

Abstract
Bacteriophage S-CRM01 has been isolated from a freshwater strain of Synechococcus and shown to be present in the upper Klamath River valley in northern California and Oregon. The genome of this lytic T4-like phage has a 178,563 bp circular genetic map with 297 predicted protein-coding genes and 33 tRNA genes that represent all 20 amino acid specificities. Analyses based on gene sequence and gene content indicate a close phylogenetic relationship to the “photosynthetic” marine cyanomyophages infecting Synechococcus and Prochlorococcus. Such relatedness suggests that freshwater and marine phages can draw on a common gene pool. The genome can be considered as being comprised of three regions. Region 1 is populated predominantly with structural genes, recognized as such by homology to other T4-like phages and by identification in a proteomic analysis of purified virions. Region 2 contains most of the genes with roles in replication, recombination, nucleotide metabolism and regulation of gene expression, as well as 5 of the 6 signature genes of the photosynthetic cyanomyophages (hli03, hsp20, mazG, phoH and psbA; cobS is present in Region 3). Much of Regions 1 and 2 are syntenous with marine cyanomyophage genomes, except that a segment encompassing Region 2 is inverted. Region 3 contains a high proportion (85%) of genes that are unique to S-CRM01, as well as most of the tRNA genes. Regions 1 and 2 contain many predicted late promoters, with a combination of CTAAATA and ATAAATA core sequences. Two predicted genes that are unusual in phage genomes are homologs of cellular spoT and nusG.
Keywords: Myoviridae, Klamath River, cyanomyophage, freshwater phage, nusG, spoT, phage tRNA, T4-like phage
INTRODUCTION
Research over recent years has brought into focus the important contribution of bacteriophages to the ecology of microbial populations and to biochemical and geochemical cycles in the environment; bacteriophages are both enormously abundant and diverse (Rohwer, 2003; Weinbauer, 2004; Breitbart and Rohwer, 2005; Rohwer et al., 2009). Marine environments have attracted the most attention (Fuhrman, 1999; Wommack and Colwell, 2000; Breitbart et al., 2002; Mann, 2003; Suttle, 2005, 2007), with relatively few studies dedicated to investigating bacteriophages present in freshwater environments (Middelboe et al., 2008; Wilhelm and Matteson, 2008). We have been interested in studying the microbial diversity and population dynamics of toxic cyanobacterial blooms (Bozarth et al., 2010), which are an increasingly common ecological dysfunction seen in a wide variety of lakes and reservoirs (Paerl and Huisman, 2009).
The only currently available genome sequence from a phage infecting freshwater cyanobacteria is that of the myophage Ma-LMM01 infecting Microcystis aeruginosa from a Japanese lake (Yoshida et al., 2008). We describe here the genome sequence and other properties of a cyanomyophage isolated from Copco Reservoir on the Klamath River in Northern California in September 2008. The phage was associated with a toxic Microcystis aeruginosa bloom, but an endemic Synechococcus lineage is the host for this phage. The genome sequence revealed close relationships to a well-studied group of “photosynthetic” exoT4-even cyanomyophages infectious to marine Synechococcus and Prochlorococcus (Mann et al., 2005; Sullivan et al., 2005; Weigele et al., 2007; Millard et al., 2009; Sullivan et al., 2010). This similarity between freshwater and marine cyanomyophages supports indications from metagenomic (Rodriguez-Brito et al., 2010) and amplicon studies with psbA and gp20 (T4 portal protein) that related phages can be found in freshwater and marine environments (Dorigo et al., 2004; Short and Suttle, 2005; Wilhelm et al., 2006; Chénard and Suttle, 2008; Sullivan et al., 2008), although there is also evidence for distinctively freshwater cyanophage lineages (Deng and Hayes, 2008; Yoshida et al., 2008; Wang et al., 2010). Our study is the first involving whole genome characterization to address the relationship between freshwater and marine cyanophages, and supports the possibility that the gene complement of water-borne phages has been shaped by gene pools in both freshwater and marine environments (Sano et al., 2004).
MATERIALS AND METHODS
Sample collection and cyanophage enrichment
Water was collected from the top 0.5 m of Copco Reservoir (mid-channel near the dam wall at latitude 41.979°N and longitude 122.333°W) during a Microcystis bloom on the Klamath River in Northern California on 10 September, 2008, and transferred to the laboratory in the dark on ice. In order to enrich for cyanophages present in the sample, a 200 mL aliquot of 0.2 μm filtered water was supplemented with 4 mL of 50x BG-11 medium (Sigma-Aldrich, St. Louis, MO) and 20 mL of Microcystis-dominated cultures derived from the Klamath River system (August 2007). The enriched water sample was incubated at 24°C under fluorescent lamps at 10 μE/m2/s for 10 days. Water quality data relevant to the collected sample are available at http://www.pacificorp.com/es/hydro/hl/kr.html#.
Cyanophage isolation and amplification
The enriched culture was treated with chloroform, and cellular material was removed by centrifugation. The 0.2 μm-filtered supernatant (Supor-200; Pall Life Sciences) was ultracentrifuged using a Ti60 rotor (Beckman) at 4°C and 177,520 g, for 90 min. Pellets were resuspended in SM (50 mM Tris-HCl, 100 mM NaCl, 10 mM MgSO4-7H20, pH 7.5) and stored at 4°C. Plaque assays were conducted using BG-11 top agar layered onto BG-11 agar plates that were incubated under the growth conditions described above until plaques were visible (~ one week). Three serial plaque isolations were performed using the Klamath River system culture, which was also used for phage amplification in liquid culture. Phage particles were collected by ultracentrifugation. During the course of phage isolation, it was noticed that the culture characteristics had changed. The identity of the resultant culture (LC16) was examined by PCR amplification of genomic DNA using the cyanobacteria-wide primers CS1F and ULR directed at the internal transcribed spacer of the rRNA operon (ITS), followed by DNA sequencing of the PCR product as described (Bozarth et al., 2010). A single derived sequence indicated culture purity. Comparison of the sequence with the GenBank database identified LC16 as a member of the Cyanobium gracile cluster that includes freshwater Synechococcus isolates (Ernst et al., 2003; Chen et al., 2006) (Fig. S1). The closest known relative has been isolated from Lake Balaton (Hungary), with other related isolates from freshwater sources in Germany and Wisconsin (USA) and brackish or saline sources in California, Baltic Sea (Denmark) and White Sea (Russia).
Genome structure analysis
Genome size was estimated using pulsed field gel electrophoresis (PFGE) after genome preparation as described (Lingohr et al., 2008), using a 1.4% agarose gel in a CHEF II PFGE unit (Bio-Rad) set to run at 6 V cm−1 for 18 h with a 0.1 sec switch time.
To determine whether the genome was linear, circular or circularly permuted, BAL-31 nuclease digestion of the phage genome was performed as described (Yoshida et al., 2008). Purified phage DNA (200ng) was incubated with 0.1 U/μl BAL-31 nuclease (NEBiolabs) at 30°C for 0, 10, 20, 40 and 60 minutes. The DNA was then extracted with phenol/chloroform, ethanol precipitated and digested overnight at 37°C with BamH1 endonuclease. The restriction products were separated on 0.8% agarose gel and visualized with ethidium bromide. 1kB DNA ladder (GeneRuler, Fermentas) and a plasmid (pGEM, Promega) were included as linear and supercoiled circular controls.
Genome sequencing
The genome (500 ng) was sequenced by Roche, Inc. (Branford, CT) using GS FLX Titanium Sequencing, accumulating 232 kbp at an average coverage of 350X. A first draft of the genome sequence was assembled using Newbler (gsAssembler) software (Roche). Five contigs larger than 5,000 bp in length were assembled, amounting to a total of 173.5 kbp. To determine the order of contigs and to fill the gaps, a multiplex PCR approach was used (Tettelin et al., 1999). Briefly, pairs of outward-oriented primers positioned about 100 bp from the two ends of each contig were used in all combinations to direct PCR amplifications using genomic DNA as template. Products visible on gels after 30 cycles were confirmed by PCR using individual primer pairs, and were extracted from agarose gels and submitted for direct Sanger sequencing (Genewiz, Inc.). Each gap was sequenced in both directions, with additional primers designed when needed.
Contigs and junction sequences were arranged and assembled in Geneious (Biomatters, Ltd., http://www.geneious.com). The gap sequences could be confirmed using smaller contigs and unassembled reads from the 454 data. The average sequencing coverage for contigs ranged between 360 and 500 with an average Phred Equivalent (Roche) quality score of 64.0.
Genome annotation and analysis
Regions of coding sequence were predicted using Glimmer 3 (Delcher et al., 1999) and Genemark S (Besemer et al., 2001); tRNAs were predicted using tRNAscan-SE (Lowe and Eddy, 1997). Annotated protein coding sequences were determined using a BLASTx search against the NCBI nr database. Annotations were made based on an E-Value cutoff <1e-5. Phage-associated ORFs were compared to a custom database of T4-like cyanophages using a BLASTx search. Genome annotation was curated in both Geneious and Artemis (Rutherford et al., 2000). Early promoters were predicted in regions upstream of ORFs by similarity to putative S-PM2 early promoters (Mann et al., 2005) and using BPROM (LDF>5; Softberry, Mount Kisco, NY). Putative terminator sequences were identified by TransTerm (http://uther.otago.ac.nz). The gene-annotated S-CRM01 sequence is available under GenBank accession HQ615693.
Phylogenomic analysis was carried out using the Hal pipeline (http://aftol.org/pages/Halweb3.htm; http://sourceforge.net/projects/bio-hal/), which consists of a set of Perl scripts that automates a series of phylogenomic analyses using existing software and sequence analysis programs (Robbertse et al., 2006); analysis was executed on a 64-bit Linux cluster operating Red Hat Linux 3.2.3, Linux version 2.4.21. The proteins encoded by 21 phage genomes were analyzed at 9 levels of “missing data” (inclusion in the analysis of genes/orthologous clusters not present in all phages), in each case using 3 gap removal methodologies (complete gap removal, and liberal and conservative gap-removal with GBlocks), resulting in a total of 27 phylogenetic trees. Inclusion of genes missing from a few taxa can improve phylogenetic analyses by increasing the number of genes analyzed (Wiens, 2006).
Transmission electron microscopy
CsCl gradient-purified phage was applied to a glow-discharged carbon-type B, 300-mesh copper grid (Ted Pella, Redding, CA) and stained with 1% phosphotungstic acid, pH 6.5. Samples were observed on a Philips CM-12 transmission electron microscope at 60 kEV.
Mass spectrometry-based proteomics
Phage was purified using a CsCl density gradient. Phage-associated proteins were prepared for SDS-PAGE by boiling for 2 minutes in SDS-PAGE protein sample loading buffer and separated on a 8–16 % gradient gel (Bio-Rad). The sample lane was cut into 11 sections, dehydrated with 50% acetonitrile in 50 mM NH4HCO3 and dried in a SpeedVac. Reduction and alkylation with DTT and iodoacetamide, and in-gel trypsin digestion were performed as described in the Protease-Max (Promega, Madison, WI) manual. A blank section of the gel was processed and included in all subsequent analyses.
