

Abstract
Background:
Inflammation and genetic instability are enabling characteristics of prostate carcinoma (PCa). Inactivation of the tumour suppressor gene phosphatase and tensin homolog (PTEN) is prevalent in early PCa. The relationship of PTEN deficiency to inflammatory signalling remains to be characterised.
Objective:
To determine how loss of PTEN functionality modulates expression and efficacy of clinically relevant, proinflammatory chemokines in PCa.
Design, setting, and participants:
Experiments were performed in established cell-based PCa models, supported by pathologic analysis of chemokine expression in prostate tissue harvested from PTEN heterozygous (Pten+/−) mice harbouring inactivation of one PTEN allele.
Interventions:
Small interfering RNA (siRNA)–or small hairpin RNA (shRNA)–directed strategies were used to repress PTEN expression and resultant interleukin-8 (CXCL8) signalling, determined under normal and hypoxic culture conditions.
Outcome measurements and statistical analysis:
Changes in chemokine expression in PCa cells and tissue were analysed by real-time polymerase chain reaction (PCR), immunoblotting, enzyme-linked immunosorbent assay (ELISA), and immunohistochemistry; effects of chemokine signalling on cell function were assessed by cell cycle analysis, apoptosis, and survival assays.
Results and limitations:
Transient (siRNA) or prolonged (shRNA) PTEN repression increased expression of CXCL8 and its receptors, chemokine (C-X-C motif) receptor (CXCR) 1 and CXCR2, in PCa cells. Hypoxia-induced increases in CXCL8, CXCR1, and CXCR2 expression were greater in magnitude and duration in PTEN-depleted cells. Autocrine CXCL8 signalling was more efficacious in PTEN-depleted cells, inducing hypoxia-inducible factor-1 (HIF-1) and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) transcription and regulating genes involved in survival and angiogenesis. Increased expression of the orthologous chemokine KC was observed in regions displaying atypical cytologic features in Pten+/− murine prostate tissue relative to normal epithelium in wild-type PTEN (PtenWT) glands. Attenuation of CXCL8 signalling decreased viability of PCa cells harbouring partial or complete PTEN loss through promotion of G1 cell cycle arrest and apoptosis. The current absence of clinical validation is a limitation of the study.
Conclusions:
PTEN loss induces a selective upregulation of CXCL8 signalling that sustains the growth and survival of PTEN-deficient prostate epithelium.
Keywords: Inflammation, Apoptosis, CXCL8, CXCR2, Hypoxia, Prostate cancer, PTEN
1. Introduction
Inactivation of phosphatase and tensin homolog (PTEN), a haplo-insufficient tumour suppressor gene (TSG), is prevalent in prostate cancer (PCa). PTEN is the most frequently mutated gene in metastatic PCa [1]: Promoter methylation or loss of heterozygosity underpin additional loss of PTEN expression in 5–27% of localised and 30–60% of metastatic PCa [2-4]. In experimental models, loss of one PTEN allele drives prostate epithelium towards hyperplasia and dysplasia and ultimately premalignant prostatic intraepithelial neoplasia (PIN) lesions. Loss of both PTEN alleles or the loss of additional TSGs leads to the development of invasive carcinoma [5,6]. Clinically, PTEN mutations are associated with aggressive, therapy-resistant tumours [7,8]. However, the molecular basis underpinning tumour progression, therapy resistance, and metastasis resulting from PTEN loss remains poorly characterised.
Chemokines underpin intercellular communication within the tumour microenvironment [9,10]. Their tissue-selective expression and chemoattractant action promotes site-selective metastasis of breast and prostate cancers [11-13]. Chemokine (C-C motif) ligand 2 (CCL2), interleukin-12 (CXCL12), and interleukin-8 (CXCL8) are three principal chemokines associated with PCa [10,12,13]. CCL2 and CXCL12 are expressed in prostate cell lines as well as primary and metastatic prostate tumours [12,14-16]. Inhibition of CXCL12/chemokine (C-X-C motif) receptor (CXCR) 4 or CCL2/chemokine (C-C motif) receptor 2 (CCR2) signalling attenuates metastasis in experimental PCa models [12,13]. Overexpression of the proinflammatory, proangiogenic chemokine CXCL8 and its receptors CXCR1 and CXCR2 is detected in prostate biopsy tissue; deregulated expression is detected in PIN foci and increases throughout disease progression [17]. Autocrine CXCL8 signalling promotes castration resistance [18] and antagonises the response to therapeutics used in the treatment of advanced disease [18-20].
CCL2, CXCL12, and CXCL8 are induced in response to stress or damage, through activation of AP-1, nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), and hypoxia-inducible factor-1 (HIF-1) [19,21,22]. Hypoxia, an environmental stress relevant to prostate tumour biology [23], induces chemokine and chemokine receptor expression in PCa cells [21,22], and elevated CXCL8 expression occurs adjacent to necrotic zones within tumours [21,24].
Inflammation is a recognised risk factor in PCa development and progression [25]. Many inflammatory cytokines and chemokines functionally underpin various “hallmarks of cancer” [26]. Our objective was to characterise whether PTEN inactivation influences chemokine expression and signalling in PCa cells and to determine its functional significance. Our study characterises a selective induction of CXCL8 signalling and its importance in maintaining the survival of PTEN-deficient PCa cells.
2. Materials and methods
2.1. Cell culture
PCa cell lines were sourced and cultured as described [19]. Hypoxic treatments (0.1% oxygen) were performed as described [22]. Where relevant, cells were treated with 3 nM recombinant human (rh)-CXCL8 (PeproTech, London, UK) or 10 μM MK2206 (Iain James, Almac Discovery, Craigavon, Northern Ireland).
2.2. Generation of PTEN-reconstituted PC3 cells and PTEN-depleted DU145 cells
PC3-PTEN–inducible cells were generated using the ViraPower T-REx Lentiviral Expression System (Life Technologies, Carlsbad, CA, USA; see Appendix). PTEN-depleted DU145 cell lines were generated using HuSH–small hairpin RNA (shRNA) constructs (see Appendix).
2.3. Quantitative real-time polymerase chain reaction
RNA was harvested and reverse-transcribed as described [22]. Primer sequences are provided in the Appendix. Quantitative real-time polymerase chain reaction (q-PCR) was performed using triplicate analysis on a LightCycler 480 instrument (Roche Diagnostics, Basel, Switzerland). Expression was normalised against 18S.
2.4. Immunoblotting
Lysates were prepared, resolved, and blotted as described [18]. Antibodies and detection reagents are as described in the Appendix.
