were localized to white matter tracts/structures using the John Hopkins University (JHU) DTI-based white matter atlas and the JHU white matter tractography atlas (53) digitally available in FSL.
Results
Demographic, clinical, temperament and behavioural measures
Subject details are presented in Table 1. There were no significant differences between the groups in terms of age, gender, handedness, or NART IQ. The groups did however differ in terms of clinical measures, where the high-risk group scored significantly higher on the HAM-D than the comparison group (p=0.05), a finding we have reported previously(6,7). There were no other significant differences between the groups in terms of the remaining clinical or temperament measures as described in Table 1.
Table 1. Participant details.
| Comparison subjects (n =62 ) |
High-risk (n =70 ) |
t/Z | p | |||
|---|---|---|---|---|---|---|
| Demographics | ||||||
|
| ||||||
| Mean age (yrs) (std dev) |
21.16 | (2.27) | 21.56 | (2.77 | 0.89 | 0.38 |
| Gender (M:F) |
30:32 | - | 34:36 | - | 0.00# | 0.98 |
| Handedness (R:L+Mixed) |
57:5 | - | 63:7 | - | 0.15# | 0.70 |
| Mean NART IQ (std dev) | 109.43 | (7.33) | 108.47 | (10.56) | 0.59 | 0.55 |
|
| ||||||
| Polygene Score | ||||||
|
| ||||||
| BD polygene Score |
0.2126 | (0.05) | 0.2359 | (0.06) | 2.55 | 0.01 |
| MDD polygene Score |
−0.1861 | (0.05) | −0.1802 | (0.05) | 0.71 | 0.48 |
|
| ||||||
| Clinical measures* (median (interquartile range)) | ||||||
|
| ||||||
| Young Mania Rating Scale score |
0 | (0) | 0 | (0) | 1.44 | 0.15 |
| Hamilton Depression Scale score |
0 | (1) | 0 | (3) | 1.94 | 0.05 |
|
| ||||||
| Temperament measures* | ||||||
|
| ||||||
| TEMPS-A (median (interquartile range)) | ||||||
|
| ||||||
| Cyclothymia | 1 | (3) | 2 | (3) | 1.66 | 0.10 |
| Depressive | 0 | (2) | 0 | (1) | 0.50 | 0.62 |
| Irritability | 1 | (2) | 1 | (2) | 0.61 | 0.54 |
| Hyperthymia | 2 | (3) | 1.5 | (3.25) | 1.22 | 0.22 |
| Anxious | 1 | (1.25) | 0 | (2) | 0.33 | 0.74 |
| Total score | 7 | (8.25) | 7 | (7.50) | 0.21 | 0.83 |
|
| ||||||
| Alcohol use | ||||||
|
| ||||||
| Alcohol (U/week) Mean (std dev) |
17.27 | (19.16) | 13.98 | (14.80) | 1.04 | 0.30 |
Chi squared test
Mann-Whitney tests, median and interquartile range presented for skewed variables. P values < 0.05 are in bold
Polygene scores
There was a significant difference between the groups in terms of the BD polygene score (t=2.55, p=0.01), however no group difference was found for the MDD polygene score (t=0.71, p=0.48). There were also no significant correlations between the BD or MDD polygene scores and clinical or temperament measures as described in Table 1.
Table 2 presents the number of loci for each polygene threshold and the percentage of shared loci. This indicated an overlap in the order of 26% for the number of loci shared between the MDD and BD polygene risk sets at the threshold used in the current study.