LC-MS/MS analyses of the extracted peptides were performed on a LTQ-FT Ultra mass spectrometer (Thermo) with an IonMax ion source. The mass spectrometer was coupled to a nanoAcquity Ultra performance LC system (Waters) equipped with a Michrom Peptide CapTrap and a C18 column (Agilent Zorbax 300SB-C18, 250 × 0.3 mm, 5 μm). A binary gradient system was used consisting of solvent A, 0.1% aqueous formic acid and solvent B, acetonitrile containing 0.1% formic acid. Peptides were trapped and washed with 1% solvent B for 3 min. Peptide separation was achieved using a linear gradient from 10% B to 30% B at a flow rate of 4 μL/min over 35 minutes.
For the LC-MS/MS analysis, the LTQ-FT mass spectrometer was operated in a data-dependent mode. A full FT-MS scan (m/z 350–2000) was alternated with collision-induced dissociation (CID) MS/MS scans of the 5 most abundant doubly or triply charged precursor ions. As the survey scan was acquired in the ICR cell, the CID experiments were performed in the linear ion trap where precursor ions were isolated and subjected to CID in parallel with the completion of the full FT-MS scan. CID was performed with helium gas at a normalized collision energy 35% and activation time of 30 ms. Automated gain control (AGC) was used to accumulate sufficient precursor ions (target value, 5 × 104/micro scan; maximum fill time 0.2 s). Dynamic exclusion was used with a repeat count of 1 and exclusion duration of 60 s. Data acquisition was controlled by Xcalibur (version 2.0.5) software (Thermo).
For the sequence database search, Thermo RAW data files were processed with Proteome Discoverer v1.0 using default parameters except for a S/N threshold setting of 10. A Mascot (v2.2.04) search of a phage-encoded protein database (299 sequences; 54263 residues) was launched from Proteome Discoverer with the following parameters: the digestion enzyme was set to Trypsin/P and two missed cleavage sites were allowed. The precursor ion mass tolerance was set to 10 ppm, while fragment ion tolerance of 0.8 Da was used. Dynamic modifications included carbamidomethyl (+57.0214 Da) for Cys and oxidation (+15.9994 Da) for Met.
PCR to detect S-CRM01 in the Klamath system
Enriched phage fractions were prepared (see above) from samples collected from the Klamath River system during August–October, 2009. Multiple plaques isolated on LC16 top agar plates from each sample were tested for the presence of S-CRM01 by PCR analysis using two primer pairs directed at g44 (gp23) and g34 (Ma-LMM01-like hypothetical protein). In all cases, reactions were either positive or negative for both primer pairs. The primer pair for detecting g34 was CRM01-g34(F) 5′ GTCAAATAGAATCCAGGATGAATTA and CRM01-g34(R) 5′ TACCATAGTCTCCACCGTTTC. The primer pair detecting g44 was CRM01-g44(F) 5′ GACGTATGTGGCGTTCAGCCAATGA and CRM01-g44(R) 5′ CGGTTGATTTCTGCAAGGATTTC. PCR reactions used High Fidelity Platinum Taq polymerase (Invitrogen) in the provided buffer with 0.2 μM of each primer, 0.2 mM dNTPs, and 2.5 mM MgSO4; after initial denaturation, 35 cycles of 0.5 min at 94°C, 0.5 min at 52°C, and 1 min at 68°C were run. These primers were designed to be specific for S-CRM01, avoiding amplification from known related cyanomyophage genomes; detection of S-CRM01 was scored as positive only when products of the expected size were amplified with both primer pairs.
RESULTS AND DISCUSSION
Isolation and physical characteristics
Phage S-CRM01 was isolated from a surface sample taken in September 2008 from a Microcystis-dominated bloom in Copco Reservoir on the Klamath River in Northern California. At 319.8 river km from the coast, this site is far removed from salt water habitats. S-CRM01 infects and lyses LC16, a culture belonging to the Cyanobium gracile cluster of mostly freshwater Synechococcus (see Materials and Methods). We have observed lytic infection of no other hosts, including the freshwater cyanobacteria Microcystis aeruginosa, Synechococcus elongatus PCC 7942 and Synechocystis sp. PCC6803. During 2009, a survey was conducted to assess the distribution of S-CRM01 between Upper Klamath Lake and the Klamath River estuary. On the basis of PCR identification of plaques on LC16 plates, S-CRM01 was present across a length of about 250 km along the Klamath River valley, from the Williamson River Delta at the northern end of Upper Klamath Lake as far downstream as Seiad Valley (Fig. 1).
Fig. 1. Distribution of S-CRM01 in the Klamath River valley, 2009.
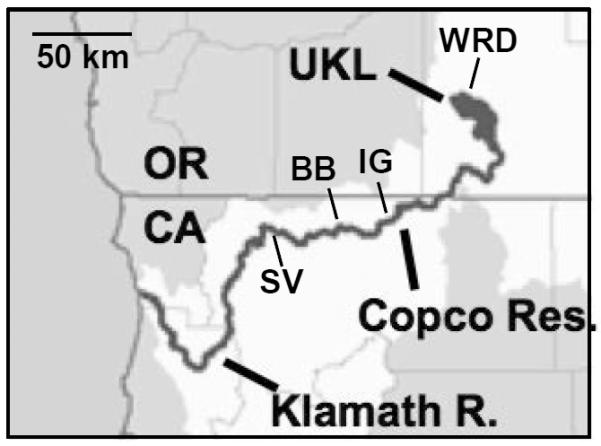
Positive identifications were made in samples from the Williamson River delta area of Upper Klamath Lake (WRD; 15 October), Copco Reservoir (near dam; 13 October), Iron Gate Reservoir (IG, near dam, 18 August & 15 September), and at two sites on the lower Klamath River: Brown Bear sampling site at Horse Creek (BB, 6 August & 15 September) and Seiad Valley (SV, 15 September).
Negative staining electron microscopy revealed a T4-like morphology, with isometric heads about 85–100 nm in diameter and rigid contractile tails 15–20 nm in diameter and 140–170 nm long (Fig. 2). Contracted tails reveal a sheath and core 20–30 nm and about 10 nm in diameter, respectively. A double-ringed baseplate and narrow neck are visible.
Fig. 2.
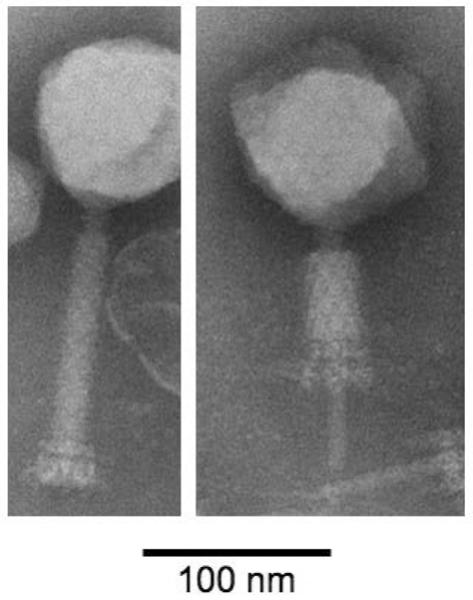
Electron micrograph of S-CRM01 phage particles negatively stained with phosphotungstic acid. An intact particle is shown at left and a contracted particle at right. Note the icosahedral head, prominent neck, two-ringed baseplate and injection tube.
The genome migrated as a 180 kbp band in PFGE. Nuclease BAL-31 digestion followed by cleavage with BamHI resulted in simultaneous loss of material from all bands, indicative of circularly permuted linear DNA (not shown). This is consistent with the presence of a gene homologous to the gp17 large terminase subunit of phage T4, which determines a circularly permuted packaging strategy (Casjens and Gilcrease, 2008).
S-CRM01 is a “photosynthetic” cyanomyophage most closely related to marine phages of Synechococcus and Prochlorococcus
Consistent with a circularly permuted organization and its migration in PFGE, the S-CRM01 genome assembled into a circular map 178,563 bp long (Fig. 3). The G+C content of the genome is 39.7%, and 297 protein-coding genes and 33 tRNA genes are predicted. Protein-coding genes have been annotated with the following conventions (Table 1): S-CRM01 genes are designated as g1 etc.; in addition, genes with homology to numbered phage T4 genes are designated as gp1, etc.; genes lacking direct homology with T4 but with similarity to other cyanomyophage genes that themselves have significant homology to T4 are designated as gp-like; other genes are designated with conventional gene names, e.g., nrdA.
Fig. 3. Circular genetic map showing the gene organization of phage S-CRM01.

The genome can be considered as comprised of Regions 1-3 (outer ring) that are dominated by structural genes, replication-related genes, and unique genes, respectively. Subsequent rings represent: positive strand ORFs, negative strand ORFs, tRNAs (maroon), and positive mass spectrometry identification (mauve). The inner rings show GC content around a 50% mid-line (black), and GC skew (G+ = cyan, C+ = blue). Putative ORF function is described by color as listed below the circular map.
Table 1.
