

Abstract
Global change in protein turnover (protein degradome) constitutes a central part of cellular responses to intrinsic or extrinsic stimuli. However, profiling protein degradome remains technically challenging. Recently, inhibition of the proteasome, e.g., by using bortezomib (BTZ), has emerged as a major chemotherapeutic strategy for treating multiple myeloma and other human malignancies, but systematic understanding of the mechanisms for BTZ drug action and tumor drug resistance is yet to be achieved. Here we developed and applied a dual-fluorescence-based Protein Turnover Assay (ProTA) to quantitatively profile global changes in human protein degradome upon BTZ-induced proteasomal inhibition. ProTA and subsequent network analyses delineate potential molecular basis for BTZ action and tumor drug resistance in BTZ chemotherapy. Finally, combined use of BTZ with drugs targeting the ProTA-identified key genes or pathways in BTZ action reduced BTZ resistance in multiple myeloma cells. Remarkably, BTZ stabilizes proteasome subunit PSMC1 and proteasome assembly factor PSMD10, suggesting a previously under-appreciated mechanism for regulating proteasome homeostasis. Therefore, ProTA is a novel tool for profiling human protein degradome to elucidate potential mechanisms of drug action and resistance, which might facilitate therapeutic development targeting proteostasis to treat human disorders.
Keywords: bortezomib, combination therapy, proteostasis, protein degradome, proteasome homeostasis, N-end rule, ubiquitin
Introduction
Much effort has been taken to identify genetic or transcriptomic changes that might underlie drug action, cellular responses and drug resistance1,2,3. Protein degradation, which represents a terminal step of regulation on protein homeostasis (proteostasis) and function, constitutes an essential but often under-explored part of cellular responses to intrinsic or extrinsic stimuli, such as stress or drug treatment4,5,6. Dysregulated protein degradation, a critical aspect of proteostasis deficiency, is known to associate with a variety of devastating human diseases including cancer and neurodegenerative diseases7,8. Profiling global protein turnover is thus essential for elucidating the molecular basis of health or disease biology, and drug action and resistance, and for the development of therapeutic intervention into cancer and other diseases9. However, monitoring global protein turnover remains challenging, mainly due to technical restraints10,11.
So far, multiple lines of techniques have emerged to monitor cellular protein turnover, each having its own strengths and pitfalls12. Immunoblotting and pulse-chasing techniques are the most widely used approaches for measuring protein abundance and turnover but only suitable for small-scale analysis. On the other hand, mass spectroscopy (MS)-based proteomics techniques, often coupled with metabolic labeling, could assess protein abundance at the proteome level, but they have limitations when applying to non-abundant but functionally important proteins10,11.
Fluorescent proteins have been widely used to non-invasively monitor protein dynamics in living cells13. Global Protein Stability assay (GPS)11,14 and “tandem Fluorescent Timers” (tFTs)15 are two of the most recent non-MS techniques for profiling protein stability. Both techniques quantitatively compare the fluorescence intensities of the co-expressed fluorescent proteins, with one fused to the protein of interest for test and the other for reference. In GPS11, a bi-cistronic mRNA was employed to sequentially encode red fluorescent protein (RFP), an IRES (internal ribosome entry site) sequence and a green fluorescent protein (GFP) fused to the N-terminus of the protein of interest. In this RIG (RFP-IRES-GFP) configuration10, the IRES sequence would ensure certain level of co-translational expression of the two fluorescent proteins. At a given time point, the ratio of the fluorescence intensity of GFP to that of RFP provides a measurement of the relative abundances of the fluorescent proteins. A concept of protein stability index (PSI) was also introduced to allow quantitative comparison of the stabilities of individual proteins11. However, efficiency of IRES is known to fluctuate as IRES-transacting factors are themselves susceptible to dynamic regulations16. Moreover, EGFP and RFP used in the GPS system tend to oligomerize and take different lengths of time to fold into fluorescent forms in vivo, which might significantly impact the dynamics of the fusion proteins12,13. In tFTs, the tandemly tagged fluorescent proteins that could switch colors during the time course of maturation were fused to individual model substrates. As different fluorophores need to be customized in tFTs to allow for monitoring the dynamics of proteins with distinct half-lives, its general applicability for high-throughput analysis is yet to be tested15. Therefore, novel tools need to be developed to overcome the limitations in the existing techniques.