Table 2. Number of loci for each polygene threshold for MDD and BD and percentage shared.
| Polygene thresholds: | ||||
|---|---|---|---|---|
| p=0.5 | p=0.1 | p=0.05 | p=0.01 | |
| MDD total |
97,923 | 31,915 | 18,259 | 4,619 |
| BD total |
95,591 | 32,677 | 19,290 | 5,401 |
| Shared (%)* |
25,807 (26.35) | 2,205 (6.90) | 781 (4.28) | 70 (1.52) |
percentage of shared loci represented as a proportion of the total number of loci for MDD
Relationship between polygenic scores and white matter integrity
There were no statistically significant polygene × group interaction effects for either the BD or MDD polygene score on FA. For the BD polygene score there was no significant association with FA, either positive or negative, across both groups. In contrast, there was a significant negative association between the MDD polygene risk score and FA values across all individuals in a large cluster encompassing both hemispheres with a peak in the right SLF, ILF and IFOF in the parietal region (35,025 voxels at pFWE<0.05, co-ordinates: x = 30, y = −48, z = 27), see Figure 1. A third of these voxels (k = 11,252) remained significant when the corrected significance level was made more stringent (pFWE<0.01) resulting in 14 smaller clusters (see supplementary Table 2). These clusters included peaks within the bilateral SLF, IFOF, ILF, posterior thalamic radiation, cingulum and splenium and body of the corpus callosum. There were no regions of positive relationship between MDD polygene score and FA.
Figure 1. Negative association between FA and MDD polygene scores.
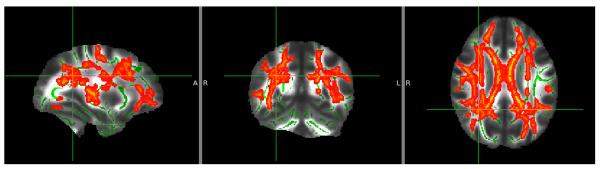
TBSS results for the effects of a negative association between the MDD polygene scores and FA values across all individuals. Images displayed at peak co-ordinate of large cluster (60, 78, 99). Threshold pFWE<0.05
To explore whether differences in group status could have been contributing to the association between FA values and polygenic loading we repeated the analysis modelling ‘group’ in the statistical design. The results of this analysis were similar to the above involving a large cluster across both hemispheres with a peak in a similar region to the above findings (39,467 voxels at pFWE<0.05, co-ordinates: x = 32, y = −46, z = 29).
Discussion
In this paper we report a significant relationship between polygenic risk for mood disorder, specifically MDD, and reduced white matter integrity in a large cluster encompassing regions previously implicated in these disorders which was not accounted for by group status. The effects were seen across all individuals, regardless of family history and in the absence of confounding disease and medication effects. Overall, therefore, these findings indicate a polygenic contribution to decreased white matter integrity reported in patients and in those at risk of mood disorders thereby providing a link between genetic predisposition to mood disorder and neuroimaging markers of illness.
One of the main findings from the current study is a reduction of FA in relation to higher polygene risk scores for MDD with a peak in the parietal portion of the SLF, a long white-matter tract strongly implicated in previous studies of MDD (34,44-46). A meta-analysis of DTI findings in MDD patients reported reductions in the SLF in parietal regions in patients versus controls using data from 7 studies involving a total of 188 patients and 221 controls(34). In addition, a recent study of unaffected individuals at familial risk of unipolar depression also identified reductions in FA in the SLF(35). It should be noted however that deficits in FA in this region are not specific to MDD or mood disorders(47,48). The SLF is a bidirectional long association fiber pathway anatomically subdivided into 4 sub-components(49). Autoradiographic studies have shown that these sub-regions connect distinct parts of tempero-parietal association corticies with frontal regions and vice versa(50). The SLF is therefore assumed to be important in a broad range of behavioural and cognitive functions, integrating regions involved in working memory, somatosensory processing, spatial attention, language and motor function and emotional regulation. It is therefore central to many associative and higher order cognitive brain functions and may relate to cognitive deficits seen in patients and inherited traits in unaffected relatives(50,51)
It is increasingly understood however that the complex disruptions in mood and cognitive functioning seen in mood disorders are unlikely to be attributable to deficits in a single white matter tract(52). Consistent with this, we report widespread regions of reduced FA that were significantly negatively associated with polygenic risk for MDD. This large cluster included the ILF, IFOF, thalamic radiations and uncinate fasciculus. Deficits in white matter integrity in these regions have previously been reported in patients with MDD(46,53), where associations between reduced FA and increasing symptom severity were also found(46). It is suggested that dysfunction in these tracts may affect cortico-limbic and thalamo-cortical networks, and that this in turn may impact cognitive control of emotional stimuli, which is considered to underlie symptoms of MDD(54). Reductions in white matter integrity could be due to a number of factors including reduced axonal density, reduced axonal diameter, dysmyelination, or indeed the crossing of fibre tracts. It should also be noted that FA values represent summary measures derived from diffusion tensor eigenvalues. They are influenced by a number of factors relating to the number of dominant fiber directions in each voxel, partial volume effects of nearby grey matter, and by the complex preprocessing steps using in analysis(55). Further study using techniques complementary to DTI, such as magnetization transfer imaging and postmortem studies, would be necessary to provide additional information regarding the nature of these deficits.