2.5. Small interfering RNA and small hairpin RNA transfections
PTEN or CXCR1/CXCR2 were downregulated using oligonucleotide pools at final concentrations of 10 nM and 2 nM, respectively (Dharmacon, Lafayette, CO, USA). CXCL8 expression was downregulated using two small interfering RNA (siRNA) sequences (0.1–20 nM; Qiagen, Crawley, West Sussex, UK). Nontargeting (NT) transfections were at the same concentrations as the siRNAs. Transfections were for 72–96 h. Target depletion was confirmed as follows: PTEN, immunoblotting; CXCR1/CXCR2, q-PCR; CXCL8, enzyme-linked immunosorbent assay (ELISA). In rescue experiments, rh-CXCL8 treatments were added every 24 h post-transfection. CXCL8 was downregulated using HuSH–shRNA plasmids (OriGene, Rockville, MD, USA) as per the manufacturer’s instructions. NT transfections were at the same concentration as the shRNA.
2.6. Clonogenic assays
Cells were seeded into six-well plates. After 10–14 d, colonies were fixed, stained with 0.4% crystal violet solution, and counted.
2.7. Luciferase reporter assays
NF-κB or HIF-1 luciferase assays were performed using the NF-κB-LUC-pGL3 [20] or HRE-LUC-pGL3 [27] plasmids, respectively, as described.
2.8. Enzyme-linked immunosorbent assay
Secreted CXCL8 in culture media was analysed by ELISA (Pelikine, Beckman Coulter, High Wycombe, UK), as described [22].
2.9. Flow cytometry
Cell surface CXCR1 or CXCR2 expression was detected using fluorescein isothiocyanate (FITC)-conjugated antibodies (R&D Systems, Abingdon, UK) and analysed by flow cytometry (Beckman Coulter, Buckinghamshire, UK), as described [22].
2.10. Cell cycle analysis
Propidium iodide staining and cell cycle analysis were performed as described [20].
2.11. Apoptosis assay
Apoptosis was measured using FITC Annexin V antibody (BD Biosciences, Oxford, UK) and analysed by flow cytometry as per the manufacturer’s instructions.
2.12. Animals
PTEN heterozygous (Pten+/−) mice, harbouring hemizygous inactivation of one PTEN allele [5], were provided by Pier Paolo Pandolfi (Memorial Sloan-Kettering Cancer Center/Beth Israel Deaconess Medical Center) and maintained according to the Institutional Animal Review Board of Georgetown University.
2.13. Immunohistochemistry
Prostate tissue sections taken from four wild type (WT) and four Pten+/− mice were deparaffinised, rehydrated, and antigen retrieval achieved by boiling (13 psi) in Tris-ethylenediaminetetraacetic acid buffer (pH 9.0). KC was detected using anti-KC antibody (1:25; Cambridge Bioscience, Cambridge, UK) and horseradish peroxidase (HRP)–labelled secondary antibody (EnVision System, Dako, Ely, UK). Sections were detected using diaminobenzidine (DAB+) substrate/chromogen system (EnVision System) and counterstained with Mayer’s haemalum
2.14. Statistical analysis
Means were compared using Mann-Whitney or a two-tailed student t test, as indicated.
3. Results
3.1. PTEN loss correlates with selective increases in CXCL8 and CXCR2
PTEN protein expression was examined in androgen-responsive (LNCaP, 22Rv1) and androgen-unresponsive (PC3, DU145) cell lines. PTEN expression was only observed in DU145 and 22Rv1 cells (Fig. 1A). Following PTEN depletion (Fig. 1B), CXCL8 mRNA expression was increased in DU145 (left panel) and 22Rv1 (right panel) cells. CCL2 or CXCL12 expression was unaffected (Fig. 1C). CXCR2 expression was also upregulated in both PTEN-expressing cell lines following PTEN loss. CCR2 or CXCR4 expression was unaffected (Fig. 1D). This selective upregulation of CXCL8 and CXCR2 suggests that autocrine CXCL8 signalling is the principal chemokine-signalling event resulting from PTEN loss in prostate epithelial cells.
Fig. 1.

Phosphatase and tensin homolog (PTEN) loss selectively increases expression of interleukin-8 (CXCL8) and its receptors in prostate cancer (PCa) cells. (A) Immunoblot characterising the expression of PTEN in four established PCa cells. Equivalent protein loading is confirmed by analysis of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) expression. (B) Immunoblots confirming the knockdown of PTEN expression in DU145 and 22Rv1 cells using gene-targeted (small interfering) oligonucleotides. Expression of PTEN is also shown for mock-transfected cells or cells transfected with a nontargeting (NT) sequence. Equal protein loading is confirmed by performing analysis of GAPDH expression. (C) Bar graphs presenting the results of quantitative polymerase chain reaction (q-PCR) analysis of chemokine (C-C motif) ligand 2, interleukin-12, or CXCL8 transcript levels in DU145 (left panel) or 22Rv1 cells (right panel) in cells transfected with NT or PTEN-targeted oligonucleotides. (D) Bar graphs presenting the results of q-PCR analysis of chemokine (C-C motif) receptor 2, chemokine (C-X-C motif) receptor (CXCR) 4, or CXCR2 transcript levels in DU145 (left panel) or 22Rv1 cells (right panel) in cells transfected with NT or PTEN-targeted oligonucleotides. Data shown is the mean plus or minus standard error of the mean value, calculated from a minimum of three independent experiments. Statistically significant differences in expression were determined using a two-tailed Mann-Whitney U test. PTEN = phosphatase and tensin homolog; GAPDH = glyceraldehyde 3-phosphate dehydrogenase; NT = nontargeting; siRNA = small interfering RNA; CCL2 = chemokine (C-C motif) ligand 2; CXCL12 = interleukin-12; CXCL8 = interleukin-8.
* p < 0.05.
3.2. Stress-induced interleukin chemokine/receptor expression correlates with PTEN status in prostate cancer cells
The relevance of PTEN status in modulating stress (hypoxia)–induced transcription of chemokine receptors and ligands was investigated. The q-PCR confirmed marked increases in CXCR1 and CXCR2 mRNA in hypoxic, PTEN-null PC3 (Fig. 2A and B, left panels) and LNCaP (Fig. 2A and B, right panels) cells. In contrast, we observed minimal increases in CXCR1/2 expression in PtenWT DU145 (Fig. 2A and B, left panels) or 22Rv1 (Fig. 2A and B, right panels) cells. Hypoxia-induced CXCL8 mRNA expression (Fig. 2C) and secretion (Fig. 2D and Supplemental Fig. 1a) were also significantly greater in PTEN-null than PTEN-expressing cells. Similarly, expression of the CXCR2-selective ligands CXCL1 (Supplemental Fig. 1b) and CXCL5 (Supplemental Fig. 1c) was more significantly upregulated in hypoxic PTEN-null cells.