In cells, the N-terminal residues of a protein usually constitute one of the major sites for modifications and processing during or after translation, which may regulate localization, stability and intracellular trafficking of the protein17. Particularly, the N-end rule, a classic pathway in the ubiquitin-proteasome system (UPS), relates the in vivo half-lives of the proteins to the identities of their N-terminal destabilizing residues (termed as N-degrons)18,19,20. The N-degrons are known to mediate degradation of substrates that regulate many fundamental biological processes including cell signaling, apoptosis, embryonic development, neurodegeneration, peptide import and protein quality control21,22,23,24,25. The functionality of N-degrons could be severely compromised if repositioned elsewhere in the proteins. Therefore, there are considerable advantages to preserve the natural N-terminal residues of the proteins when studying protein turnover.
Bortezomib (BTZ, N-acyl-pseudo dipeptidyl boronic acid) reversibly binds to PSMB526, the chymotrypsin-like β5 subunit of the catalytic chamber of the 20S proteasome27. BTZ was the first clinically approved proteasome inhibitor and is currently in use for treating multiple myeloma and other human malignancies. BTZ exhibits strong clinical efficacy, but complications, particularly the high-rate relapse of drug-resistant tumors, have also been frequently reported in BTZ chemotherapy28,29. A system-level understanding of the mechanism for BTZ efficacy as well as tumor resistance to BTZ remains yet elusive30. As BTZ primarily functions as a proteasome inhibitor, it would be critical to characterize BTZ-induced changes in protein turnover at the system level.
In this work, we have developed a Protein Turnover Assay (ProTA) that maintains the natural N-terminal residues of 15 000 human proteins and ensures strict co-translational expression of the dual monomeric fluorescent proteins, thus permitting accurate and facile snapshot profiling of global protein stability at the system level. We next applied ProTA to profile changes in human protein degradome induced by BTZ, aiming to elucidate the potential molecular basis for BTZ action and tumor drug resistance.
Results
The ProTA
To develop ProTA, monomeric forms of EGFP and RFP (mEGFP and mRFP) were chosen over the most widely used EGFP or its other derivatives, as oligomerization of the fluorescent proteins would alter their intracellular dynamics and turnover rates of the fusion proteins13. Additionally, in mammalian cells, both nascent mEGFP and mRFP are known to fold into their fluorescent forms within ∼2-3 h with comparable stability13.
ProTA reporter vector is lentivirus based and uses cytomegalovirus (CMV) promoter for transcription of a mono-cistronic mRNA, which sequentially encodes mEGFP, double FLAG tags, lysine-less ubiquitin (Ubk0), mRFP and double FLAG tags (Figure 1A). This allows translation of the fusion protein, mEGFP-(FLAG)2-Ubk0-mRFP-(FLAG)2 (abbreviated as mEGFPfu-mRFPf), within a single reading frame. Both mEGFP and mRFP were C-terminally tagged with double FLAG epitopes to facilitate detection and affinity purification if ever desired. As demonstrated for the ubiquitin fusion technique31,32, when ubiquitin is fused to the N-terminus of a protein, the approximately 100 deubiquitinating enzymes in human cells would warrant efficient cleavage after the Gly76 residue in the ubiquitin moiety to generate mEGFPfu and mRFPf as two separate proteins. Meanwhile, as any lysine (Lys, K) residue in the ubiquitin moiety could become potential site for further ubiquitin conjugation, Lys-less human ubiquitin (Ubk0) generated by replacing all the Lys residues in ubiquitin with arginine (Arg, R) is inserted as a linker between mEGFPf and mRFPf. This would not only ensure production of mEGFPfu and mRFPf as separate molecules during or after translation, but also prevent unwanted Ub conjugation on the Ubk0 linker, which could otherwise artificially alter the dynamics of the mEGFPfu fusion protein in the ProTA system.
Figure 1.