One of the main issues in general for psychiatric research has been the lack of robust, validated endophenotypic markers of illness. Endophenotypes are described as measurable traits that are more proximal to the underlying genetic cause of a disorder than the clinical manifestations of illness. They are therefore more stable and more sensitive to genetic variation than the clinical phenotype. Their role in identifying risk associated genetic variants, and in understanding aetiological processes, has long been emphasised(56). Although there are varying definitions, the properties of an endophenotype are generally that the trait must be associated with illness, must be heritable and co-segregate with illness in families, and must occur irrespective of clinical status(56). In the current study we have demonstrated reductions in white matter integrity in regions previously implicated in mood disorders(53), that were significantly related to genetic liability for MDD, and which occurred in unaffected relatives, therefore fulfilling 3 of the 4 criteria above. Previous studies have indeed reported that whole brain white matter integrity is under strong genetic control(8), and several studies of various risk genes for BD have reported reduced FA values in those carrying risk variants(57-59). Further, quantitative trait linkage analysis studies of FA maps in healthy controls have indicated linkage with genetic markers previously implicated in MDD(8). Overall therefore these findings suggest that these disruptions in white matter integrity may be a suitable candidate as an endophenotypic marker of genetic susceptibility for mood disorder, specifically MDD.
An important consideration in terms of the current results is that we did not report any association between FA and BD polygene scores, either across all subjects, or as an interaction. This may seem initially surprising, given our previous finding of reduced FA in BD and their unaffected relatives(6). Also, given previous studies indicating disrupted structural connectivity in bipolar patients(60), and alterations in FA in carriers of various risk associated variants for BD, including ANK3, NRG1 and ErbB4, notably in the anterior limb of the internal capsule(57-59). It is however the case that the majority of the heritability of BD remains obscured from current GWAS(17). This is in apparent contrast to MDD where a recently published study indicated that up to 80% of the heritability to MDD could be attributed to common SNPs(18). This may explain why the search for imaging biomarkers of MDD was apparently more successful in the current study than for BD.
We also do not report interaction effects with regard to associations of white matter integrity with the MDD polygene score. Our results therefore suggest that effects of these multiple and common alleles on white matter integrity were similar across both groups irrespective of background familial liability, i.e. that there was no evidence for a fundamentally different action in each of the groups. We also did not report differences in polygenic loading for MDD between the groups. However, this study concerned individuals at familial risk for mood disorder, hence a composite of individuals who will become ill (with MDD or indeed BD) along with others who will remain well. It may be the case therefore that group differences in polygenic loading for MDD, or associations with BD loading, will emerge once final clinical group status is established after completion of longitudinal assessments.