Fig. 2.

Stress-induced expression of interleukin-8 (CXCL8)–related genes is potentiated in phosphatase and tensin homolog (PTEN)–null prostate cancer cells. Bar graphs presenting the results of quantitative polymerase chain reaction analysis determining (A) chemokine (C-C motif) receptor (CXCR) 1, (B) CXCR2, or (C) CXCL8 transcript levels in androgen-independent PC3 and DU145 cells (left panel) or androgen-dependent LNCaP and 22Rv1 cells (right panel) following exposure to hypoxia (0.1% oxygen) over a 24-h time course. Data points shown are the mean plus or minus standard error of the mean (SEM) value, calculated from a minimum of three independent experiments. Statistically significant differences in expression were determined using a two-tailed Mann-Whitney U test (* p < 0.05; ** p < 0.01). (D) Bar graph presenting enzyme-linked immunosorbent assay (ELISA) data measuring the concentration of CXCL8 in the cell media in androgen-independent PC3 cells (left panel) or androgen-dependent LNCaP cells (right panel). Values illustrated are mean plus or minus SEM (n = at least 5) for time-matched samples taken from cells cultured in normal culture conditions or those continuously exposed to hypoxia (0.1% oxygen) over the 24-h time course. Differences in ELISA values were determined by performing a two-tailed student t test (* p < 0.05; ** p < 0.01).
CXCL8 = interleukin-8.
We examined the effect of hypoxia upon CCL2/CCR2 expression. Hypoxia-induced increases in CCR2 mRNA levels were equivalent in PC3 and DU145 cells, while expression was unchanged in LNCaP or 22Rv1 cells (Supplemental Fig. 2a). Similarly, there was no significant difference in CCL2 mRNA expression between hypoxic PTEN-expressing and PTEN-null cells (Supplemental Fig. 2b). Therefore, although the stress-induced regulation of CXCL8 and related genes is clearly subject to PTEN status, this is not the case for the other principal PCa-associated chemokines.
3.3. PTEN loss regulates transcription factor activation and CXCL8/CXCL8 receptor expression in prostate cancer cells
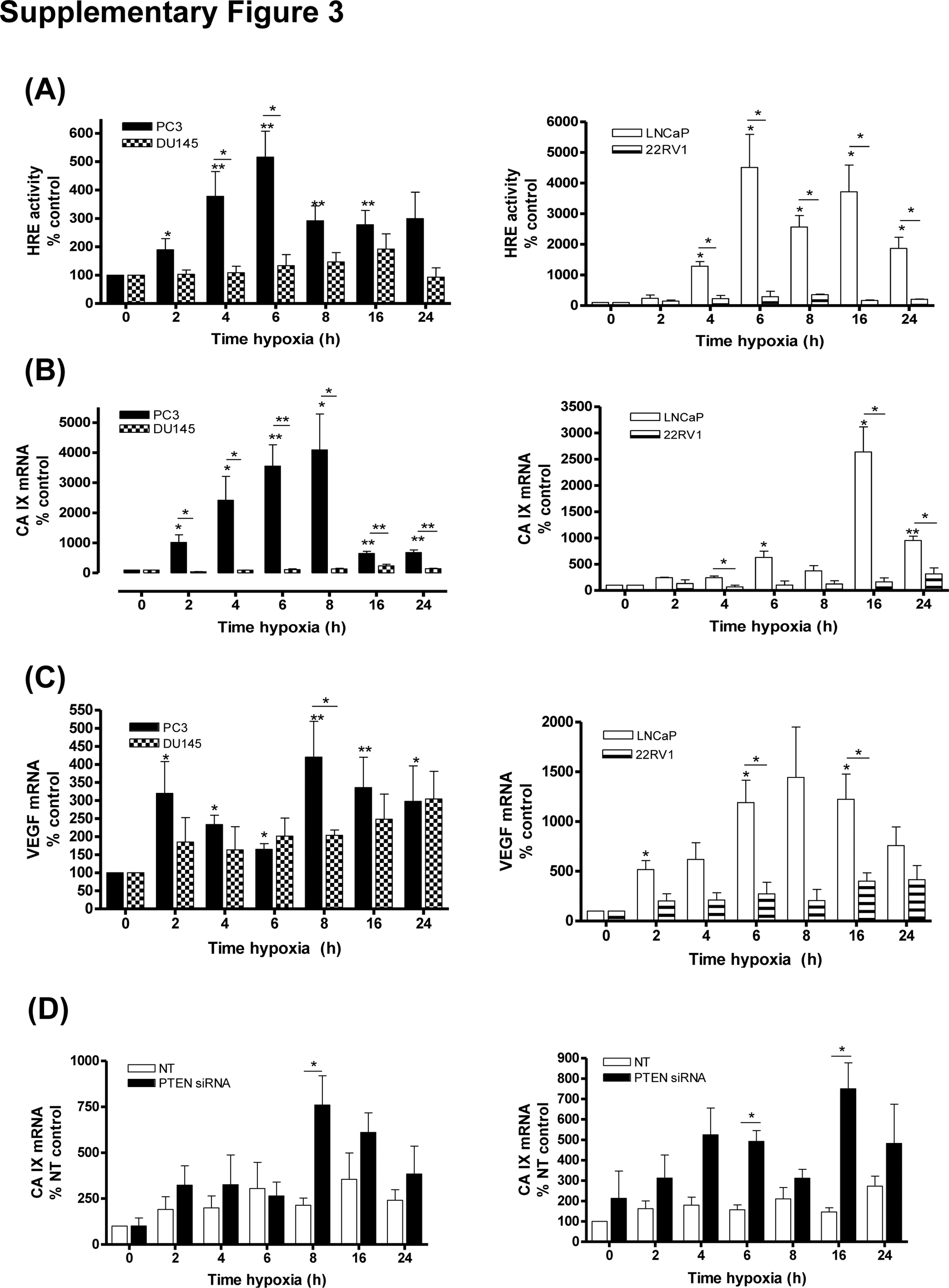
HIF-1 and NF-κB regulate CXCR1 and CXCR2 transcription [22]. Under hypoxia, HIF-1 activity (Supplemental Fig. 3a) and HIF-1 target gene expression (Supplemental Fig. 3b and c) were markedly increased only in PTEN-null cells; NFkB activity (Supplemental Fig. 4a) and NF-κB target gene expression (Supplemental Fig. 4b) were also induced in PTEN-null backgrounds (Supplemental Fig. 4a). In PTEN-depleted DU145 and 22Rv1 cells, hypoxia-induced carbonic anhydrase IX (CAIX; Supplemental Fig. 3d) or B-cell CLL/lymphoma 2 (Bcl-2; Supplemental Fig. 4c) mRNA expression was significantly greater than in PTEN-expressing cells. Hypoxia-induced changes in CAIX (Supplemental Fig. 5a) and Bcl-2 (Supplemental Fig. 5b) mRNA expression were attenuated when PTEN-null cells were pretreated with the Akt-inhibitor MK2206.