Predicted protein-coding genes of S-CRM01
| ORF# | Strand | Size (aa) | T4 similarity1 | Other genes | Mass spec ID | E-value | Annotation from best hit2 | Accession number of best hit | T4Gene Clusters3 |
|---|---|---|---|---|---|---|---|---|---|
| Region 1 | |||||||||
| Region 1A | |||||||||
| 1 | + | 143 | gp25 | 8.E−20 | Base plate wedge - P-SSM2 | YP_214341 | 105 | ||
| 4 | + | 621 | gp6 | X | 3.E−105 | Base plate wedge subunit - S-PM2 | YP_195114 | 106 | |
| 5 | + | 1455 | gp 7-like | X | 1.E−31 | Base plate wedge initiator - S-ESM4 | YP_003097385 | 826 | |
| 6 | + | 318 | X | ||||||
| 7 | + | 505 | X | ||||||
| 8 | + | 495 | gp8-like | X | 2.E−58 | Base plate wedge - P-SSM4 | YP_214644 | 108 | |
| 9 | + | 2485 | X | 5.E−180 | Putative structural protein SRSMp1494 - S-ESM4 | YP_195118 | 828 | ||
| 10 | + | 69 | 8.E−08 | Hypothetical protein PSSM2p116 - P-SSM2 | YP_214348 | 112 | |||
| 11 | + | 437 | gp37 | gp 12-like | X | 7.E−25 | Tail collar domain protein similar to Desulfovibrio salexigens DSM 2638 | YP_002991291 | 1672/1598 |
| 12 | + | 205 | X | ||||||
| 13 | + | 237 | X | 2.E−10 | Collagen triple helix repeat domain protein Burkholderia phymafum STM815 | YP_001859603 | |||
| 14 | + | 250 | X | 5.E−10 | Phage phiJL001 g88 | YP_224012 | |||
| 16 | + | 255 | 3.E−41 | Phage tail collar domain protein Ralstonia phage RSL1 | YP_001950074 | ||||
| 17 | + | 174 | 3.E−12 | 3-deoxy-D-arabino-heptulosonate 7-phosphate synthase from Campylobacter gracilis RM3268 | ZP_05623792 | ||||
| 18 | + | 326 | x | 1.E−09 | Hypothetical protein RRSLp02437 Ralstonia solanacearum UW551 | ZP_00944779 | |||
| 19 | + | 133 | 1.E−05 | Hypothetical protein PSSM2p216 - P-SSM2 | YP_214447 | ||||
| 20 | + | 579 | X | 3.E−78 | Collagen triple helix repeat domain protein Bacillus cereus W | ZP_03101444 | |||
| 22 | + | 512 | X | ||||||
| 23 | + | 377 | X | 2.E−30 | Collagen triple helix repeat domain protein Bacillus cereus G9842 | YP_002448294 | 25 | ||
| 24 | + | 272 | X | ||||||
| 26 | + | 100 | X | ||||||
| 29 | + | 382 | gp13 | X | 3.E−31 | Neck protein - S-PM2 | YP_195127 | 116 | |
| 30 | + | 352 | gp14 | X | 5.E−34 | Neck protein - Syn9 | YP_717777 | 117 | |
| 31 | + | 283 | gp15 | X | 1.E−57 | Tail sheath stabilizer - Syn9 | YP_717778 | 118 | |
| 32 | + | 141 | gp16-like | 9.E−26 | DNA terminase - S-ESM4 | YP_003097360 | 120 | ||
| 33 | + | 219 | X | ||||||
| 34 | + | 517 | X | 3.E−09 | Hypothetical protein from Microcystis phage MaLMM01p164 | YP_851178 | 1348 | ||
| 35 | + | 568 | gp17 | 0.E+00 | Large terminase protein - S-ESM4 | YP_717790 | 124 | ||
| 36 | + | 805 | gp18 | X | 6.E−138 | Tail sheath stabilizer - P-SSM4 | YP_214663 | 125 | |
| 37 | + | 200 | gp19 | X | 8.E−43 | Tail tube protein - P-SSM2 | YP_214362 | 126 | |
| 38 | + | 583 | gp20 | X | 4.E−173 | Portal vertex head protein - S-ESM4 | YP_003097343 | 127 | |
| 41 | + | 215 | gp21 | 1.E−79 | Prohead core scaffolding protein and protease - S-PM2 | YP_195140 | 129 | ||
| 42 | + | 637 | X | 6.E−91 | PrE−peptidase C-terminal domain-contain ing protein Acaryochlons | YP_001517120 | |||
| 43 | + | 361 | gp22 | X | 4.E−54 | Prohead core scaffold protein - P-SSM2 | YP_214366 | 130 | |
| 44 | + | 470 | gp23 | X | 7.E−170 | Major capsid protein - P-SSM2 | YP_214367 | 131 | |
| 45 | + | 225 | gp3 | X | 1.E−28 | Tail completion and sheath stabilizer - P-SSM2 | YP_214369 | 133 | |
| Region 1B | |||||||||
| 46 | + | 142 | uvsY | 9.E−29 | UvsY - -P-SSM2 | YP_214370 | 134 | ||
| 47 | + | 498 | uvsW | 4.E−141 | UvsW - P-SSM2 | YP_214374 | 138 | ||
| 48 | + | 139 | 2.E−12 | Hypothetical Protein S-PM2p113 - S-PM2 | YP 195147 | 139 | |||
| 50 | + | 165 | gp55 | 1.E−5 | Late transcription sigma factor - P-SSM2 | YP_717814 | 140 | ||
| 51 | + | 692 | X | ||||||
| 52 | + | 349 | gp47 | 5.E−82 | Endonuclease - P-SSM2 | YP_214377 | 141 | ||
| 53 | + | 99 | 4.E−10 | Hypothetical protein PSSM2p146 - P-SSM2 | YP_214378 | 142 | |||
| 55 | + | 575 | gp46 | 8.E−124 | Endonuclease - PSSM2 | YP_214379 | 143 | ||
| Region 1C | |||||||||
| 56 | − | 98 | 9.E−12 | PAAR repeat-contain ing protein Aminobacterium colombiense DSM 12261 | ZP_02190607 | 505 | |||
| 58 | − | 302 | X | ||||||
| 59 | − | 736 | gP5 | 2.E−21 | Putative base plate hub subunit and tail lysozyme - S-PM2 | YP_195245 | 340 | ||
| 60 | − | 424 | X | 1.E−23 | Hypothetical cyanophage protein - S-RSM4 | YP_003097461 | 15 | ||
| 61 | − | 865 | X | 4.E−10 | Hypothetical protein PSSM4p016 - P-SSM4 | YP_214577 | 847 | ||
| 62 | − | 61 | gp51-like | 2.E−09 | Similar to base plate hub assembly catalyst gp51 S-PM2p206 - S-PM2 | YP_195241 | 12 | ||
| 63 | − | 235 | gp26-like | 1.E−3 | Baseplate hub subunit - P-SSM2 | YP_214246 | 11 | ||
| 64 | − | 140 | gp4 | 2.E−21 | Head completion protein - S-PM2 | YP_195238 | 9 | ||
| 65 | + | 197 | X | ||||||
| 66 | + | 114 | 8.E−24 | Hypothetical protein PSSM2p011 - P-SSM2 | YP_214243 | 8 | |||
| 67 | + | 283 | gp48-like | X | 1.E−16 | Baseplate tail tube cap - S-RSM4 | YP_003 097468 | 1152 | |
| 68 | + | 219 | gp53 | 2.E−18 | Baseplate wedge protein - P-SSM4 | YP_214571 | 6 | ||
| Region 2 | |||||||||
| 69 | − | 302 | gp32 | 2.E−81 | ssDNA binding protein - S-RSM4 | YP_003097471 | 5 | ||
| 74 | − | 204 | gp59 | 1.E−40 | Loader of gp41 DNA hehcase - P-SSM2 | YP_214238 | 3 | ||
| 75 | − | 95 | gp33-like | 1.E−21 | Late promoter transcriptional accessory protein - P-SSM2 | YP_214233 | 326 | ||
| 76 | − | 178 | 2.E−27 | Putative Exonuclease PSSM2p329 - P-SSM2 | YP_214232 | 325 | |||
| 77 | − | 252 | phoH | 3.E−26 | PhosphatE−starvation inducible protein - P-SSM2 | YP_214558 | 322 | ||
| 78 | 82 | 1.E−12 | Hypothetical protein PSSM2p325 - P-SSM2 | YP_214557 | 321 | ||||
| 79 | − | 61 | 2.E−18 | Hypothetical protein Syn9 g97 - Syn9 | YP_717764 | 160 | |||
| 80 | − | 229 | td | 1.E−69 | FAD - dependent thymidilate synthase - P-SSM2 | YP_214554 | 318 | ||
| 82 | − | 170 | 1.E−28 | Hypothetical protein PSSM4p 192 - P-SSM4 | YP_214753 | 313 | |||
| 83 | − | 281 | rnh | 4.E−67 | RnaseH - S-PM2 | YP_195220 | 870 | ||
| 84 | − | 85 | 4.E−07 | Hypothetical protein SRSM4p016 - S-RSM4 | YP_003097250 | 312 | |||
| 86 | − | 81 | nrdC | 3.E−10 | Glutaredoxin - S-PM2 | YP_195197 | 311 | ||
| 87 | − | 64 | hli03 | 6.E−15 | High light inducible protein Prochlorococcus marinuc | YP_001016892 | 267 | ||
| 90 | − | 418 | 2.E−30 | Glycosyl transferase family protein Flavobacterium johnsoniae | YP_001194249 | ||||
| 95 | − | 81 | X | ||||||
| 96 | − | 364 | psbA | 0.E+00 | psbA photosystem II D1 protein S-PM2 | YP_195211 | 280 | ||
| 97 | − | 246 | 2.E−24 | Hypothetical protein AFEp1218 Acidithiobacillus ferrooxidans | YP_002425655 | ||||
| 98 | − | 124 | 4.E−10 | Hypothetical protein S-PM2p013 - S-PM2 | YP_195047 | 759 | |||
| 101 | − | 55 | 2.E−07 | Hypothetical cyanophage protein - S-RSM4 | YP_003097410 | 751 | |||
| 104 | − | 194 | musG | 2.E−21 | NusG antitermination factor Cyanothece sp. PCC 7425 | YP_002484689 | |||
| 105 | − | 68 | 1.E−06 | Hypothetical Protein SSM2p188 - S-SM2 | ???????? | 249 | |||
| 108 | − | 87 | 3.E−06 | Hypothetical protein WH5701p16051 Synechococcus sp WE 5701 | ZP_01084255 | ||||
| 109 | − | 126 | 3.E−34 | RlpA-like lipoprotein precursor Microcystis aeruginosa NIES-843 | YP_001659233 | 851 | |||
| 110 | − | 74 | 1.E−05 | Hypothetical protein PSSM2p201 - P-SSM2 | YP_214432 | 424 | |||
| 112 | − | 251 | 3.E−56 | Hypothetical protein AM1p0818 Acaryochloris manna MBIC11017 | YP_001515176 | ||||
| 113 | − | 186 | 4.E−24 | Putative lysin - S-PM2 | YP_195189 | 848 | |||
| 114 | − | 116 | 2.E−19 | Hypothetical protein S-PM2p152 - S-PM2 | YP_195187 | 846 | |||
| 115 | − | 59 | 2.E−12 | Hypothetical protein S-PM2p190 - S-PM2 | YP_195225 | 872 | |||
| 116 | − | 382 | nrdB | 3.E−170 | Ribonucleotide reductase subunit B - Syn9 | YP_717864 | 204 | ||
| 118 | 132 | 4.E−10 | Hypothetical protein S-PM2p009 - S-PM2 | YP_195043 | 755 | ||||
| 119 | − | 771 | nrdA | 0.E+00 | Ribonucleotide reductase sub unit A - S-PM2 | YP_195185 | 203 | ||
| 120 | − | 339 | gp61 | 4.E−87 | Primase - P-SSM2 | YP_214438 | 202 | ||
| 122 | − | 124 | 2.E−36 | Hypothetical protein PSSM2p203 - P-SSM2 | YP_214434 | 198 | |||
| 123 | − | 202 | 6.E−30 | Putative calcium binding hemolysin protein Microcystis aerugmosa NIES-843 | YP_001655361 | ||||
| 124 | − | 109 | X | ||||||
| 125 | − | 242 | X | ||||||
| 127 | − | 286 | X | 2.E−51 | Hypothetical protein PSSM2p194 - P-SSM2 | YP_214426 | 190 | ||
| 130 | − | 822 | X | 3.E−09 | Serine/threonine kinase domain PKN8 Plesiocystis pacifica SIR-1 | ZP_01911869 | |||
| 132 | − | 398 | 1.E−62 | Hypothetical protein P700755 27646 Psychroflexus torquis ATCC 700755 | YP_003097298 | 71 | |||
| 136 | − | 137 | mazG | 1.E−46 | MazG - P-SSM2 | YP_214420 | 184 | ||
| 138 | − | 459 | gp41 | 6.E−151 | HelicasE− P-SSM2 | ACY76067 | 182 | ||
| 140 | − | 193 | 3.E−22 | 2OG-Fe(H) oxygenase superfamily-dioxygenase - P-SSM4 | YP_717846 | 104 | |||
| 141 | − | 338 | uvsX | 3.E−142 | UvsX - recA like recombination protein - P-SSM2 | YP_214417 | 181 | ||
| 142 | 838 | gp43 | 0.E+00 | DNA Polymerase - P-SSM2 | YP_214414 | 178 | |||
| 145 | 136 | hsp20 | 6.E−30 | Small heat shock protein - S-PM2 | YP_195165 | 170 | |||
| 146 | − | 135 | regA | 8.E−53 | RegA translation repressor - P-SSM4 | YP_214692 | 168 | ||
| 148 | − | 382 | 2.E−70 | DNA-cytosine methyltransferase Ochrobactntm anthropi | YP_001370055 | 2562 | |||
| 149 | − | 232 | 1.E−32 | Methyltransferase domain-containing protein Bacteroides sp 2p2p4 | ZP_04553186 | ||||