Alternatively, although this study is relatively large in terms of neuroimaging, it is relatively small in terms of genetic studies. It may be the case therefore that differences in MDD polygene scores are not apparent due to reduced power related to sample sizes. The difficulties of recruiting individuals at high familial risk of mood disorders unfortunately also result in there being no replication dataset on which to independently test these findings. Finally, it should be considered that the polygene risk scoring method may not fully account for the effects of rare variants, or for epistasic effects, although there is little positive evidence for these effects in MDD or BD in whole genome studies. Overall, however, the association between genetic risk and biological markers of illness progressively informs models of risk prediction. Whole genome assessment, in conjunction with intermediate quantitative traits that reflect the impact of susceptibility variants on brain structure or function, may eventually enable risk stratification and personalised treatment.
In conclusion, we have presented evidence for the aggregate effect of multiple SNPs associated with risk for mood disorder on white matter integrity in individuals at familial risk. These findings lend weight to the hypothesis that MDD is a disorder of neurocircuitry, where impaired connectivity is considered to underlie affective dysregulation. It further extends this model to indicate a genetic origin of these deficits. These findings may in turn help to identify mechanistic biological pathways involved in the illness.
Supplementary Material
Acknowledgements
We would like to thank all of the participants and their families who took part in the study and the radiographers who acquired the MRI scans. This study was conducted at the Brain Research Imaging Centre (http://www.bric.ed.ac.uk) which is supported by SINAPSE (Scottish Imaging Network, a Platform for Scientific Excellence; http://www.sinapse.ac.uk). The investigators also acknowledge the financial support of National Health Service (NHS) Research Scotland, through the Scottish Mental Health Research Network (http://www.smhrn.org.uk) who provided assistance with subject recruitment and cognitive assessments. All imaging aspects also received financial support from the Dr Mortimer and Theresa Sackler Foundation. We also acknowledge the Psychiatric GWAS Consortium Bipolar Disorder Working Group and Dr Shaun Purcell for his help and advice.
Footnotes
Financial Disclosures
The author HCW is supported by a Dorothy Hodgkin Fellowship from the Royal Society (DH080018). JH is supported by a Scottish Senior Clinical Fellowship. JES is supported by a Clinical Research Training Fellowship from the Wellcome Trust. AMM was supported by the Health Foundation through a Clinician Scientist Fellowship (Ref: 2268/4295), by the Brain and Behaviour Research Foundation through a NARSAD Independent Investigator Award and by a Scottish Funding Council Senior Clinical Fellowship. The investigators also acknowledge the financial support of National Health Service (NHS) Research Scotland, through the Scottish Mental Health Research Network (http://www.smhrn.org.uk) who provided assistance with subject recruitment and cognitive assessments.
HCW, ES, JH, SML and AMM have received financial support from Pfizer (formerly Wyeth) in relation to imaging studies of people with schizophrenia and bipolar disorder. SML and AMM have done consultancy work for Roche Pharmaceuticals in connection with a possible new treatment for schizophrenia. SML has also received honoraria for lectures, chairing meetings, and consultancy work from Janssen in connection with brain imaging and therapeutic initiatives for psychosis. The authors SH, DHB, DCG, MB, and JES have no competing interests to declare.