The Protein Turnover Assay (ProTA), a novel tool to monitor protein degradation. (A) A schematic representation of the parental reporter construct in the ProTA system. (B) The abundance of ORF-mEGFPfu in relative to mRFPf, X-mEGFPfu/mRFPf, is determined by the stabilities of protein (X) encoded by the specific ORF and the fluorescent proteins. mRFPf at steady state level serves as a reference for translation. Notably, the molar ratio, X-mEGFPfu/mRFPf, is co-related with, but not equal to, the ratio of the fluorescence intensities of mEGFPfu and mRFPf, [mEGFPfu]/[mRFPf]. (C) Flow cytometry analysis of HEK293FT cells that stably express the mEGFPfu-mRFPf reporter. The clustering of cells in proximity to the diagonal indicates a linear correlation of [mEGFPfu] and [mRFPf]. (D) Comparative assessment of BTZ-induced changes in turnover of selected proteins with immunoblotting and flow cytometry analyses. HEK293FT cells stably expressing proteins mEGFPfu-mRFPf, ANXA1-mEGFPfu-mRFPf or RGS4-mEGFPfu-mRFPf were treated with or without BTZ (1.0 μM) for 6 h. The x axis indicates the fraction numbers sorted according to [mEGFPfu]/[mRFPf] (see Materials and Methods for more details); the y axis denotes the relative abundance of the cells of the indicated [mEGFPfu]/[mRFPf], presented in relative to the maximal number of the cells scored in the flow cytometry histogram. (E) Fusion of RGS4 to the N-terminus of mEGFP in the ProTA system preserves the functionality of the N-degrons. HEK293FT cells or UBR1−/−UBR2−/− MEF cells that expressed RGS4-mEGFPfu-mRFPf or pcDNA3.1-EGFP-RGS4 were subjected to BTZ treatment. The expression levels of RGS4-mEGFPfu, EGFP-RGS4 or GAPDH were determined with anti-RGS4 or anti-GAPDH.
In the ProTA system, open reading frames (hORFs) of human genes are fused to the N-terminus of mEGFPfu, allowing generation of human ORF-encoded proteins fused to mEGFP-(FLAG)2-UBk0 (mEGFPfu). Depending on the identity and strength of the degron(s) in a specific ORF-coded protein (X), the molar ratio of the steady state levels of the fluorescent proteins, X-mEGFPfu to mRFPf, could vary from 0 to 1 (Figure 1B). To validate the ProTA system, we first monitored the ratio of the fluorescence intensity of mEGFPfu to that of mRFPf, [mEGFPfu]/[mRFPf]. In the ProTA reporter HEK293FT cells that stably expressed mEGFPfu-mRFPf, where no human ORF was fused to the N-terminus of mEGFP, [mEGFPfu] and [mRFPf] showed a linear coorelation on flow cytometry diagram (Figure 1C). Data of immunoblotting analysis confirmed that the steady state levels of both mEGFPfu and mRFPf were constant, regardless of the treatment with the proteasome inhibitor, BTZ (Figure 1D). Similar results were obtained with the cells that stably expressed ANXA1-mEGFPfu-mRFPf fusion, consistent with the previous observation that ANXA1 protein was highly stable in vivo33. RGS4 (regulator of G-protein signaling 4) protein, a bona fide substrate that we have identified for the N-end rule pathway and proteasome34, is highly unstable. As shown in Figure 1D, the ratio of fluorescence intensities of RGS4-mEGFPfu and mRFPf, [RGS4-mEGFPfu]/[mRFPf], was low in cells that stably expressed the fluorescence-tagged RGS4 (RGS4-mEGFPfu-mRFPf), but significantly increased upon BTZ treatment, indicating that degradation of RGS4 was proteasome dependent.
Using the above RGS4-ProTA reporter cells as a model, we went on to ask whether this ProTA system is suitable for studying protein degradation in a specific setup, e.g., when a protein degradation pathway is genetically manipulated. In UBR1−/−UBR2−/− mouse embryonic fibroblast (MEF) cells, both UBR1 and UBR2, the two major E3 ubiquitin ligases, were genetically ablated to impair the functionality of the N-end rule pathway, thus stabilizing RGS435,36. Indeed, protein RGS4-mEGFPfu was unstable in HEK293FT cells, but significantly accumulated in UBR1−/−UBR2−/− MEF cells (Figure 1E). Remarkably, BTZ treatment stabilized RGS4-mEGFPfu in HEK293FT cells, but not in UBR1−/−UBR2−/− cells (Figure 1E). However, EGFP-RGS4, with EGFP tagged to the N-terminus of RGS4, similar to that in the GPS system11, was stable in both HEK293FT and UBR1−/−UBR2−/− MEF cells, regardless of BTZ treatment (Figure 1E). This observation strongly underscored the importance of preserving the natural N-terminus of a protein when studying its turnover dynamics. Altogether, these results demonstrated the applicability of the ProTA system in monitoring changes in protein stability, caused by either drug treatment or genetic manipulation. Of note, cautions should be taken for those proteins whose degrons reside in their C-termini, as the current configuration of the ProTA fusion reporters might impact the functionality of these C-degrons in an undesirable way.