The role of PTEN in dampening stress-induced CXCL8 and receptor expression in PCa cells was also studied using PTEN-targeted siRNA. Increases in CXCR1 and CXCR2 mRNA expression were clearly detectable in hypoxic, PTENdepleted DU145 and 22Rv1 cells (Fig. 3A and B). Similarly, PTEN loss coincided with sustained increases in CXCL8 mRNA in hypoxic DU145 and 22Rv1 cells (Fig. 3C). Therefore, downregulation of PTEN promotes a stress-induced HIF-1 and NF-κB response to hypoxia, resulting in upregulation of CXCL8, CXCR1, and CXCR2.
Fig. 3.

Knockdown of phosphatase and tensin homolog (PTEN) potentiates hypoxia-driven transcription of interleukin-8 (CXCL8), chemokine (C-C motif) receptor (CXCR) 1, and CXCR2 in prostate cancer cells. Expression of PTEN in DU145 or 22Rv1 cells was depleted using small interfering RNA (siRNA) prior to the cells being exposed to hypoxia. Bar graphs present the results of quantitative polymerase chain reaction analysis determining (A) CXCR1, (B) CXCR2, or (C) CXCL8 transcript levels in androgen-independent DU145 cells (left panel) or androgen-dependent 22Rv1 cells (right panel) following exposure to hypoxia (0.1% oxygen) over a 24-h time course. Data show the relative expression of the gene in nontargeting-transfected or PTEN siRNA-transfected conditions. Data points shown are the mean plus or minus the standard error of the mean value, calculated from a minimum of four independent experiments. The student t test was used to calculate the statistical significance of any observed differences.
NT = nontargeting; PTEN = phosphatase and tensin homolog; siRNA = small interfering RNA; CXCL8 = interleukin-8.
* p < 0.05.
** p < 0.01.
3.4. PTEN loss increases the efficacy of CXCL8 signalling in prostate cancer cells
To examine whether PTEN modulates sensitivity of PCa cells to CXCL8 stimulation, we measured CXCL8-induced transcription factor (TF) activation. Stimulation with rh-CXCL8 selectively increased HIF-1 activity in PTEN-null but not PtenWT cells (Fig. 4A), accompanied by a significant increase in vascular endothelial growth factor (VEGF) mRNA levels in PTEN-null cells (Supplemental Fig. 6a). The previously absent CXCL8-induced increase in VEGF expression was unmasked following attenuation of PTEN expression in DU145 (Fig. 4B, left panel) and 22Rv1 cells (Fig. 4B, right panel).
Fig. 4.

The efficacy of interleukin-8 (CXCL8) signalling is potentiated under phosphatase and tensin homolog (PTEN)–null or PTEN-depleted conditions. (A) Bar graphs presenting the relative change in hypoxia-responsive element–dependent luciferase activity in androgen-independent PC3 and DU145 cells (left panel) or androgen-dependent LNCaP and 22Rv1 cells (right panel) following exposure to 3 nM recombinant human (rh) CXCL8 over an 8-h time course. (B) Expression of PTEN in DU145 or 22Rv1 cells was depleted using small interfering RNA (siRNA) prior to the cells being treated with 3 nM rh-CXCL8. Bar graphs illustrate the quantitative polymerase chain reaction (q-PCR) analysis determining the expression of vascular endothelial growth factor transcript levels in androgen-independent DU145 cells (left panel) or androgen-dependent 22Rv1 cells (right panel) in nontargeting (NT) oligonucleotide-transfected cells or PTEN-targeted siRNA-transfected cells. (C) Bar graphs presenting the relative change in nuclear factor kappa-light-chain-enhancer of activated B cells–dependent luciferase activity in androgen-independent PC3 and DU145 cells (left panel) or androgen-dependent LNCaP and 22Rv1 cells (right panel) following exposure to 3 nM rh-CXCL8 over an 8-h time course. (D) Expression of PTEN in DU145 or 22Rv1 cells was depleted using siRNA prior to the cells being treated with 3 nM rh-CXCL8. Bar graphs illustrate the q-PCR analysis determining the expression of B-cell CLL/lymphoma 2 transcript levels in androgen-independent DU145 cells (left panel) or androgen-dependent 22Rv1 cells (right panel) in NT oligonucleotide-transfected cells or PTEN-targeted siRNA-transfected cells. Data points shown are the mean plus or minus standard error of the mean value, calculated from a minimum of three independent experiments. Statistically significant differences in expression were determined using a two-tailed Mann-Whitney U test. HRE = hypoxia-responsive element; CXCL8 = interleukin-8; VEGF = vascular endothelial growth factor; NT = nontargeting; PTEN = phosphatase and tensin homolog; si = small interfering; NF-κB = nuclear factor kappa-light-chain-enhancer of activated B cells.
* p < 0.05.
** p < 0.01.
*** p < 0.001.
CXCL8-induced NF-κB-luciferase activity was detected in both PTEN-null cell lin es (Fig. 4C), consistent with significant, sustained increases in Bcl-2 mRNA levels (Supplemental Fig. 6b). A dampened effect on NF-κB activity was observed in PtenWT cells (Fig. 4C). However, following attenuation of PTEN expression, CXCL8 induced a greater upregulation of Bcl-2 mRNA expression in DU145 and 22Rv1 cells (Fig. 4D).
3.5. Stable PTEN loss correlates with increased CXCL8 expression in vitro and KC expression in vivo
DU145 cells are heterozygous for PTEN [28]. DU145 clones displaying different degrees of further PTEN knockdown were generated. DU145-sh10.06 and DU145-sh11.02 showed approximately 50% and >95% knockdown of PTEN expression relative to parental or DU145-NT01 cells (Fig. 5A). PTEN loss correlated with increased CXCL8 expression (Fig. 5B) and secretion (Fig. 5C). CCL2 or CXCL12 expression was equivalent in the PTEN-depleted and DU145-NT01 clones (Fig. 5B). Flow cytometry demonstrated that PTEN loss corresponded with marked increases in CXCR1 and CXCR2 but not CXCR4 expression in PTEN-depleted clones (Supplemental Table 1).