References
- 1.Schulze TG, Akula N, Breuer R, Steele J, Nalls MA, Singleton AB, et al. Molecular genetic overlap in bipolar disorder, schizophrenia, and major depressive disorder. World J Biol Psychiatry. 2012 doi: 10.3109/15622975.2012.662282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schosser A, Gaysina D, Cohen-Woods S, Chow PC, Martucci L, Craddock N, et al. Association of DISC1 and TSNAX genes and affective disorders in the depression case-control (DeCC) and bipolar affective case-control (BACCS) studies. Mol Psychiatry. 2010;15:844–849. doi: 10.1038/mp.2009.21. [DOI] [PubMed] [Google Scholar]
- 3.Green EK, Grozeva D, Jones I, Jones L, Kirov G, Caesar S, et al. The bipolar disorder risk allele at CACNA1C also confers risk of recurrent major depression and of schizophrenia. Mol Psychiatry. 2010;15:1016–1022. doi: 10.1038/mp.2009.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu Y, Blackwood DH, Caesar S, de Geus EJ, Farmer A, Ferreira MA, et al. Meta-analysis of genome-wide association data of bipolar disorder and major depressive disorder. Mol Psychiatry. 2011;16:2–4. doi: 10.1038/mp.2009.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kloiber S, Czamara D, Karbalai N, Muller-Myhsok B, Hennings J, Holsboer F, et al. ANK3 and CACNA1C - Missing genetic link for bipolar disorder and major depressive disorder in two German case-control samples. J Psychiatr Res. 46:973–979. doi: 10.1016/j.jpsychires.2012.04.017. [DOI] [PubMed] [Google Scholar]
- 6.Sprooten E, Sussmann JE, Clugston A, Peel A, McKirdy J, Moorhead TWJ, et al. White matter integrity in individuals at high genetic risk of bipolar disorder. Biol Psychiatry. 2011;70:350–356. doi: 10.1016/j.biopsych.2011.01.021. [DOI] [PubMed] [Google Scholar]
- 7.Whalley HC, Sussmann JE, Chakirova G, Mukerjee P, Peel A, McKirdy J, et al. The neural basis of familial risk and temperamental variation in individuals at high risk of bipolar disorder. Biol Psychiatry. 2011;70:343–349. doi: 10.1016/j.biopsych.2011.04.007. [DOI] [PubMed] [Google Scholar]
- 8.Kochunov P, Glahn DC, Lancaster JL, Winkler AM, Smith S, Thompson PM, et al. Genetics of microstructure of cerebral white matter using diffusion tensor imaging. Neuroimage. 2010;53:1109–1116. doi: 10.1016/j.neuroimage.2010.01.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ferreira MA, O’Donovan MC, Meng YA, Jones IR, Ruderfer DM, Jones L, et al. Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nat Genet. 2008;40:1056–1058. doi: 10.1038/ng.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sklar P, Ripke S, Scott LJ, Andreassen OA, Cichon S, Craddock N, et al. Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat Genet. 43:977–983. doi: 10.1038/ng.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baum AE, Akula N, Cabanero M, Cardona I, Corona W, Klemens B, et al. A genome-wide association study implicates diacylglycerol kinase eta (DGKH) and several other genes in the etiology of bipolar disorder. Mol Psychiatry. 2008;13:197–207. doi: 10.1038/sj.mp.4002012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sklar P, Smoller JW, Fan J, Ferreira MA, Perlis RH, Chambert K, et al. Whole-genome association study of bipolar disorder. Mol Psychiatry. 2008;13:558–569. doi: 10.1038/sj.mp.4002151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith EN, Bloss CS, Badner JA, Barrett T, Belmonte PL, Berrettini W, et al. Genome-wide association study of bipolar disorder in European American and African American individuals. Mol Psychiatry. 2009;14:755–763. doi: 10.1038/mp.2009.