The pipeline for ProTA
ProTA consists of four major steps: (1) construction of dual fluorescence-based cell library; (2) cell sorting; (3) DNA microarray hybridization; (4) calculation of PSI.
Firstly, through Gateway reactions, approximately, 15 000 human ORFs (from human ORFeome Version 5.137) were fused to the N-terminus of the mEGFPfu in a lentivirus-based expression vector. A lentivirus-based library was thus generated, and used to infect human HEK293FT cells at a low titer, which allows single-copy integration of the viral vector-based ORF into the chromosomal DNA (see Supplementary information, Data S1 for details). As a common problem in library manipulation, nonsense mutations in some human ORFs could prematurely terminate translation of the fluorescence-tagged proteins. The virus-infected cells were then sorted with fluorescence-activated cell sorting (FACS) to remove cells that did not bear fluorescence signals. This step is critical for handling an expression library as high content as the ProTA system, highlighting another advantage of fusing the ORFs to the N-terminus of mEGFP in our ProTA system. This finally brought about the ProTA reporter library in which each cell was chromosomally integrated with one of the 15 000 human ORFs fused in a typical ProTA configuration (Figure 1A). Microarray analysis was also performed with the extracted and amplified genomic DNAs (see Materials and Methods for details) to check the diversity and coverage of ORFs using customized probes targeting all the ORFs. The data indicated that 94.3% of the ORFs were preserved (no less than 3-fold above background signal in microarray analysis) in the library. All the cells in the ProTA library were thus barcoded for subsequent flow cytometry and microarray analyses.
Secondly, FACS was employed to sort the library into eight different fractions (subpopulations), R1, R2,...R8, according to their ascending ratios of the fluorescence intensities of mEGFPfu to mRFPf, [mEGFPfu]/[mRFPf] (Figures 1B and 2A). Obviously, the mEGFPfu-tagged proteins in cells of R1 fraction are the most unstable in contrast to the most stable fluorescent fusions in R8 fraction.
Figure 2.

Profiling human protein degradome with ProTA. Schematic diagram of the four experimental steps in ProTA. (A) Reporter cells with different [mEGFPfu]/[mRFPf] ratios are shown in different colors. The library cells were first sorted into eight fractions (R1 to R8) with ascending [mEGFPfu]/[mRFPf] ratios using FACS. The barcoded ORFs for cells of each fraction were amplified with PCR, using the genomic DNAs extracted from the corresponding fractions as templates. The resulted PCR products were used as templates for in vitro transcription of complementary RNAs (cRNAs). cRNAs were then labeled with Cy5 and subjected to microarray hybridizations. Eight hybridizations were performed with the cRNAs transcribed from the PCR-amplified ORFs from each FACS-isolated subpopulation. Protein stability index (PSI) of an individual protein was calculated using the algorithm, 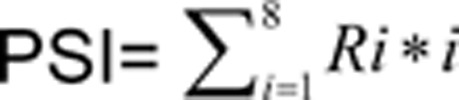 , integrating the relative abundance of the gene product in each FACS fraction and the [mEGFPfu]/[mRFPf] ratio of the fraction. (B) Representative data of PSIs of individual ORF products.
, integrating the relative abundance of the gene product in each FACS fraction and the [mEGFPfu]/[mRFPf] ratio of the fraction. (B) Representative data of PSIs of individual ORF products.
Subsequently, genomic DNAs were extracted from the cells in each subpopulation and served as templates for subsequent PCR analysis to amplify the barcoded ORFs for microarray hybridization. Since none of the ORFs in the ProTA library retains their natural 3-UTRs (untranslated regions), we customized the microarray with probes specially targeting each ORF sequence in the human ORFoeme V5.1 library. Hereby, the identities and relative abundances of the individual transcripts of the barcoded human ORFs could be reliably assessed.