Fig. 5.

Effects of dose-dependent phosphatase and tensin homolog (PTEN) loss on chemokine expression in vitro and in vivo models. (A) Immunoblot illustrating the expression of PTEN in the parental and clonal DU145 cells. The NT01 clone was selected following transfection with a nontargeting (NT) small hairpin RNA (shRNA), while Sh10.06 and Sh11.02 clones were selected following transfection with a PTEN-targeted shRNA vector. Expression of PTEN is shown relative to expression of glyceraldehyde 3-phosphate dehydrogenase. (B) Bar graphs presenting the results of quantitative polymerase chain reaction analysis examining basal interleukin-8 (CXCL8), chemokine (C-C motif) ligand 2 (CCL2), or interleukin-12 (CXCL12) transcript levels in the clonal DU145 cells. (C) Bar graph presenting enzyme-linked immunosorbent assay (ELISA)–based determination of CXCL8 secretion from each of the DU145 clones. Data shown in Fig. 5B and 5C are the mean plus or minus standard error of the mean value calculated from three independent experiments. (D) Characterisation of chemokine expression in wild type PTEN (PtenWT) and PTEN heterozygous (Pten+/−) murine prostate tissue. KC protein expression was determined by immunohistochemical analysis of murine prostate tissue harvested from PtenWT and Pten+/− mice. Images are as follows: (i, ii): normal epithelium in PtenWT (×400); (iii, iv): normal epithelium in Pten+/− (×630 and ×400, respectively); (v, vi): Pten+/− with cytologic atypia (×400); (vii, viii): Pten+/− with cytologic and architectural atypia (×400); and (ix, x): Pten+/− with high-grade prostatic intraepithelial neoplasia (×400). Semiquantitative analysis of the percentage of positively stained cells is shown.
PTEN = phosphatase and tensin homolog; NT = nontargeting; CXCL8 = interleukin-8; CCL2 = chemokine (C-C motif) ligand 2; CXCL12 = interleukin-12; PtenWT = wild type PTEN; Pten+/− = PTEN heterozygous; HG-PIN = high-grade prostatic intraepithelial neoplasia.
To validate our in vitro observations, we performed immunohistochemistry on prostate tissue from PtenWT or heterozygous Pten+/− mice. PtenWT prostate tissue exhibited negligible to low levels of the CXCL8-orthologous murine chemokine, KC (<1% in a semiquantitative fashion; Fig. 5D[i] and 5D[ii]). However, in Pten+/− mice, KC expression increased gradually from normal epithelium (<1%; Fig. 5D[iii] and 5D[iv]) through increasing degrees of architectural and cytological atypia (10–20%, Fig. 5D[v] and 5D[vi]; 50%, Fig. 5D[vii]and 5D[viii]) to well-established high-grade PIN (80%, Fig. 5D[ix] and 5D[x]).
3.6. Reconstitution of PTEN represses CXCL8 expression in PC3 cells
We further validated our findings using a novel cell model, exploiting tetracycline-driven expression of PTEN in PC3 cells (Supplemental Fig. 7a). Re-expression of PTEN resulted in a diminution of CXCL8 mRNA expression (Supplemental Fig. 7b) and secretion (Supplemental Fig. 7c).
3.7. Abrogating CXCL8 signalling is synthetically lethal in PTEN-deficient cells
The role of elevated CXCL8 signalling in maintaining viability of PTEN-depleted cells was examined using siRNA to attenuate CXCL8 expression in DU145 clones (Supplemental Fig. 8). PTEN-depleted clones displayed reduced viability following downregulation of CXCL8 (Fig. 6A). Experiments in PTEN-deficient PC3 cells confirmed a concentration-dependent reduction in viability following siRNA-mediated down-regulation of CXCL8 (Fig. 6B), and shRNA-mediated downregulation of CXCL8 (Supplemental Fig. 9a) or siRNA-mediated downregulation of CXCR1/2 (Supplemental Fig. 9b) also resulted in reduced viability in PTEN-null cells. This reduction in viability was partially rescued by the addition of exogenous rh-CXCL8 (Fig. 6C). The depletion of CXCL8 signalling induced an initial G1 arrest (Fig. 6D), followed by increased apoptosis (Fig. 6E) and the concurrent down-regulation of XIAP, Bcl-2, and c-FLIP mRNA expression (Fig. 6F) at later time points. These experiments indicate that the induction of autocrine CXCL8 signalling plays a central role in maintaining cell survival following PTEN loss.
Fig. 6.

Selective induction of interleukin-8 (CXCL8) signalling promotes cell survival in PTEN-depleted prostate cancer cells. (A) Expression of CXCL8 was repressed in clonal DU145 cells using two independent small interfering RNA (siRNA) oligonucleotides siIL8-1 and siIL8-2. Viability was determined by colony count assays. Bar graphs demonstrate the effect of attenuating CXCL8 expression on the viability of DU145-NT, DU145-sh10.06, and DU145-sh11.02 cells. Data shown are the mean plus or minus standard error of the mean (SEM) value calculated from four independent clonogenic experiments. (B) Bar graph showing the viability of PC3 cells following transient transfection with increasing concentrations of the CXCL8 siRNA, siIL8-1. (C) Bar graph illustrating the effect of exogenous recombinant human (rh)-CXCL8 administration upon the survival of CXCL8-depleted PC3 cells. In Fig. 6B and 6C, viability was determined by MTT assay. (D) Bar graph showing the cell cycle distribution of PC3 cells 72 h post-transfection with the CXCL8 siRNA, siIL8-1. Cell cycle distribution was determined by propidium iodide staining. (E) Bar graph illustrating the percentage of apoptotic cells 120 h post-transfection of PC3 cells with siIL8-1. Apoptosis was assessed by annexin V/PI staining. (F) Bar graph illustrating results from quantitative polymerase chain reaction analysis, demonstrating the expression of antiapoptotic genes following attenuation of CXCL8 expression in PC3 cells. For all experiments, data shown are the mean plus SEM value calculated from at least three independent experiments. Statistically significant differences were determined using a two-tailed student t test.
NT = nontargeting; IL-8 = interleukin-8; siRNA = small interfering RNA; CXCL8 = interleukin-8.
* p < 0.05.