| 153 | − | 124 | gp62-like | 4.E−21 | Clamp loader subunit - S-RSM4 | YP_003097312 | 167 | ||
| 155 | 311 | gp44 | 1.E−92 | Clamp loader subunit - P-SSM2 | YP_214393 | 157 | |||
| 156 | − | 223 | ep45 | 7.E−59 | Sliding clamp - P-SSM2 | YP_214389 | 153 | ||
| 161 | − | 76 | 6.E−21 | GDSL family lipase Serratia proteamaculans 568 | YP 001478498 | ||||
| Region 1 | |||||||||
| 166 | + | 714 | 3.E−97 | Peptidase S-PM2p119 - S-PM2 | YP_195154 | 146 | |||
| 167 | + | 422 | cobS | 9.E−120 | CobS - cobalamin phosphate synthase like protein - S-PM2 | YP_195155 | 150 | ||
| 171 | + | 157 | 6.E−27 | Hypothetical protein Switp2129 Sphingomonas wittichii RW1 | YP_001262626 | ||||
| 172 | + | 175 | X | ||||||
| 176 | + | 58 | 3.E−12 | Hypothetical protein S-PM2p038 - S-PM2 | YP_195072 | 783 | |||
| 185 | + | 132 | 7.E−07 | Acetyltransferase Acaryochloris marina MBIC11017 | YP_001520833 | ||||
| 194 | + | 288 | lig | 3.E−63 | DNA Ligase Chlorella Virus | YP_001497930 | 1627 | ||
| 201 | + | 183 | 4.E−26 | Conserved hypothetical protein Streptomyces clavuligerus ATCC 27064 | ZP_05005O46 | 287 | |||
| 205 | + | 157 | 2.E−35 | HNH endonuclease Cyanothece sp. PCC 7425 | ZP_002482562 | 287 | |||
| 206 | + | 85 | gp39.1 | X | 1.E−15 | gp39.1 hypothetical protein Enterobacteria phage JS10 | YP_002922353 | 1465 | |
| 208 | + | 162 | 9.E−33 | Redox protein Prochlorococcus marmus str. MIT 9211 and P-SSM4 | YP_001551391 | 395 | |||
| 209 | + | 116 | speD | 3.E−19 | S-adenosylmethionine decarboxylase S-PM2 | YP_753372 | 411 | ||
| 215 | + | 97 | spoT | 1.E−12 | Penta-phosphate guanosinE−3'-pyrophosphohydrolase (spoT) Helicobacter pylori 52 | ACX99173 | 1803 | ||
| 220 | + | 261 | 8.E−37 | Metallophosphoesterase - delta proteobactenum MLMS-1 | ZP_01289066 | 2146 | |||
| 230 | + | 115 | 2E−09 | Hypothetical protein S-PM2p156 - S-PM2 | YP_195090 | 68 | |||
| 234 | + | 184 | 2.E−55 | Hypothetical protein PSSM2p151 - P-SSM2 | YP_214383 | 147 | |||
| 249 | + | 110 | 5.E−22 | Hypothetical protein S-PM2p016 - S-PM2 | YP_195050 | 66 | |||
| 255 | + | 79 | 1.E−05 | Hypothetical protein S-PM2p043 - S-PM2 | YP_195077 | ||||
| 281 | + | 54 | 1.E−09 | Hypothetical protein PretD1p03444 Providencia rettgeri DSM 1131 | ZP_06124080 | ||||
| 291 | + | 201 | 7.E−06 | Hypothetical protein PSSM7p193 - P-SSM7 | ?????? | 422 | |||
| 296 | + | 79 | 4.E−07 | Hypothetical protein syn9 g98 - Syn9 | YP 717765 | 916 | |||
ORF number, gene orientation (strand) and protein size (number of amino acids) are noted for predicted protein-coding genes that either have a database match or were detected by mass spectrometry. ORFs were identified with BLASTP run against the NCBI nr database with a threshold E value of≤10−5. The best-hit E−values, homologous gene, and NCBI accession numbers are noted.
T4 similarity identifies genes (by T4 name) with BLASTP hits to the phage T4 genome, T4-like genes are homologous to cyanophage genes annotated with T4 gene names.
Gene annotation based on sequence homology and color coded as in Fig 2: Structural genes (inci assembly catalyst), yellow, Replication, recombination, nucleotide metabolism and gene expression control genes, blue; (marine) Cyanophage signature genes (Millard et al, 2009), olive, Novel S-CRM01 genes, pink; Genes of proteins identified by mass spectrometry, purple.
T4 Gene Clusters (T4-GC) numbers are based on seduence homology with a threshold E value <10−5 from Sullivan et al (2010)
The genome contains 34 genes with homology to T4, and a total of 86 ORFs (29% of total) with homology to ORFs found in at least one of a group of 17 closely related myophages that lytically infect marine Synechococcus or Prochloroccus (Mann et al., 2005; Sullivan et al., 2005; Weigele et al., 2007; Millard et al., 2009; Sullivan et al., 2010)(Tables 1, S1). Phage S-PM2 from this group shares the largest number of genes with S-CRM01 (75), while the others have between 60 and 73 genes in common with S-CRM01 (Fig. S2). In contrast, the S-CRM01 genome shares a mere 4 genes with the only other known genome from a freshwater cyanomyophage, that of Ma-LMM01, which infects Microcystis aeruginosa (Yoshida et al., 2008).
The S-CRM01 genome encodes all six genes proposed as (marine) cyanophage signature genes (Millard et al., 2009): cobS, hli03, hsp20, mazG, phoH and psbA. The psbA gene, encoding the D1 protein of photosystem II, and hli03 (high light inducible) gene are characteristic “photosynthetic phage” genes that are thought to function in augmenting the photosynthetic capacity of infected cells or in protecting against oxidative stress resulting from high light intensities (Lindell et al., 2005; Mann et al., 2005; Clokie et al., 2006).
The S-CRM01 genome shows extensive synteny with the marine cyanomyophage genomes, but a 64 kbp part of the syntenous region is in an inverted orientation (Fig. 4) with most genes between g56 and g165 expressed from the negative strand. Together with the GC-skew profile (Fig. 3, innermost ring; the leading strand in prokaryotic genomes is enriched in G; Lobry, 1996), this is suggestive of bidirectional replication with an origin near the gene inversion boundary (nt 123K). No such pronounced GC-skew patterns are evident in the marine cyanomyophage genomes (not shown).
Fig. 4. Extensive synteny between S-CRM01 and marine cyanomyophages.

A. Dot plots comparing gene order between S-CRM01 and phage T4 and indicated cyanomyophage genomes. The S-CRM01 genome regions 1-3 (see Fig. 3) are indicated by colored shading and are labeled at right. Syntenous segments I-IV are gp13–gp46 (SCRM01 g29–g55), gp5-td (g59–g80), nrdB-gp45 (g116–g156), and g166–167 (cobS), respectively. B. Diagram indicating the location of the inversion between the S-CRM01 and S-PM2 genomes. Note that the P-SSM2 and S-PM2 genomes have insertions between syntenous segments II and III, and that P-SSM2 and Syn9 genome numbering convention places the regions syntenous with Region 1C at the start of the genomes.
The S-CRM01 genome possesses an unusually large number (182) of ORFs with no significant homologs in the GenBank database (as of 30 August, 2010), representing 61% of predicted ORFs (genes shown in black in Fig. 3). This is far higher than the number of unique genes in the marine cyanomyophage genomes of similar size (about 60–100) (Millard et al., 2009; Sullivan et al., 2010).
Based on both gene content and sequence relatedness, S-CRM01 is a member of a discrete clade that encompasses all 17 currently sequenced marine cyanomyophages, but it is the most divergent member of this group (Fig. 5). The phylogenetic relationships among the cyanomyophages were explored using the Hal phylogenomics pipeline (Robbertse et al., 2006), which produces whole genome phylogenies using single-copy protein coding genes. The pipeline can be configured for inclusion of different numbers of orthologous clusters to allow the analysis to be expanded by including genes missing from a few genomes (e.g., Fig. 5D; see methods). As shown in the consensus tree in Fig. 5A, the cyanomyophages (including S-CRM01) form a monophyletic group supported by high bootstrap values. However, the group is quite diverse, with S-CRM01 being the most divergent member and the marine phages partitioning into at least 4 clades. To examine the possibility of long branch attraction artifacts, the same analysis was run without the T4, Aeh1 and KVP40 genomes, with the overall topology matching that of the consensus tree in Fig. 5A. Phylogenetic analysis based on gene content (using a subset of the genomes analyzed here) showed similar overall relationships (Millard et al., 2009).
Fig. 5. Phylogenetic relationship of S-CRM01 to other myophages.

A. Consensus tree for the most highly supported topology across all tested values of included non-universal genes (missing data). Values at nodes indicate the average bootstrap value for all trees with that node/the number of trees for which that node occurred out of a total of 27 trees (see Materials and Methods). The marine cyanomyoviruses have been grouped into the clades MC1-4 as indicated. B and C. Consensus trees showing alternative topologies among clades MC1-3 when including 0–10% and 15–40% included non-universal genes, respectively. D. The number of orthologous clusters analyzed for each phage as a function of % included non-universal genes are indicated and color-matched. The genes used for each level of analysis are indicated in Table S2.
With different numbers of genes (orthologous clusters) included in the analysis, two additional alternative tree topologies with differences in the relative positions of MC1, MC2 and MC3 were observed (Fig. 5B, C). Analysis of individual gene phylogenies produced a similar range of topologies, although the S-CRM01 branch (typically the longest) was at times placed among the MC1-3 clades. No pattern between tree topology and gene type or location in the genome could be discerned. The variable relationships indicated above suggest high levels of horizontal gene exchange among the cyanomyophages, analogous to that proposed among the Synechococcus or Prochlorococcus hosts of these phage (Zhaxybayeva et al., 2009). Evidence for gene exchange at individual loci among the marine cyanophages has been reported by Zeidner et al. (2005), Sullivan et al. (2006) and Bryan et al. (2008). Note that the analysis of Fig. 5 does not support the classification of phages based on host (Synechococcus or Prochlorococcus), since both hosts are represented in clades MC2 and MC4 (c.f. Sullivan et al., 2010).