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scott LJ, Muglia P, Kong XQ, Guan W, Flickinger M, Upmanyu R, et al. Genome-wide association and meta-analysis of bipolar disorder in individuals of European ancestry. Proc Natl Acad Sci U S A. 2009;106:7501–7506. doi: 10.1073/pnas.0813386106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.The Wellcome Trust Case Control Consortium Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wray NR, Pergadia ML, Blackwood DH, Penninx BWJH, Gordon SD, Nyholt DR, et al. Genome-wide association study of major depressive disorder: new results, meta-analysis, and lessons learned. Mol Psychiatry. 2012;17:36–48. doi: 10.1038/mp.2010.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Purcell SM, Wray NR, Stone JL, Visscher PM, O’Donovan MC, Sullivan PF, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lubke GH, Hottenga JJ, Walters R, Laurin C, de Geus EJ, Willemsen G, et al. Estimating the Genetic Variance of Major Depressive Disorder Due to All Single Nucleotide Polymorphisms. Biol Psychiatry. 2012;72:707–709. doi: 10.1016/j.biopsych.2012.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gottesman II, Shields J. A polygenic theory of schizophrenia. Proc Natl Acad Sci U S A. 1967;58:199–205. doi: 10.1073/pnas.58.1.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lubke GH, Hottenga JJ, Walters R, Laurin C, de Geus EJ, Willemsen G, et al. Estimating the Genetic Variance of Major Depressive Disorder Due to All Single Nucleotide Polymorphisms. Biol Psychiatry. doi: 10.1016/j.biopsych.2012.03.011. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sullivan PF, Consortium MDDWGotPG A mega-analysis of genome-wide association studies for major depressive disorder. Mol Psychiatry. 2012;1:1–15. doi: 10.1038/mp.2012.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Whalley HC, Papmeyer M, Sprooten E, Romaniuk L, Blackwood DH, Glahn DC, et al. The influence of polygenic risk for bipolar disorder on neural activation assessed using fMRI. Translational Psychiatry. doi: 10.1038/tp.2012.60. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Derks EM, Vorstman JAS, Ripke S, Kahn RS, Consortium TSPG, Ophoff RA. Investigation of the Genetic Association between Quantitative Measures of Psychosis and Schizophrenia: A Polygenic Risk Score Analysis. PLoS One. 2012;7:e37852. doi: 10.1371/journal.pone.0037852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McGuffin P, Rijsdijk F, Andrew M, Sham P, Katz R, Cardno A. The heritability of bipolar affective disorder and the genetic relationship to unipolar depression. Arch Gen Psychiatry. 2003;60:497–502. doi: 10.1001/archpsyc.60.5.497. [DOI] [PubMed] [Google Scholar]
- 25.Goodwin FK, Jamison KR. Manic-depressive illness: Bipolar disorders and recurrent depression. Second Edition ed Oxford University Press; Oxford: 2007. [Google Scholar]
- 26.Schmierer K, Wheeler-Kingshott CA, Boulby PA, Scaravilli F, Altmann DR, Barker GJ, et al. Diffusion tensor imaging of post mortem multiple sclerosis brain. Neuroimage. 2007;35:467–477. doi: 10.1016/j.neuroimage.2006.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith SM, Jenkinson M, Johansen-Berg H, Rueckert D, Nichols TE, Mackay CE, et al. Tract-based spatial statistics: voxelwise analysis of multi-subject diffusion data. Neuroimage. 2006;31:1487–1505. doi: 10.1016/j.neuroimage.2006.02.024. [DOI] [PubMed] [Google Scholar]
- 28.Behrens TE, Woolrich MW, Jenkinson M, Johansen-Berg H, Nunes RG, Clare S, et al. Characterization and propagation of uncertainty in diffusion-weighted MR imaging. Magn Reson Med. 2003;50:1077–1088. doi: 10.1002/mrm.10609. [DOI] [PubMed] [Google Scholar]
- 29.Zalesky A. Moderating registration misalignment in voxelwise comparisons of DTI data: a performance evaluation of skeleton projection. Magn Reson Imaging. 2011;29:111–125. doi: 10.1016/j.mri.2010.06.027. [DOI] [PubMed] [Google Scholar]