** p < 0.01.
*** p < 0.001.
4. Discussion
Loss of PTEN function, through mutation or reduced expression, is prevalent in PCa [1] and underpins the initiation and early progression of prostate tumour development in mice [5,6]. Here, we demonstrate that PTEN loss promotes a selective increase in expression of the proin-flammatory chemokine CXCL8 in PCa cell lines and tissue. Our data confirm that reduced PTEN expression is sufficient to increase CXCL8 expression and secretion in PCa cells, consistent with prior observations in glioblastoma [29]. Our in vitro results are supported by the increased expression of KC, the murine ortholog of CXCL1 and CXCL8, in the prostate epithelium of Pten+/− mice. Importantly, increasing KC staining corresponded with the morphologic changes that occur during the transition from normal epithelium to premalignant lesions. In contrast, expression of CCL2/CCR2 and CXCL12/CXCR4 was independent of PTEN status, suggesting that these chemokines are not essential to the early stages of PCa development associated with PTEN loss. Instead, they may contribute to later stages of malignant progression. Although our data show that PTEN loss increases CXCL8 secretion, we do observe basal CXCL8 secretion in PTEN-expressing PCa cells, suggesting that additional factors (eg, AP-1) may regulate basal CXCL8 expression under PtenWT conditions but that gene expression is further induced upon PTEN loss.
The relevance of potentiating CXCL8 signalling was demonstrated in a series of in vitro experiments. Silencing CXCL8 in PTEN-null cells promoted a dramatic decrease in viability. Thus, we conclude that CXCL8 signalling contributes to cell survival during the earliest stages of tumour development arising from PTEN loss. CXCL8 signalling promotes activation of survival pathways [10], leading to upregulation of antiapoptotic proteins [19,20]. This study demonstrates that repressing autocrine CXCL8 signalling in PTEN-depleted cells triggers G1 cell cycle arrest and downregulation of antiapoptotic genes and subsequently promotes apoptosis in PTEN-depleted cells.
PTEN status also modulated the response of PCa cells to CXCL8. CXCL8-promoted NF-κB or HIF-1 activation was greater in magnitude and duration in PTEN-deficient cells. Increased CXCL8 signalling in PTEN-null cells may be partly the result of the observed increase in CXCR1/2 density. Moreover, increased CXCL8 signalling under PTEN-null conditions may result from the loss of regulation of PI3K, one of the major signalling pathways downstream of chemokine receptors [10]. Because CXCL8 signalling activates many TFs, including AR [18], NF-κB [19,20], and HIF-1, the resulting global gene expression changes may significantly affect disease progression. This coupling of CXCL8 signalling with TFs associated with cell survival is consistent with our observation of growth arrest and apoptosis following attenuation of CXCL8 signalling in PTEN-depleted cells. Moreover, because CXCL8 signalling modulates castration-resistant transition in PCa and decreases the efficacy of therapeutic agents [18-20], the induction of CXCL8 signalling may contribute to the development of treatment-resistant tumours.
Although this study has characterised the importance of autocrine CXCL8 signalling, the contribution of paracrine CXCL8 signalling should not be overlooked. CXCR1 and CXCR2 are expressed by tumour-associated stromal cells, rendering these cells responsive to tumour-derived CXCL8, CXCL1, or CXCL5 [10]. A sustained and elevated secretion of these chemokines will influence the microenvironment of PTEN-deficient tumours (eg, CXCL8 recruits neutrophils to initiate MMP-9–associated angiogenesis in prostate tumour xenografts) [30]. Attenuation of autocrine and paracrine CXCL8 signalling in PTEN-deficient prostate tumours may therefore have a significant impact on preventing tumour development or retarding tumour progression.
5. Conclusions
PTEN loss promotes a selective increase in the expression and efficacy of CXCL8 in prostate epithelium. The resultant increase in autocrine CXCL8 signalling significantly contributes to cell viability following the loss of PTEN in prostate epithelium. Targeting the autocrine and paracrine effects of CXCL8 may therefore provide a novel therapeutic approach to treating PTEN-null PCa.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgement statement
The authors would like to thank Mr. Ken Arthur (Tissue Core Technology Unit, CCRCB) for assistance with tissue-based studies.
Funding/Support and role of the sponsor: This work was supported by research grants to Dr. Waugh from Cancer Research UK [C11512/A11825 and C212/A11342], the Medical Research Council [MR/J007641/1], the McClay Foundation (Dr. Neisen), the Ulster Cancer Foundation, Friends of the Cancer Centre, and the Department of Employment and Learning in Northern Ireland (Dr. McKechnie) as well as a grant from the National Institutes of Health R01 CA129003 to Dr. Albanese. These sponsors contributed to the design and conduct of the study as well as the collection, analysis, and interpretation of the data.
Appendix A. Supplemental materials and methods
A.1. Generation of PTEN-knockdown DU145 clones
The DU145-NT01, DU145-sh10.06, and DU145-sh11.02 clones were generated by stable downregulation of PTEN using commercially available shRNA plasmids (HuSH shRNA constructs in pGFP-V-RS vector; OriGene), according to the manufacturers’ instructions. Briefly, DU145 cells (5 × 105) were incubated in a transfection mix comprising plasmid (4 μg), Lipofectamine 2000, and Opti-MEM medium (Life Technologies). A separate transfection with a nontargeting sequence was included in these experiments at the same concentration as the shRNA sequence used. At 24-h post-transfection, cells were trypsinised, counted, and reseeded, and cells that stably incorporated the shPTEN plasmids were selected using puromycin (0.5 μg/ml). Individual colonies were selected and expanded, and PTEN expression was validated by Western blot. Cells were maintained in RPMI-1640 media, supplemented with foetal calf serum (FCS) and L-glutamine, and maintained in 0.5 μg/ml puromycin.
A.2. Generation of the PC3-PTEN–inducible cell line
The PC3-PTEN–inducible cell line was generated using the ViraPower T-REx Lentiviral Expression System. Briefly, a PTEN expression construct was acquired from Genart in the Gateway vector pDONR221. This was used to generate a tetracycline-inducible construct in the pLenti4.0/TO/V5-DEST vector using the Gateway cloning system from Invitrogen, as per the manufacturers’ instructions. 293FT cells were transfected with pLenti/tetracycline repressor (TR) and PTEN-pLenti4.0/TO/V5-DEST using Lipofectamine 2000 and viral particles harvested. PC3 cells were sequentially infected with the TR (Plenti6/TR) followed by the PTEN-pLenti4.0/TO/V5-DEST viral particles 24 h later. Cells were allowed to recover for a day, and clones were selected using 10 μg/ml blasticidin and 300 μg/ml Zeocin (Life Technologies) to select for PTEN-pLenti4.0/TO/V5-DEST and the TR, respectively. Cells were selected for 2 wk and colonies picked and induced for PTEN expression using 1 μg/ml tetracycline. Cells were maintained in F-12K nutrient mixture, Kaighn’s modification supplemented with FCS, penicillin-streptomycin, and L-glutamine and maintained in 10 μg/ml blasticidin and 300 μg/ml Zeocin.