A structural gene cluster mostly on the plus strand
The bioinformatically identifiable structural genes are all present in a 72 kbp segment of the genome (Region 1), comprised of two clusters: g1 through g45, covering nts 1–51,380 (genome Region 1A), and g59 through g68, covering nts 62,104–72,062 (Region 1C). Region 1A includes strong synteny with the genomes of marine exoT-even cyanomyophages and phage T4 (Fig. 4). This region encodes most of the recognizable structural genes, with all but one gene expressed from the plus strand. Expression is predicted to be dominated by the activity of late promoters (Table S4), as appropriate for structural protein genes.
Both transcriptional directions are represented in Region 1C, with transcription again predicted to be dominated by late promoters. This region is also syntenous, although in inverted orientation, to the marine cyanomyovirus genomes (Fig. 4), but is not syntenous to T4.
As has been observed for marine cyanomyophages, the conserved genes that define synteny are variably interspersed with additional genes. Twenty-three ORFs with no homologs in the GenBank database are located within Region 1; other genes are most similar to phage or bacterial proteins (Table 1). Millard et al. (2009) have described a hyperplastic region between gp15 and gp18 in S-RSM4 and other cyanomyophages. In S-CRM01 this segment contains only one non-conserved gene, but several genes of varied apparent origins are located upstream of gp13.
Virion proteomics
Mass spectrometry was used to identify phage proteins present in a preparation of S-CRM01 virions purified through a CsCl density gradient. Forty-three proteins were identified with high certainty (Fig. 3, Tables 1 and S3). All of these, except two proteins encoded in Region 3 of the genome, are encoded by genes that are closely associated with late promoters (Table S4). Most of the identified proteins are encoded in Regions 1A and 1C, emphasizing the clustering of structural protein genes in these parts of the genome. Thirteen of these proteins correspond to structural proteins of T4 (gp or gp-like genes), 13 correspond to genes with BLAST hits to other phage or bacterial genes, while 9 are encoded by genes unique to S-CRM01. These results indicate that the S-CRM01 virion is composed of proteins with a variety of origins: homologs of T4 proteins, homologs of proteins from the related marine cyanomyophages, proteins with closest BLAST hits in other phage or in bacterial genomes, and proteins that have no currently known homologs.
An additional 8 proteins encoded by genes not clustered with structural genes were identified by our proteomic study. These genes are located in Regions 1B, 2 and 3 (Fig. 3, Table 1). Electron microscopy suggested that host material attached to phage baseplates may have been present in the phage preparation made for proteomic analysis. Consequently, it is uncertain whether these proteins are truly virion-associated (structural) proteins or phage-encoded proteins that have been inserted into host structural components. Their identification does prove the expression of the respective genes during viral infection, indicating tht these proteins (4 of which are unique to S-CRM01) are functionally relevant.
As expected, abundant mass spectrometry signals were registered for gp23 major capsid protein (g44), gp18 contractile tail sheath protein (g36) and gp19 tail tube protein (g37), which are present in multiple copy number in the T4 capsid (Miller et al., 2003a). Abundant signals were also observed for peptides from g9, at 271 kDa the largest protein encoded by the S-CRM01 genome. This huge protein has a predicted strong β-strand character and no identifiable sequence motifs, but has BLAST matches to a wide range of phage proteins (including putative tail protein) and to glycosyl hydrolase bacterial neuraminidase repeat (BNR) proteins. An internal dot plot analysis shows numerous different internal repeat elements 10 to 20 residues long, mostly repeated only once. These properties suggest that g9 is a tail fiber gene (Weigele et al., 2007), although we have not observed tail fibers under the electron microscope.
Three proteins with collagen-like triple helix repeat domains were detected by mass spectrometry: g13 with 36 GXY repeats, g20 with 85 GXQ repeats, and g23 with 40 GXQ repeats. Collagen-like proteins have been reported from some phage, participating in spike formation in phage PRD1 (Caldentey et al., 2000) and suggested to be found in tail fibers (Smith et al., 1998; Sullivan et al., 2005). More careful virion characterization will be needed to determine whether S-CRM01 possesses the tail fibers or whiskers that are predicted by the presence of these proteins.
T4-like nonstructural genes predominantly on the minus strand
A second major region of synteny with the exoT-even marine cyanomyophage genomes is the 51 kbp segment between genes g69 and g165 (Region 2, nts 72,094–123,515) (Figs. 3, 4; Table 1)(c.f. Sullivan et al., 2010). These genes, all of which are on the minus strand, include core T4-like genes with roles in DNA replication, recombination and repair (e.g., gp43 DNA polymerase, gp61 primase, gp45 sliding clamp, uvsX), nucleotide metabolism (nrdA, nrdB, nrdC, td thymidylate synthase) and 5 of the 6 marine cyanophage signature genes (phoH, hli03, psbA, mazG, hsp20). Other significant genes also present in Region 2 are: a member of the 2OG-FeII oxygenase superfamily that is present in nearly all marine cyanomyophages, a nusG transcription anti-termination factor homolog (not found in T4-related phages), regA regulator of early gene translation (common to all marine cyanomyo and T4-like phages), and RNase H (rnh, common to T4-like phages but found in only S-PM2 and Syn19 among the marine cyanomyophages).
This segment of the genome is also predicted to be expressed predominantly via late transcription, though some putative early promoters have been identified, notably for the expression of gp45, gp44, gp62 and gp33, all of which are involved in establishing and maintaining T4 late gene transcription (Miller et al., 2003a). The other key gene needed for late transcription, gp55 alternative sigma factor, is located in Region 1B between the two structural gene segments and on the plus strand, and it also appears to be expressed from an early promoter (Table S4). The recombination genes uvsY, uvsW, gp47 and gp46 are also located in Region 1B, all on the plus strand in another region of synteny with marine cyanomyophage genomes (Figs. 3, 4).
As in the structural gene region, but even more so, the conserved genes in Region 2 are interspersed with additional genes at multiple sites. Forty-eight (49%) of ORFs in Region 2 are unique to S-CRM01.
49 kbp (27%) of the genome contains 132 ORFs, 85% of which are unique genes
The remainder of the genome (Region 3, genes g166–g297) possesses no overall similarity to the genomes of other phages apart from g166 and g167 (Fig. 4) and contains only 20 genes with homology to previously identified ORFs. Fifteen of these have homologs in at least one T4-like or cyanomyo phage. This part of the genome also contains all but two of the 33 tRNA genes. The significant genes present in this segment are cobS (g167), DNA ligase (g194), speD (g109), spoT (g215) and HNH endonuclease (g205). cobS, involved in cobalamin biosynthesis, may support the synthesis of deoxynucleotides (Sullivan et al., 2005) and is one of the marine cyanophage signature genes (Millard et al., 2009). DNA ligase is common in T4-like phage genomes, though not found in the marine cyanomyophages; the S-CRM01 DNA ligase gene is most closely related to Chorella virus and PB1-like myophage ligases. speD encodes S-adenosylmethionine decarboxylase, which is involved in polyamine synthesis and found in a few marine cyanomyophage genomes (Mann et al., 2005). spoT, which encodes ppGpp synthetase and hydrolase, has not previously been recognized in myophage genomes but is present in the Aeromonas myophages Aeh1, 44RR, PHG25 and PHG31. HNH endonuclease is possibly a member of a family of homing endonucleases, although no introns have been detected in the S-CRM01 genome.
All but one of the genes (g186) in Region 3, including the tRNA genes, reside on the plus strand. Region 3 genes are atypical in a number of ways. Identifiable promoters are sparse; the G+C content is generally high (Fig. 3) and the ORFs are more likely to have a higher G+C% in the third codon position; the ORFs are shorter and less tightly spaced. A similarly extensive array of unique genes is also present in the Aeh1 genome (Comeau et al., 2007) and to a lesser extent in other myophages, including the marine cyanomyophages (e.g., S-PM2; Mann et al., 2005).
Genome transcription
The predicted transcription control signals—start sites of 34 early promoters and 81 late promoters, and coordinates of 17 terminator hairpins — are listed in Table S4. As for T4-like phages such as RB49 (Desplats et al., 2002) and marine cyanomyophages such as S-PM2 (Mann et al., 2005), S-CRM01 gene expression lacks the middle phase of transcription found in T4. Either early or late (in a few instances both) promoters are predicted for genes that are middle-transcribed in T4. Thus, early promoters are predicted upstream of the genes involved in establishing late transcription: gp55 (sigma factor), gp44, gp45, gp46 and gp33 (sliding clamp and clamp loader proteins). DNA replication and repair genes that are either middle or late expressed in T4, such as gp43 (DNA polymerase), gp32 (ssDNA binding protein), gp41 (helicase), uvsX, uvsW, and gp46 and gp47 endonucleases, are associated with putative late promoters. This is also true of nucleotide metabolism genes td, nrdA and nrdC, as well as each of the marine cyanomyophage signature genes except cobS. A few of these genes — rnh, td, phoH, hsp20 — are associated with both predicted early and late promoters. This is also true of some, though not all, tRNA genes.
All identifiable structural genes are associated with putative late promoters, although in several cases (gp8, gp13 & gp14, gp17, gp18 & gp19, gp22, gp5) early promoters have unexpectedly also been predicted with good prediction scores. Transcription patterns will need to be experimentally assessed to verify these predictions.
Late promoters are predicted to fall into two categories, 46 with a CTAAATA core sequence and 35 with an ATAAATA core sequence (Fig. 6, Table S4). ACTAAATA is the most frequent promoter sequence. ATAAATA promoters are most common with T4 (Miller et al., 2003a) and the marine cyanomyophages (Mann et al., 2005; Weigele et al., 2007; Sullivan et al., 2010). The CTAAATA sequence is largely absent from the latter genomes, indicating that these promoters have not merely been overlooked in previous annotations. CTAAATA promoters are unusual among T4-like phages, previously reported for RB49 (Desplats et al., 2002) and Aeh1 (Nolan et al., 2006). Like RB49, S-CRM01 late promoters are a combination of CTAAATA and ATAAATA types, with no evident specialization within gene groups that might suggest separate regulation. We have also only detected a single late transcription gp55 sigma factor, which may thus have unusually broad promoter recognition. Interestingly, S-CRM01 gp55 is clearly more closely related to marine cyanomyophage gp55 genes (which recognize ATAAATA promoters) than to RB49 or Aeh1 gp55.
Fig. 6. Late promoter consensus sequence.
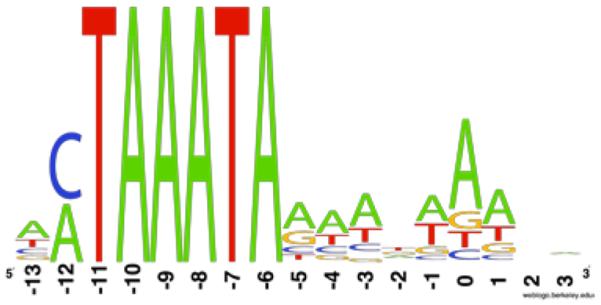
Weblogo representation of the sequences of 81 predicted late promoters (Table S3), indicating the prevalence of both CTAAATA and ATAAATA core sequences. Nucleotide 0 marks the predicted start of transcription based on T4 late promoters (Miller et al., 2003a).