- 30.Smith SM, Johansen-Berg H, Jenkinson M, Rueckert D, Nichols TE, Miller KL, et al. Acquisition and voxelwise analysis of multi-subject diffusion data with tract-based spatial statistics. Nat Protoc. 2007;2:499–503. doi: 10.1038/nprot.2007.45. [DOI] [PubMed] [Google Scholar]
- 31.Vederine FE, Wessa M, Leboyer M, Houenou J. A meta-analysis of whole-brain diffusion tensor imaging studies in bipolar disorder. Prog Neuropsychopharmacol Biol Psychiatry. 35:1820–1826. doi: 10.1016/j.pnpbp.2011.05.009. [DOI] [PubMed] [Google Scholar]
- 32.Sexton CE, Mackay CE, Ebmeier KP. A systematic review of diffusion tensor imaging studies in affective disorders. Biol Psychiatry. 2009;66:814–823. doi: 10.1016/j.biopsych.2009.05.024. [DOI] [PubMed] [Google Scholar]
- 33.Chaddock CA, Barker GJ, Marshall N, Schulze K, Hall MH, Fern A, et al. White matter microstructural impairments and genetic liability to familial bipolar I disorder. Br J Psychiatry. 2009;194:527–534. doi: 10.1192/bjp.bp.107.047498. [DOI] [PubMed] [Google Scholar]
- 34.Murphy ML, Frodl T. Meta-analysis of diffusion tensor imaging studies shows altered fractional anisotropy occurring in distinct brain areas in association with depression. Biol Mood Anxiety Disord. 1:3. doi: 10.1186/2045-5380-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang H, Fan X, Williamson DE, Rao U. White matter changes in healthy adolescents at familial risk for unipolar depression: a diffusion tensor imaging study. Neuropsychopharmacology. 2011;36:684–691. doi: 10.1038/npp.2010.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.First MB, Spitzer RL, Gibbon M, Williams JBW. Structured Clinical Interview for DSM-IV-TR Axis I Disorders, Research Version, Patient Edition with Psychotic Screen. Biometrics Research, New York State Psychiatric Institute; New York: 2002. [Google Scholar]
- 37.McGuffin P, Farmer A, Harvey I. A polydiagnostic application of operational criteria in studies of psychotic illness. Development and reliability of the OPCRIT system. Archives of General Psychiatry. 1991;48:764–770. doi: 10.1001/archpsyc.1991.01810320088015. [DOI] [PubMed] [Google Scholar]
- 38.Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol. 2010;34:816–834. doi: 10.1002/gepi.20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Team RDC . R: A language and environment for statistical computing. R Foundation for Statistical Computing. Vienna, Austria: 2010. http://www.R-project.org/ [Google Scholar]
- 41.Young RC, Biggs JT, Ziegler VE, Meyer DA. A rating scale for mania: reliability, validity and sensitivity. Br J Psychiatry. 1978;133:429–435. doi: 10.1192/bjp.133.5.429. [DOI] [PubMed] [Google Scholar]
- 42.Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23:56–62. doi: 10.1136/jnnp.23.1.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Akiskal HS, Mendlowicz MV, Jean-Louis G, Rapaport MH, Kelsoe JR, Gillin JC, et al. TEMPS-A: validation of a short version of a self-rated instrument designed to measure variations in temperament. J Affect Disord. 2005;85:45–52. doi: 10.1016/j.jad.2003.10.012. [DOI] [PubMed] [Google Scholar]
- 44.Wu F, Tang Y, Xu K, Kong L, Sun W, Wang F, et al. Whiter matter abnormalities in medication-naive subjects with a single short-duration episode of major depressive disorder. Psychiatry Res. 2011;191:80–83. doi: 10.1016/j.pscychresns.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zou K, Huang X, Li T, Gong Q, Li Z, Ou-yang L, et al. Alterations ofwhitematter integrity in adults withmajor depressive disorder: a magnetic resonance imaging study. J Psychiatry Neurosci. 2008;33:525–530. [PMC free article] [PubMed] [Google Scholar]