A.3. Real-time polymerase chain reaction primer sequences
Primer sequences for q-PCR were as follows: 18S: Forward; 5′-CATTCGTATTGCGCCGCT-3′; Reverse; 5′-CGACGGTATCT-GATCGTC-3′: Bcl2: Forward 5′-AAAGGACCTGATCATTGGGG-3; Reverse: 5′-CAACTCTTTTCCTCCCACCA-3′: Carbonic anhydrase IX: Forward: 5′-AGGGACAAAGAAGGGGATGA-3′; Reverse: 5′-AGTTGCACACTGTGGCCATT-3′: CCL2: forward: 5′-AAAGTCTCTGCCGCCCTTCTGTG-3′; Reverse: 5′-AACAG-CAGGTGACTGGGGCAT-3′: CCR2: Forward: 5′-GGTCATCTGC-TACTCGGGAATC-3′; Reverse: 5′-TGCCTCTTCTTCTCGTTTC-GA-3′: c-FLIPL: Forward: 5′-CCTAGGAATCTGCCTGATAAT-CGA-3′; Reverse: 5′-TGCGATATACCATGCATACTGAGA-3: c-FLIPS: Forward: 5′-GCAGCAATCCAAAAGAGTCTCA-3′; Reverse: 5′-ATTTCCAAGAATTTTCAGATCAGG-3′: CXCL-8: Forward: 5′-ATGACTTCCAAGCTGGCCGTGG-3′; Reverse: 5′-CATAATTTCTGTTTGGCGCAGTGTGG-3′: CXCL1: Forward: 5′-CGCCCAAACCGAAGTCAT-3′; Reverse: 5′-GCAGGATT-GAGGCAAGCTTTC-3′: CXCL5: Forward: 5′-CAGACCACG-CAAGGAGTTCA-3′; Reverse: 5′-GGGCCTATGGCGAACACTT-3′: CXCL12: Forward: 5′-GCCCTTCAGATTGTAGCCCGGCT-3′; Reverse: 5′-TCCACTTTAGCTTCGGGTCAATGCA-3′: CXCR1: Forward: 5′-TGCATCAGTGTGGACCGTTA-3′; Reverse: 5′-TG-TCATTTCCCAGGACCTCA-3′: CXCR2: Forward: 5′-TGCATCA-GTGTGGACCGTTA-3′; Reverse: 5′-CCGCCAGTTTGCTGTAT-TG-3′: CXCR4: Forward: 5′-AAGTCCACGTTCCTCAAGCA-3′; Reverse: 5′-TTCTCGAAGGCCATCAGGA-3′: HIF-1α: Forward; 5′-TGGCCTTGTGAAAAAGGGT-3′; Reverse; 5′-TTGATGGGT-GAGGAATGGGT-3′: VEGF: Forward: 5′-AGCTACTGCCATC-CAATCGA; Reverse: 5′-GGTGAGGTTTGATCCGCATA-3′.
A.4. Immunoblotting
Anti-PTEN, anti-AKT, and anti-phosphoserine-473-AKT antibodies (Cell Signaling, Beverly, MA, USA) were used to detect the corresponding proteins. Anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) confirmed equal loading (Biogenesis, Dorset, UK). In all experiments, the secondary antibody was a HRP-labelled secondary antibody (GE Healthcare UK, Buckinghamshire, UK), and the detection reagent was ECL Plus reagents (GE Healthcare).
Footnotes
Study concept and design: Waugh, Maxwell, Albanese, Kennedy, Salto-Tellez.
Acquisition of data: Maxwell, Coulter, Walker, McKechnie, Neisen, McCabe.
Analysis and interpretation of data: Maxwell, Salto-Tellez.
Drafting of the manuscript: Maxwell, Waugh.
Critical revision of the manuscript for important intellectual content: Albanese, Kennedy.
Statistical analysis: Maxwell.
Obtaining funding: Waugh.
Administrative, technical, or material support: None.
Supervision: Maxwell, Waugh.
Other (specify): None.
Financial disclosures: David J.J. Waugh certifies that all conflicts of interest, including specific financial interests and relationships and affiliations relevant to the subject matter or materials discussed in the manuscript (eg, employment/affiliation, grants or funding, consultancies, honoraria, stock ownership or options, expert testimony, royalties, or patents filed, received, or pending), are the following: None.
Appendix B. Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.eururo.2012.08.032.