A full set of tRNAs
The 33 tRNA genes in the S-CRM01 genome (Table 2) is more than found in some bacteria and includes all of the 20 amino acid specificities, although there is no initiator tRNAMet as found in some phage genomes. KVP40 also has all 20 specificities represented, though some are thought to be pseudogenes that may not be functional (Miller et al., 2003b). The S-PM2 genome contains 24 tRNA genes representing all but the cysteine and phenylalanine specificities, but the other marine cyanomyophage genomes have much smaller subsets of these. tRNAHis is the only S-CRM01 tRNA gene with an encoded 3′ CCA terminus, while in S-PM2 five tRNA genes (including tRNAHis) include the 3′ CCA. Three tRNA genes with a CAU anticodon are present. Based on features in the anticodon loop (C32 and A38) that are recognized by the TilS enzyme, which modifies C34 in the anticodon to lysidine (Nakanishi et al., 2009), the t2 and t31 genes are proposed to be tRNAIle2 genes, leaving one tRNAMet gene (t22). Gene t2 has multiple additional nucleotides that are proposed to be characteristic of lysidine-containing tRNAIle in cyanobacteria (Freyhult et al., 2007)(see Table S5).
Table 2.
tRNA genes in the S-CRM01 genome
| tRNA gene | Identity | Anticodon | Nt positions | Strand |
|---|---|---|---|---|
| t1 | Leu | UAA | 88617-544 | − |
| t2 | Ile2 | CAU (LysAU)1 | 88643-715 | − |
| t3 | Gly | UCC | 127547-618 | + |
| t4 | Arg | UCU | 127623-697 | + |
| t5 | Lys | UUU | 128953-9025 | + |
| t6 | Pro | UGG | 129165-236 | + |
| t7 | Tyr | GUA | 129535-619 | + |
| t8 | Gly | GCC | 130519-590 | + |
| t9 | Asp | GUC | 130857-930 | + |
| t10 | Trp | CCA | 130985-1055 | + |
| t11 | Asn | GUU | 131716-787 | + |
| t12 | Glu | UUC | 131866-941 | + |
| t13 | Glu | cue | 132043-115 | + |
| t14 | Pro | UGG | 134674-748 | + |
| t15 | Ile | GAU | 134944-5014 | + |
| t16 | Ile | GAU | 135055-128 | + |
| t17 | Phe | GAA | 135201-274 | + |
| t18 | Ala | UGC | 135313-385 | + |
| t19 | Val | GAC | 135762-833 | + |
| t20 | Ser | GCU | 136579-663 | + |
| t21 | Ser | GGA | 136668-759 | + |
| t22 | Met | CAU | 139590-660 | + |
| t23 | His | GUG | 139662-7382 | + |
| t24 | Thr | GGU | 138739-810 | + |
| t25 | Thr | UGU | 141268-341 | + |
| t26 | Gln | UUG | 141517-587 | + |
| t27 | Lys | CUU | 141839-912 | + |
| t28 | Cys | GCA | 142050-123 | + |
| t29 | Leu | CAA | 145010-084 | + |
| t30 | Arg | ACG | 149226-300 | + |
| t31 | Ile2 | CAU (LysAU)1 | 149448-521 | + |
| t32 | Leu | GAG | 149554-627 | + |
| t33 | Val | UAC | 149712-783 | + |
Probable modification to lysidine-A-U;
Includes 3' CCA
Multiple genes: Arg (2x): t4, t30; Glu (2x): t12, t13; Gly (2x): t3, t8; He (2x): t15, t16; Ile2 (2x): t2, t31; Leu (3x): t1, t29, t32; Lys (2x): t5, t21; Pro (2x): t6, t14; Ser (2x): t20, t21; Thr (2x): t24, t25; Val (2x):t19, t33.
Tightly juxtaposed genes are t3/t4, t20/t21, t22/t23/t24.
It cannot be determined without experimentation whether all predicted tRNAs are functional, although key identity elements and conserved features (Giegé et al., 1998) are generally present (Table S5). Some of the predicted tRNAs do have highly unusual features (see Table S5 and refer to tRNA database; Juhling et al., 2009) that may compromise function: e.g., U73 and long variable loop in tRNAGlu t12, A1 in tRNAGlu t13, U73 in tRNAIle2 t31, U10:U25 in tRNASer t21 (probable cause for identification as possible pseudogene by tRNAScan-SE). The tRNAHis gene t23, like other phage tRNAHis occurrences (e.g., S-PM2), lacks the additional 5′ residue G-1 that is in most systems critical for histidine identity (Giegé et al., 1998). G-1 would be either replaced with U-1 (adjacent nucleotide in the genome) or lacking in the mature S-CRM01 tRNAHis, a situation that in E. coli allows only partial translational function (Yan and Francklyn, 1994). Another possibility is that G-1 could be added post-transcriptionally by a Thg1-like activity (Heinemann et al., 2010) or that a variant histidyl-tRNA synthetase that does not rely on G-1 is present in host cells (Wang et al., 2007). No Thg1 or histidyl-tRNA synthetase is evident among the S-CRM01 genes. Establishing the functionality of phage tRNA genes is important in determining their role during infection and the selective forces that act on them.
A connection between the suite of S-CRM01 tRNA genes and codon usage in phage ORFs or in those ORFs expected to be expressed late could not be discerned. It is thought that phage tRNAs serve to optimize expression of proteins by allowing more efficient decoding of codons in phage mRNAs that are under-represented in host mRNAs (Bailly-Bechet et al., 2007). In the absence of the host genome sequence, this hypothesis cannot be tested for S-CRM01. However, there must be a different reason for the presence of tRNAs for the two specificities represented by single codons (methionine and tryptophan) or of tRNA genes with unusual features that could be expected to decrease decoding efficacy (see Table S5); perhaps the viral tRNAs provide an advantage by being more flexible in their accommodation to the interactions on the ribosome in a stressed (infected) cell.
In several cyanomyophage and T4-like genomes, some tRNA genes are located in the general region downstream of the nrdA and nrdB genes. Only two of the 33 tRNA genes in S-CRM01 are found in this location, the remainder being loosely spaced across a 22 kbp segment of Region 3 among mostly genes that are unique to S-CRM01 (Fig. 3). The S-CRM01 genome includes homologs of 22 of the 24 tRNA genes from S-PM2, with nucleotide identity ranging from 42% to 81%, with an average of 68%. tRNAs among the MC1 clade (S-PM2, Syn1 and S-RSM4) share average identities of 76–80%, consistent with their close relationship on the basis of protein-coding genes. This would suggest vertical inheritance of tRNAs, but the lack of extensive synteny, wide range in nucleotide identity, and wide variety of tRNA gene content presents a complex picture of tRNA gene evolution in the cyanomyophages; the MC1 clade members possess 24, 12 and 6 tRNA genes, and the MC2 clade members possess 10, 5 and 1 tRNA genes.
Noteworthy genes encoded by S-CRM01
Two predicted genes, spoT and nusG (indicated in pink in Fig. 3) are unusual genes in T4-like phages. SpoT is a pyrophosphohydrolase that in E. coli removes the alarmone (p)ppGpp, which accumulates as a result of RelA action in cells when the stringent response is triggered by amino acid starvation (Srivatsan and Wang, 2008). SpoT is also capable of synthesizing (p)ppGpp under certain conditions, such as fatty acid starvation. Through a network of interactions with other proteins, it seems to sense the physiological status of the cell and modulate (p)ppGpp levels (Potrykus and Cashel, 2008; Srivatsan and Wang, 2008). The effect of elevated (p)ppGpp is a down-regulation of normal macromolecular synthesis and a switch to gene expression governed by alternative sigma factors, turning gene expression to pathways such as amino acid biosynthesis. The introduction into a cell of a replicating phage can be expected to deplete cellular nutrients, with the risk of inducing elevated (p)ppGpp and shutting off ribosome synthesis, general transcription and replication. Indeed, one may speculate that host cells utilize (p)ppGpp to establish an antiviral state that inhibits viral replication and amplification, perhaps until nucleases can attack the viral nucleic acid. Specific effects on viral gene expression are also possible, as illustrated by the ability of (p)ppGpp to shut down some lambda phage promoters (Potrykus and Cashel, 2008). It could thus be advantageous for an infecting phage to counter this type of innate defense, and a virally expressed SpoT enzyme could do that by hydrolyzing (p)ppGpp or otherwise altering the regulation of (p)ppGpp levels.
The predicted S-CRM01 SpoT possesses most of the key amino acids needed for (p)ppGpp hydrolysis (Hogg et al., 2004), but it is a very small protein, suggesting activity as a (p)ppGpp hydrolase with little or no regulatory control. Phage-encoded spoThas not been previously recognized, although homologous genes are present in the Aeromonas phages AehI, 44RR, PHG25 and PHG31, which were assigned to T4 gene cluster (T4-GC) 1803 by Sullivan et al. (2010). All of the marine cyanomyophage genomes possess a mazG gene (as does S-CRM01), which has been postulated to hydrolyze (p)ppGpp and avoid stationary phase conditions in the cell (Clokie et al., 2010; Bryan et al., 2008). However, MazG proteins have varied nucleotide substrate specificities (Galperin et al., 2006) and it has been cautioned that they may not function in (p)ppGpp hydrolysis (Sullivan et al., 2010). In fact, it may be that the broad substrate specificity of MazG (Lu et al., 2010; Zhang and Inouye, 2002; Zhang et al., 2003) involves this protein both in the removal of mutagenic nucleotides that are produced as a result of oxidative stress (Lu et al., 2010) associated with photosynthesis as well as suppression of (p)ppGpp levels.
S-CRM01 may be the first phage genome to contain a putative nusG gene. The NusG protein associates with elongating RNA polymerase to modulate transcription in various ways. Based mainly on E. coli studies, NusG is considered a transcription elongation factor because it increases transcription elongation rates (Squires and Zaporojets, 2000; Yakhnin et al., 2008), although in some bacteria the opposite is true (Sevostyanova and Artsimovitch, 2010). In addition, NusG can promote or suppress pausing at different sites (and thereby facilitate attenuation control) (Sevostyanova and Artsimovitch, 2010; Yakhnin et al., 2008), promote transcript release at termination sites (Chalissery et al., 2007), and may exert effects on translation (Squires and Zaporojets, 2000). S-CRM01 may benefit by influencing the elongation and termination phases of transcription of the phage, or even host, genome through the action of viral NusG. Additionally, Cardinale et al. (2008) have shown that NusG functions in concert with Rho to decrease doubling time and prevent expression of the cryptic rac prophage in E. coli MG1655. Over-expression of NusG by S-CRM01 may be able to increase the metabolic activity of the infected host and prevent interference or competition for transcriptional machinery from prophages endemic to the host. If such benefits do exist, the absence of identifiable nusG from other phage genomes might suggest that unrecognized transcriptional regulators are encoded by other phages.