- 46.Cole J, Chaddock CA, Farmer AE, Aitchison KJ, Simmons A, McGuffin P, et al. White matter abnormalities and illness severity in major depressive disorder. Br J Psychiatry. 2012;201:33–39. doi: 10.1192/bjp.bp.111.100594. [DOI] [PubMed] [Google Scholar]
- 47.Guo W, Liu F, Liu Z, Gao K, Xiao C, Chen H, et al. Right lateralized white matter abnormalities in first-episode, drug-naive paranoid schizophrenia. Neurosci Lett. 2012;531:5–9. doi: 10.1016/j.neulet.2012.09.033. [DOI] [PubMed] [Google Scholar]
- 48.Baur V, Hanggi J, Rufer M, Delsignore A, Jancke L, Herwig U, et al. White matter alterations in social anxiety disorder. J Psychiatr Res. 2011;45:1366–1372. doi: 10.1016/j.jpsychires.2011.05.007. [DOI] [PubMed] [Google Scholar]
- 49.Schmahmann JD, Pandya DN, Wang R, Dai G, D’Arceuil HE, de Crespigny AJ, et al. Association fibre pathways of the brain: parallel observations from diffusion spectrum imaging and autoradiography. Brain. 2007;130:630–653. doi: 10.1093/brain/awl359. [DOI] [PubMed] [Google Scholar]
- 50.Makris N, Kennedy DN, McInerney S, Sorensen AG, Wang R, Caviness VS, Jr., et al. Segmentation of subcomponents within the superior longitudinal fascicle in humans: a quantitative, in vivo, DT-MRI study. Cereb Cortex. 2005;15:854–869. doi: 10.1093/cercor/bhh186. [DOI] [PubMed] [Google Scholar]
- 51.Glahn DC, Curran JE, Winkler AM, Carless MA, Kent JW, Jr., Charlesworth JC, et al. High dimensional endophenotype ranking in the search for major depression risk genes. Biol Psychiatry. 2011;71:6–14. doi: 10.1016/j.biopsych.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mayberg HS. Limbic-cortical dysregulation: a proposed model of depression. J Neuropsychiatry Clin Neurosci. 1997;9:471–481. doi: 10.1176/jnp.9.3.471. [DOI] [PubMed] [Google Scholar]
- 53.Kieseppa T, Eerola M, Mantyla R, Neuvonen T, Poutanen VP, Luoma K, et al. Major depressive disorder and white matter abnormalities: a diffusion tensor imaging study with tract-based spatial statistics. J Affect Disord. 2010;120:240–244. doi: 10.1016/j.jad.2009.04.023. [DOI] [PubMed] [Google Scholar]
- 54.Phillips ML, Drevets WC, Rauch SL, Lane R. Neurobiology of emotion perception II: Implications for major psychiatric disorders. Biol Psychiatry. 2003;54:515–528. doi: 10.1016/s0006-3223(03)00171-9. [DOI] [PubMed] [Google Scholar]
- 55.Zhan L, Leow AD, Zhu S, Baryshev M, Toga AW, McMahon KL, et al. A novel measure of fractional anisotropy based on the tensor distribution function. Med Image Comput Comput Assist Interv. 2009;12:845–852. doi: 10.1007/978-3-642-04268-3_104. [DOI] [PubMed] [Google Scholar]
- 56.Gottesman II, Gould TD. The endophenotype concept in psychiatry: etymology and strategic intentions. Am J Psychiatry. 2003;160:636–645. doi: 10.1176/appi.ajp.160.4.636. [DOI] [PubMed] [Google Scholar]
- 57.Linke J, Witt SH, King AV, Nieratschker V, Poupon C, Gass A, et al. Genome-wide supported risk variant for bipolar disorder alters anatomical connectivity in the human brain. Neuroimage. 2012;59:3288–3296. doi: 10.1016/j.neuroimage.2011.10.083. [DOI] [PubMed] [Google Scholar]
- 58.Zuliani R, Moorhead TW, Bastin ME, Johnstone EC, Lawrie SM, Brambilla P, et al. Genetic variants in the ErbB4 gene are associated with white matter integrity. Psychiatry Res. 2011;191:133–137. doi: 10.1016/j.pscychresns.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McIntosh AM, Moorhead TWJ, Job DE, Lymer GK, Munoz Maniega S, McKirdy J, et al. The effects of neuregulin 1 variant on white matter density and integrity. Mol Psychiatry. 2008;13:1054–1059. doi: 10.1038/sj.mp.4002103. [DOI] [PubMed] [Google Scholar]
- 60.Bellani M, Brambilla P. Diffusion imaging studies of white matter integrity in bipolar disorder. Epidemiol Psychiatr Sci. 20:137–140. doi: 10.1017/s2045796011000229. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.