References
- [1].Zhao H, Dupont J, Yakar S, Karas M, LeRoith D. PTEN inhibits cell proliferation and induces apoptosis by down-regulating cell surface IGF-1R expression in CaP cells. Oncogene. 2004;23:786–94. doi: 10.1038/sj.onc.1207162. [DOI] [PubMed] [Google Scholar]
- [2].Feilotter HE, Nagai MA, Boag AH, Eng C, Mulligan LM. Analysis of PTEN and the 10q23 region in primary prostate carcinomas. Oncogene. 1998;16:1743–8. doi: 10.1038/sj.onc.1200205. [DOI] [PubMed] [Google Scholar]
- [3].Suzuki H, Freije D, Nusskern DR, et al. Interfocal heterogeneity of PTEN/MMAC1 gene alterations in multiple metastatic CaP tissues. Cancer Res. 1998;58:204–9. [PubMed] [Google Scholar]
- [4].Wang SI, Parsons R, Ittmann M. Homozygous deletion of the PTEN tumor suppressor gene in a subset of prostate adenocarcinomas. Clin Cancer Res. 1998;4:811–5. [PubMed] [Google Scholar]
- [5].Trotman LC, Niki M, Dotan ZA, et al. PTEN dose dictates cancer progression in the prostate. PloS Biol. 2003;1:E59. doi: 10.1371/journal.pbio.0000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Chen Z, Trotman LC, Shaffer D, et al. Crucial role of p53-dependent cellular senescence in suppression of PTEN-deficient tumorigenesis. Nature. 2005;436:725–30. doi: 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Huang H, Cheville JC, Pan Y, Roche PC, Schmidt LJ, Tindall DJ. PTEN induces chemosensitivity in PTEN-mutated CaP cells by suppression of Bcl-2. J Biol Chem. 2001;276:38830–6. doi: 10.1074/jbc.M103632200. [DOI] [PubMed] [Google Scholar]
- [8].Anai S, Goodison S, Shiverick K, Iczkowski K, Tanaka M, Rosser CJ. Combination of PTEN gene therapy and radiation inhibits the growth of human CaP xenografts. Hum Gene Ther. 2006;17:975–84. doi: 10.1089/hum.2006.17.975. [DOI] [PubMed] [Google Scholar]
- [9].Allevena P, Germano G, Marchesi F, Mantovani A. Chemokines in cancer related inflammation. Exp Cell Res. 2011;317:664–73. doi: 10.1016/j.yexcr.2010.11.013. [DOI] [PubMed] [Google Scholar]
- [10].Waugh DJJ, Wilson C. The interleukin-8 signaling pathway in cancer. Clin Cancer Res. 2008;14:6735–41. doi: 10.1158/1078-0432.CCR-07-4843. [DOI] [PubMed] [Google Scholar]
- [11].Müller A, Homey B, Soto H, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50–6. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- [12].Sun X, Cheng G, Hao M, et al. CXCL12/CXCR4/CXCR7 chemokine axis and cancer progression. Cancer Metastasis Rev. 2010;29:709–22. doi: 10.1007/s10555-010-9256-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Zhang J, Lu Y, Pienta KJ. Multiple roles of chemokine (C-Cmotif) ligand 2 in promoting CaP growth. J Nat Cancer Inst. 2010;102:522–8. doi: 10.1093/jnci/djq044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Loberg RD, Day LL, Harwood J, et al. CCL2 is a potent regulator of CaP cell migration and proliferation. Neoplasia. 2006;8:578–86. doi: 10.1593/neo.06280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sun YX, Wang J, Shelburne CE, et al. Expression of CXCR4 and CXCL12 (SDF-1) in human CaPs (PCa) in vivo. J Cell Biochem. 2003;89:462–73. doi: 10.1002/jcb.10522. [DOI] [PubMed] [Google Scholar]
- [16].Begley LA, Kasina S, MacDonald J, Macoska JA. The inflammatory microenvironment of the aging prostate facilitates cellular proliferation and hypertrophy. Cytokine. 2008;43:194–209. doi: 10.1016/j.cyto.2008.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Murphy C, McGurk M, Pettigrew J, et al. Nonapical and cytoplasmic expression of interleukin-8, CXCR1 and CXCR2 correlates with cell proliferation and microvessel density in CaP. Clin Cancer Res. 2005;11:4117–27. doi: 10.1158/1078-0432.CCR-04-1518. [DOI] [PubMed] [Google Scholar]
- [18].Seaton A, Scullin P, Maxwell PJ, et al. Interleukin-8 signaling promotes androgen-independent proliferation of CaP cells via induction of androgen receptor expression and activation. Carcinogenesis. 2008;29:1148–56. doi: 10.1093/carcin/bgn109. [DOI] [PubMed] [Google Scholar]
- [19].Wilson C, Purcell C, Seaton A, et al. Chemotherapy-induced CXC-chemokine/CXC-chemokine receptor signaling in metastatic CaP cells confers resistance to oxaliplatin through potentiation of nuclear factor-kappaB transcription and evasion of apoptosis. J Pharm Exp Ther. 2008;327:746–59. doi: 10.1124/jpet.108.143826. [DOI] [PubMed] [Google Scholar]
- [20].Wilson C, Wilson T, Johnston PG, Longley DB, Waugh DJ. Interleukin-8 signaling attenuates TRAIL- and chemotherapy-induced apoptosis through transcriptional regulation of c-FLIP in CaP cells. Mol Cancer Ther. 2008;7:2649–61. doi: 10.1158/1535-7163.MCT-08-0148. [DOI] [PubMed] [Google Scholar]
- [21].Xu L, Pathak PS, Fukumura D. Hypoxia-induced activation of p38 mitogen-activated protein kinase and phosphatidylinositol-3 kinase signaling pathways contributes to expression of interleukin-8 in human ovarian cancer cells. Clin Cancer Res. 2004;10:701–7. doi: 10.1158/1078-0432.ccr-0953-03. [DOI] [PubMed] [Google Scholar]
- [22].Maxwell PJ, Gallagher R, Seaton A, et al. HIF-1 and NF-κB-mediated pregulation of CXCR1 and CXCR2 expression promotes cell survival in hypoxic CaP cells. Oncogene. 2007;26:7333–45. doi: 10.1038/sj.onc.1210536. [DOI] [PubMed] [Google Scholar]
- [23].Zhong H, Semenza GL, Simons JL, DeMarzo A. Up-regulation of hypoxia-inducible factor-1alpha is an early event in prostate carcinogenesis. Cancer Detect Prev. 2004;28:88–93. doi: 10.1016/j.cdp.2003.12.009. [DOI] [PubMed] [Google Scholar]
- [24].Kunz M, Hartmann A, Flory E, et al. Anoxia-induced up-regulation of interleukin-8 in human malignant melanoma. A potential mechanism for high tumor aggressiveness. Am J Pathol. 1999;155:753–63. doi: 10.1016/S0002-9440(10)65174-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].De Marzo AM, Platz EA, Sutcliffe S, et al. Inflammation in prostate carcinogenesis. Nat Rev Cancer. 2007;7:256–69. doi: 10.1038/nrc2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Hanahan D, Weinberg R. Hallmarks of cancer—the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- [27].Brown LM, Cowen RL, Debray C, et al. Reversing hypoxic cell chemoresistance in vitro using genetic and small molecule approaches targeting hypoxia inducible factor-1. Mol Pharmacol. 2006;69:411–8. doi: 10.1124/mol.105.015743. [DOI] [PubMed] [Google Scholar]
- [28].Fraser M, Zhao H, Luoto KR, et al. PTEN deletion in prostate cancer cells does not associated with loss of RAD51 function: implications for radiotherapy and chemotherapy. Clin Cancer Res. 2012;18:1015–27. doi: 10.1158/1078-0432.CCR-11-2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].De La Inglesia N, Konopka G, Lim KL, et al. Deregulation of a STAT3-interleukin-8 signaling pathway promotes human glioblastoma cell proliferation and invasiveness. J Neurosci. 2008;28:5870–8. doi: 10.1523/JNEUROSCI.5385-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Bekes EM, Schweighofer B, Kupriyanova TA, et al. Tumor recruited neutrophils and neutrophil TIMP-free MMP-9 regulate co-ordinately the levels of tumor angiogenesis and efficiency of malignant cell intravasation. Am J Pathol. 2011;179:1455–70. doi: 10.1016/j.ajpath.2011.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.