Relationship between freshwater and marine phages
There has not been enough data to derive a clear picture of the genetic relationships between phages in freshwater and marine environments. Some early observations with podophages (Breitbart et al., 2004) and myophages (Short and Suttle, 2005) emphasized the discovery of very similar sequences in the viral populations of the two aquatic environments. For myophages, additional phylogenetic studies with the gp20, psbA and psbD genes have verified this observation, but have also shown that most (though not all) freshwater sequences map at high resolution to clades that do separate from marine isolates (Wilhelm et al., 2006; Chénard and Suttle, 2008; Sullivan et al., 2008; Wang et al., 2010). SCRM01 seems to represent both of these scenarios, with the closest currently known gp20 sequence found in the Atlantic Ocean isolate P-ShM1 (Sullivan et al., 2008)(Fig. S3). On the other hand, the psbA gene locates to a unique branch between freshwater and marine clades (Fig. S3).
The complete S-CRM01 genome sequence allows more meaningful consideration of the relationship between related marine and freshwater phages. The data in Fig. 5 show the relationships to be complex. On the basis of synteny (Fig. 4A) and protein-coding (Fig. 5D) and tRNA gene content, S-CRM01 is most similar to S-PM2, while on the basis of some phylogenomic comparisons, P-SSM2, S-SM2 and S-SSM7 can be considered more similar (Fig. 5D). It thus seems likely that there have been multiple gene exchanges between the S-CRM01 and marine cyanomyophage lineages during evolution. Brackish estuarine waters are likely connections between freshwater and marine environments, where genetic exchanges could occur between phages that principally exist in one or the other aquatic habitat, especially for phages of bacteria such as Synechococcus, which have broad distributions. Highly unique phages do seem to exist in freshwater, however, such as the Microcystis-infecting Ma-LMM01 (Yoshida et al., 2008), and it will be interesting to learn in the future whether this is the pattern for phages infecting hosts that do not exist in the oceans.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge the assistance of Susan Corum of the Karuk Tribe of California, Richard Raymond and PacifiCorp Inc., and Mary Lindenberg and Tamara Wood of the US Geological Survey for sample collections from the Klamath valley. We thank Elizabeth Kutter for advice on initial experiments, Josh Powell and Theresa Sawyer for assistance with electron microscopy, Andrea McHugh for assisting in the laboratory, Michael Schwalbach for assistance with PFGE, Joey Spatafora for discussions, Susan Golden and Roger Ely for providing cultures, and Roger Hendrix for comments on the manuscript. The OSU mass spectrometry facility and services core is supported in part by NIH/NIEHS grant P30 ES000210. This work was supported by Oregon Sea Grant award NA060AR4170010 and by Oregon State University Agricultural Experiment Station.
REFERENCES
- Bailly-Bechet M, Vergassola M, Rocha E. Causes for the intriguing presence of tRNAs in phages. Genome Res. 2007;17:1486–1495. doi: 10.1101/gr.6649807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besemer J, Lomsadze A, Borodovsky M. GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001;29:2607–2618. doi: 10.1093/nar/29.12.2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozarth CS, Schwartz AD, Shepardson JW, Colwell FS, Dreher TW. Population turnover in a Microcystis bloom results in predominantly non-toxigenic variants late in the season. Appl Environ Microbiol. 2010;76:5207–5213. doi: 10.1128/AEM.00001-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitbart M, Miyake JH, Rohwer F. Global distribution of nearly identical phage-encoded DNA sequences. FEMS Microbiol Letts. 2004;236:249–256. doi: 10.1016/j.femsle.2004.05.042. [DOI] [PubMed] [Google Scholar]
- Breitbart M, Rohwer F. Here a virus, there a virus, everywhere the same virus? Trends Microbiol. 2005;13:278–284. doi: 10.1016/j.tim.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Breitbart M, Salamon P, Andresen B, Mahaffy JM, Segall AM, Mead D, et al. Genomic analysis of uncultured marine viral communities. Proc Natl Acad Sci U S A. 2002;99:14250–14255. doi: 10.1073/pnas.202488399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan MJ, Burroughs NJ, Spence EM, Clokie MR, Mann NH, Bryan SJ. Evidence for the intense exchange of MazG in marine cyanophages by horizontal gene transfer. PLoS One. 2008;3:e2048. doi: 10.1371/journal.pone.0002048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldentey J, Tuma R, Bamford DH. Assembly of bacteriophage PRD1 spike complex: Role of the multidomain protein P5. Biochemistry. 2000;39:10566–73. doi: 10.1021/bi000711+. [DOI] [PubMed] [Google Scholar]
- Cardinale CJ, Washburn RS, Tadigotla VR, Brown LM, Gottesman ME, Nudler E. Termination factor Rho and its cofactors NusA and NusG silence foreign DNA in E. coli. Science. 2008;320:935–938. doi: 10.1126/science.1152763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casjens SR, Gilcrease EB. Determining DNA Packaging Strategy by Analysis of the Termini of the Chromosomes in Tailed-Bacteriophage Virions. In: Clokie MR, Kropinski AM, editors. Bacteriophages: Methods and Protocols, Volume 2: Molecular and applied aspects. Humana Press; New York: 2008. pp. 91–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalissery J, Banerjee S, Bandey I, Sen R. Transcription termination defective mutants of Rho: role of different functions of Rho in releasing RNA from the elongation complex. J. Mol Biol. 2007;371:855–872. doi: 10.1016/j.jmb.2007.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Wang K, Kan J, Suzuki MT, Wommack KE. Diverse and unique picocyanobacteria in Chesapeake Bay, revealed by 16S-23S rRNA internal transcribed spacer sequences. Appl Environ Microbiol. 2006;72:2239–2243. doi: 10.1128/AEM.72.3.2239-2243.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chénard C, Suttle CA. Phylogenetic diversity of sequences of cyanophage photosynthetic gene psbA in marine and freshwaters. Appl Environ Microbiol. 2008;74:5317–5324. doi: 10.1128/AEM.02480-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clokie MR, Millard AD, Mann NH. T4 genes in the marine ecosystem: studies of the T4-like cyanophages and their role in marine ecology. Virol J. 2010;7:291. doi: 10.1186/1743-422X-7-291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clokie MR, Shan J, Bailey S, Jia Y, Krisch HM, West S, Mann NH. Transcription of a 'photosynthetic' T4-type phage during infection of a marine cyanobacterium. Environ Microbiol. 2006;8:827–835. doi: 10.1111/j.1462-2920.2005.00969.x. [DOI] [PubMed] [Google Scholar]
- Comeau AM, Bertrand C, Letarov A, Tetart F, Krisch HM. Modular architecture of the T4 phage superfamily: a conserved core genome and a plastic periphery. Virology. 2007;362:384–396. doi: 10.1016/j.virol.2006.12.031. [DOI] [PubMed] [Google Scholar]
- Delcher AL, Harmon D, Kasif S, White O, Salzberg SL. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 1999;27:4636–4641. doi: 10.1093/nar/27.23.4636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L, Hayes PK. Evidence for cyanophages acrive against bloom-forming freshwater cyanobacteria. Freshwater Biology. 2008;53:1240–1252. [Google Scholar]
- Desplats C, Dez C, Tetart F, Eleaume H, Krisch HM. Snapshot of the genome of the pseudo-T-even bacteriophage RB49. J. Bacteriol. 2002;184:2789–2804. doi: 10.1128/JB.184.10.2789-2804.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorigo U, Jacquet S, Humbert JF. Cyanophage diversity, inferred from g20 gene analyses, in the largest natural lake in France, Lake Bourget. Appl Environ Microbiol. 2004;70:1017–1022. doi: 10.1128/AEM.70.2.1017-1022.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst A, Becker S, Wollenzien UI, Postius C. Ecosystem-dependent adaptive radiations of picocyanobacteria inferred from 16S rRNA and ITS-1 sequence analysis. Microbiology. 2003;149:217–228. doi: 10.1099/mic.0.25475-0. [DOI] [PubMed] [Google Scholar]
- Freyhult E, Cui Y, Nilsson O, Ardell DH. New computational methods reveal tRNA identity element divergence between Proteobacteria and Cyanobacteria. Biochimie. 2007;89:1276–1288. doi: 10.1016/j.biochi.2007.07.013. [DOI] [PubMed] [Google Scholar]
- Fuhrman JA. Marine viruses and their biogeochemical and ecological effects. Nature. 1999;399:541–548. doi: 10.1038/21119. [DOI] [PubMed] [Google Scholar]
- Galperin MY, Moroz OV, Wilson KS, Murzin AG. House cleaning, a part of good housekeeping. Mol Microbiol. 2006;59:5–19. doi: 10.1111/j.1365-2958.2005.04950.x. [DOI] [PubMed] [Google Scholar]
- Giegé R, Sissler M, Florentz C. Universal rules and idiosyncratic features in tRNA identity. Nucleic Acids Res. 1998;26:5017–5035. doi: 10.1093/nar/26.22.5017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- Heinemann IU, Randau L, Tomko RJ, Jr., Soll D. 3'–5' tRNAHis guanylyltransferase in bacteria. FEBS Lett. 2010;584:3567–3572. doi: 10.1016/j.febslet.2010.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogg T, Mechold U, Malke H, Cashel M, Hilgenfeld R. Conformational antagonism between opposing active sites in a bifunctional RelA/SpoT homolog modulates (p)ppGpp metabolism during the stringent response [corrected] Cell. 2004;117:57–68. doi: 10.1016/s0092-8674(04)00260-0. [DOI] [PubMed] [Google Scholar]
- Juhling F, Morl M, Hartmann RK, Sprinzl M, Stadler PF, Putz J. tRNAdb 2009: compilation of tRNA sequences and tRNA genes. Nucleic Acids Res. 2009;37:D159–162. doi: 10.1093/nar/gkn772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindell D, Jaffe JD, Johnson ZI, Church GM, Chisholm SW. Photosynthesis genes in marine viruses yield proteins during host infection. Nature. 2005;438:86–89. doi: 10.1038/nature04111. [DOI] [PubMed] [Google Scholar]
- Lingohr E, Frost S, Johnson R. Determination of bacteriophage genome size by Pulsed-Field Gel Electrophoresis. In: Clokie MR, Kropinski AM, editors. Bacteriophages: Methods and Protocols, Volume 2: Molecular and applied aspects. Humana Press; New York: 2008. pp. 19–25. [DOI] [PubMed] [Google Scholar]