Finally, for quantitative indexing of protein stability, we adopted the concept of PSI that was previously introduced in the GPS system11, and 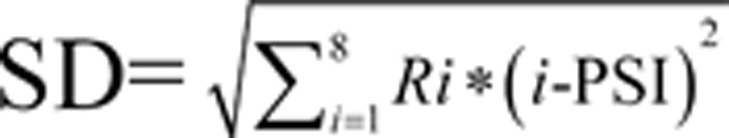 , where i denoted the fraction number of the subpopulation in FACS sorting and Ri was the fraction of the signal present for a gene in that given subpopulation i (Figure 2A). The larger PSI a protein scores in the ProTA assay, the more stable it is (Figure 2B). Thereby, the whole proteome could be clustered into different subgroups based on the distinct PSIs of individual proteins (Supplementary information, Table S2), which made possible a snapshot profiling of protein turnover at the system level (protein degradome) (Figure 2B).
, where i denoted the fraction number of the subpopulation in FACS sorting and Ri was the fraction of the signal present for a gene in that given subpopulation i (Figure 2A). The larger PSI a protein scores in the ProTA assay, the more stable it is (Figure 2B). Thereby, the whole proteome could be clustered into different subgroups based on the distinct PSIs of individual proteins (Supplementary information, Table S2), which made possible a snapshot profiling of protein turnover at the system level (protein degradome) (Figure 2B).
Profiling BTZ-induced changes in global protein turnover
When the ProTA cell library was treated with BTZ (1.0 μM, for 6 h), there was a global increase in ratios of [mEGFPfu]/[mRFPf] of the whole ProTA cell library (Figure 3A), suggesting that many human ORF-encoded proteins were stabilized upon BTZ treatment. This is in agreement with the general notion that, under normal conditions, degradation of most cellular proteins goes through proteasome system. Following the established pipeline (Figure 2A), the PSIs were generated for each protein in the cells treated with or without BTZ (PSIBTZ and PSI0, respectively), and increases in PSIs (ΔPSI = PSIBTZ − PSI0) were calculated for each fluorescence-labeled protein (Figure 3B). Then, relative increases in stabilities of these fusion proteins were indexed by ranking their individual ΔPSI/PSI0 values. Hereby, BTZ-induced changes in stabilities of the human proteome could be grouped according to their individual ΔPSI/PSI0 values (Figure 3C and see Supplementary information, Table S2 for the complete ProTA-BTZ data set), which altogether constituted a global profile of BTZ-induced changes in human protein degradome.
Figure 3.

Using ProTA to profile BTZ-induced changes in human protein degradome. (A) BTZ treatment induced global shifts in [mEGFPfu]/[mRFPf] ratio of the ProTA reporter cell library. (B) Representative data of ProTA-BTZ data set. Results for individual ORFs from microarrays and the percentages of cells in different FACS subpopulations were compared between those from cells with or without BTZ treatment. PSIBTZ and PSI0 refer to the PSIs of a specific protein in cells treated with BTZ and DMSO, respectively. ΔPSI = PSIBTZ – PSI0. (C) Distribution of ORFs according to ΔPSI/PSI0 values. (D-F) FACS and western blotting analyses of HEK293FT cells expressing the TP53-mEGFPfu-mRFPf (D), NF-КbIB-mEGFPfu-mRFPf (E) or ARC-mEGFPfu-mRFPf (F) that were treated with or without BTZ (1.0 μM 6 h). The x axis indicates the fraction numbers sorted according to [mEGFPfu]/[mRFPf]; the y axis denotes the relative abundance of the cells of indicated [mEGFPfu]/[mRFPf], presented in relative to the maximal number of the cells scored in the flow cytometry histogram.