- Lobry JR. Asymmetric substitution patterns in the two DNA strands of bacteria. Mol Biol Evol. 1996;13:660–665. doi: 10.1093/oxfordjournals.molbev.a025626. [DOI] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu LD, Sun Q, Fan XY, Zhong Y, Yao YF, Zhao GP. Mycobacterial MazG is a novel NTP pyrophosphohydrolase involved in oxidative stress response. J Biol Chem. 2010;285:28076–28085. doi: 10.1074/jbc.M109.088872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann NH. Phages of the marine cyanobacterial picophytoplankton. FEMS Microbiol Rev. 2003;27:17–34. doi: 10.1016/S0168-6445(03)00016-0. [DOI] [PubMed] [Google Scholar]
- Mann NH, Clokie MR, Millard A, Cook A, Wilson WH, Wheatley PJ, et al. The genome of S-PM2, a “photosynthetic” T4-type bacteriophage that infects marine Synechococcus strains. J Bacteriol. 2005;187:3188–3200. doi: 10.1128/JB.187.9.3188-3200.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middelboe M, Jacquet S, Weinbauer M. Viruses in freshwater ecosystems: an introduction to the exploration of viruses in new aquatic habitats. Freshwater Biology. 2008;53:1069–1075. [Google Scholar]
- Millard AD, Zwirglmaier K, Downey MJ, Mann NH, Scanlan DJ. Comparative genomics of marine cyanomyoviruses reveals the widespread occurrence of Synechococcus host genes localized to a hyperplastic region: implications for mechanisms of cyanophage evolution. Environ Microbiol. 2009;11:2370–2387. doi: 10.1111/j.1462-2920.2009.01966.x. [DOI] [PubMed] [Google Scholar]
- Miller ES, Kutter E, Mosig G, Arisaka F, Kunisawa T, Ruger W. Bacteriophage T4 genome. Microbiol Mol Biol Rev. 2003a;67:86–156. doi: 10.1128/MMBR.67.1.86-156.2003. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller ES, Heidelberg JF, Eisen JA, Nelson WC, Durkin AS, Ciecko A, et al. Complete genome sequence of the broad-host-range vibriophage KVP40: comparative genomics of a T4-related bacteriophage. J Bacteriol. 2003b;185:5220–5233. doi: 10.1128/JB.185.17.5220-5233.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi K, Bonnefond L, Kimura S, Suzuki T, Ishitani R, Nureki O. Structural basis for translational fidelity ensured by transfer RNA lysidine synthetase. Nature. 2009;461:1144–1148. doi: 10.1038/nature08474. [DOI] [PubMed] [Google Scholar]
- Nolan JM, Petrov V, Bertrand C, Krisch HM, Karam JD. Genetic diversity among five T4-like bacteriophages. Virol J. 2006;3:30. doi: 10.1186/1743-422X-3-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paerl HW, Huisman J. Climate change: a catalyst for global expansion of harmful cyanobacterial blooms. Environ Microbiol Rep. 2009;1:27–37. doi: 10.1111/j.1758-2229.2008.00004.x. [DOI] [PubMed] [Google Scholar]
- Potrykus K, Cashel M. (p)ppGpp: still magical? Annu Rev Microbiol. 2008;62:35–51. doi: 10.1146/annurev.micro.62.081307.162903. [DOI] [PubMed] [Google Scholar]
- Robbertse B, Reeves JB, Schoch CL, Spatafora JW. A phylogenomic analysis of the Ascomycota. Fungal Genet Biol. 2006;43:715–725. doi: 10.1016/j.fgb.2006.05.001. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Brito B, Li L-L, Wegley L, et al. Viral and microbial community dynamics in four aquatic environments. ISME J. 2010;4:739–51. doi: 10.1038/ismej.2010.1. [DOI] [PubMed] [Google Scholar]
- Rohwer F. Global phage diversity. Cell. 2003;113:141. doi: 10.1016/s0092-8674(03)00276-9. [DOI] [PubMed] [Google Scholar]
- Rohwer F, Prangishvili D, Lindell D. Roles of viruses in the environment. Environ Microbiol. 2009;11:2771–2774. doi: 10.1111/j.1462-2920.2009.02101.x. [DOI] [PubMed] [Google Scholar]
- Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream MA, Barrell B. Artemis: sequence visualization and annotation. Bioinformatics. 2000;16:944–945. doi: 10.1093/bioinformatics/16.10.944. [DOI] [PubMed] [Google Scholar]
- Sano E, Carlson S, Wegley L, Rohwer F. Movement of viruses between biomes. Appl Environ Microbiol. 2004;70:5842–5846. doi: 10.1128/AEM.70.10.5842-5846.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevostyanova A, Artsimovitch I. Functional analysis of Thermus thermophilus transcription factor NusG. Nucleic Acids Res. 2010 doi: 10.1093/nar/gkq623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Short CM, Suttle CA. Nearly identical bacteriophage structural gene sequences are widely distributed in both marine and freshwater environments. Appl Environ Microbiol. 2005;71:480–486. doi: 10.1128/AEM.71.1.480-486.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MC, Burns N, Sayers JR, Sorrell JA, Casjens SR, Hendrix RW. Bacteriophage collagen. Science. 1998;279:1834. doi: 10.1126/science.279.5358.1831g. [DOI] [PubMed] [Google Scholar]
- Squires CL, Zaporojets D. Proteins shared by the transcription and translation machines. Annu Rev Microbiol. 2000;54:775–798. doi: 10.1146/annurev.micro.54.1.775. [DOI] [PubMed] [Google Scholar]
- Srivatsan A, Wang JD. Control of bacterial transcription, translation and replication by (p)ppGpp. Curr Opin Microbiol. 2008;11:100–105. doi: 10.1016/j.mib.2008.02.001. [DOI] [PubMed] [Google Scholar]
- Sullivan MB, Coleman ML, Weigele P, Rohwer F, Chisholm SW. Three Prochlorococcus cyanophage genomes: signature features and ecological interpretations. PLoS Biol. 2005;3:e144. doi: 10.1371/journal.pbio.0030144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan MB, Lindell D, Lee JA, Thompson LR, Bielawski JP, Chisholm SW. Prevalence and evolution of core photosystem II genes in marine cyanobacterial viruses and their hosts. PLoS Biol. 2006;4:e234. doi: 10.1371/journal.pbio.0040234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan MB, Coleman ML, Quinlivan V, Rosenkrantz JE, DeFrancesco AS, Tan G, Fu R, Lee JA, Waterbury JB, Bielawski JP, Chisholm SW. Portal protein diversity and phage ecology. Environ. Microbiol. 2008;10:2810–23. doi: 10.1111/j.1462-2920.2008.01702.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan MB, Huang KH, Ignacio-Espinoza JC, Berlin AM, Kelly L, Weigele PR, et al. Genomic analysis of oceanic cyanobacterial myoviruses compared with T4-like myoviruses from diverse hosts and environments. Environ Microbiol. 2010 doi: 10.1111/j.1462-2920.2010.02280.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suttle CA. Viruses in the sea. Nature. 2005;437:356–361. doi: 10.1038/nature04160. [DOI] [PubMed] [Google Scholar]
- Suttle CA. Marine viruses--major players in the global ecosystem. Nat Rev Microbiol. 2007;5:801–812. doi: 10.1038/nrmicro1750. [DOI] [PubMed] [Google Scholar]
- Tettelin H, Radune D, Kasif S, Khouri H, Salzberg SL. Optimized multiplex PCR: efficiently closing a whole-genome shotgun sequencing project. Genomics. 1999;62:500–507. doi: 10.1006/geno.1999.6048. [DOI] [PubMed] [Google Scholar]
- Wang C, Sobral BW, Williams KP. Loss of a universal tRNA feature. J Bacteriol. 2007;189:1954–1962. doi: 10.1128/JB.01203-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Murase J, Asakawa S, Kimura M. Unique viral capsid assemply protein gene (g20) of cyanophages in the floodwater of a Japanese paddy field. Biol Fertil Soils. 2010;46:93–102. [Google Scholar]
- Weigele PR, Pope WH, Pedulla ML, Houtz JM, Smith AL, Conway JF, et al. Genomic and structural analysis of Syn9, a cyanophage infecting marine Prochlorococcus and Synechococcus. Environ Microbiol. 2007;9:1675–1695. doi: 10.1111/j.1462-2920.2007.01285.x. [DOI] [PubMed] [Google Scholar]
- Weinbauer MG. Ecology of prokaryotic viruses. FEMS Microbiol Rev. 2004;28:127–181. doi: 10.1016/j.femsre.2003.08.001. [DOI] [PubMed] [Google Scholar]
- Wiens JJ. Missing data and the design of phylogenetic analyses. J Biomed Informatics. 2006;39:34–42. doi: 10.1016/j.jbi.2005.04.001. [DOI] [PubMed] [Google Scholar]
- Wilhelm SW, Matteson AR. Freshwater and marine virioplankton: a brief overview of commonalities and differences. Freshwater Biology. 2008;53:1076–1089. [Google Scholar]
- Wilhelm SW, Carberry MJ, Eldridge ML, Poorvin L, Saxton MA, Doblin MA. Marine and freshwater cyanophages in a Laurentian Great Lake: evidence from infectivity assays and molecular analyses of g20 genes. Appl Environ Microbiol. 2006;72:4957–4963. doi: 10.1128/AEM.00349-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wommack KE, Colwell RR. Virioplankton: viruses in aquatic ecosystems. Microbiol Mol Biol Rev. 2000;64:69–114. doi: 10.1128/mmbr.64.1.69-114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakhnin AV, Yakhnin H, Babitzke P. Function of the Bacillus subtilis transcription elongation factor NusG in hairpin-dependent RNA polymerase pausing in the trp leader. Proc Natl Acad Sci U S A. 2008;105:16131–16136. doi: 10.1073/pnas.0808842105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan W, Francklyn C. Cytosine 73 is a discriminator nucleotide in vivo for histidyl tRNA in Escherichia coli. J Biol Chem. 1994;269:10022–10027. [PubMed] [Google Scholar]
- Yoshida T, Nagasaki K, Takashima Y, Shirai Y, Tomaru Y, Takao Y, et al. Ma-LMM01 infecting toxic Microcystis aeruginosa illuminates diverse cyanophage genome strategies. J Bacteriol. 2008;190:1762–1772. doi: 10.1128/JB.01534-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeidner G, Bielawski JP, Shmoish M, Scanlan DJ, Sabehi G, Béjà O. Potential photosynthesis gene recombination between Prochlorococcus and Synechococcus via viral intermediates. Environ. Microbiol. 2005;7:1505–13. doi: 10.1111/j.1462-2920.2005.00833.x. [DOI] [PubMed] [Google Scholar]
- Zhang J, Inouye M. MazG, a nucleoside triphosphate pyrophosphohydrolase, interacts with Era, an essential GTPase in Escherichia coli. J Bacteriol. 2002;184:5323–5329. doi: 10.1128/JB.184.19.5323-5329.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Zhang Y, Inouye M. Thermotoga maritima MazG protein has both nucleoside triphosphate pyrophosphohydrolase and pyrophosphatase activities. J Biol Chem. 2003;278:21408–21414. doi: 10.1074/jbc.M213294200. [DOI] [PubMed] [Google Scholar]
- Zhaxybayeva O, Doolittle WF, Papke RT, Gogarten JP. Intertwined evolutionary histories of marine Synechococcus and Prochlorococcus marinus. Genome Bio Evol. 2009;1:325–39. doi: 10.1093/gbe/evp032. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.