Validation of the ProTA hits
To examine whether ProTA could faithfully capture changes in the stability of individual proteins upon BTZ treatment, HEK293FT cell lines were generated to stably express TP53, NF-КbIB or ARC in a typical ProTA format. Remarkably, FACS analyses with these individual stable cell lines well recapitulated the profile of corresponding flow cytometry patterns in the high-throughput ProTA assay (Figure 3D-3F). Data of immunoblotting analysis further confirmed that stabilities of these proteins increased upon BTZ treatment. We then selected another 16 hits for validation; these selected ORFs were fused in a typical ProTA configuration and the resulting ProTA constructs were introduced into HEK293FT cells. Immunoblotting analyses indicated that BTZ treatment stabilized 13 out of 16 proteins (Figures 4B, 5A, Supplementary information, Figure S1 and data not shown). Moreover, for another 11 ProTA hits, immunoblotting analysis indicated that the endogenous levels of 10 proteins in HEK293FT cells were indeed increased upon BTZ treatment (Supplementary information, Figure S2). Taken together, we conclude that application of ProTA could reliably profile BTZ-induced changes in global protein stabilities.
Figure 4.

Pathway and sub-network analysis of BTZ-induced changes in global protein turnover. (A) Protein-protein interactions (PPI) network analysis with STRING identified the sub-networks or modules (see Materials and Methods for the definition) in BTZ action. (B) FASN and SOST that were fused to mEGFPfu-mRFPf were stabilized upon BTZ treatment (1.0 μM, 6 h). Stabilities of FASN and SOST were determined through immunoblotting analysis in HEK293FT cells using anti-FLAG, with mRFPf as references. (C) Levels of endogenous FASN and SOST proteins in CZ-1 cells were elevated upon BTZ treatment (1.0 μM, 12 h). (D) The basal levels of endogenous FASN and SOST proteins were higher in drug-resistant CZ-1/R cells than in CZ-1 cells. Levels of endogenous proteins were examined through immunoblotting analysis with the indicated antibodies.
Figure 5.

BTZ stabilizes proteasome subunit, PSMC1, and proteasome assembly factor, PSMD10. (A) PSMC1 and PSMD10 were stabilized upon BTZ treatment (1.0 μM, 6 h). HEK293FT cells expressing PSMC1-mEGFPfu-mRFPf or PSMD10-mEGFPfu-mRFPf were treated with or without BTZ before cell lysis and immunoblotting analysis using anti-FLAG. Stabilities of each protein were assessed by comparing the abundances of the mEGFPfu fusions and the individual mRFPf references. (B) CHX chase analysis was performed with HEK293FT cells expressing PSMC1 or PSMD10 in the presence or absence of BTZ (1.0 μM) at the indicated time points. (C) After BTZ treatment (1.0 μM, 12 h), levels of endogenous PSMC1 or PSMD10 proteins in CZ-1 cells were assessed. (D) Levels of endogenous PSMC1 and PSMD10 proteins were higher in drug-resistant CZ-1/R cells than in CZ-1 cells. The expression levels of endogenous proteins were examined with immunoblotting analysis, using the indicated antibodies. (E) A schematic view of the feedback regulations of the proteasome homeostasis in responses to BTZ treatment.
Gene ontology and pathway analyses
The top 1 500 ProTA hits were subjected to the gene ontology (GO) analysis (biological processes), and processed using the Database for Annotation, Visualization and Integrated Discovery (DAVID)38 and Protein Analysis Through Evolutionary Relationship (PANTHER) database for further functional categorization39. Our data suggest that genes enriched upon BTZ treatment predominantly fall into the categories of cell proliferation and differentiation, cell cycle, apoptosis, as well as the effectors of cytotoxicity (Supplementary information, Figure S3).
Subsequently, the top 750 ProTA hits were subjected to pathway analyses using BioCarta and KEGG databases that are specialized in signaling and metabolic pathways, respectively40. Results from BioCarta analysis suggest that BTZ stabilized CD36, MAPK14 and FOS in the pathway of “TSP-1 induced apoptosis in microvascular endothelial cell (mvEC)” (Table 1). CD36, MAPK14 and FOS are known proteasome substrates, whose stabilization might account for the cytotoxicity of BTZ against tumor cells including multiple myeloma41. In the cytokines and inflammatory response pathway (CSF1, CSF2, IFNB1, IL6), IL6 is a known major growth and survival factor for multiple myeloma42. BTZ treatment also enriched the bone remodeling pathway involving IFNB1, TNFSF11 and FOS (Table 1). This is in agreement with a previous report43 that BTZ might promote synovial osteoclastogenesis and bone destruction through upregulating the expression of c-FOS and RANKL/TNFSF11. Our data showed that SOST, a downstream target in the BMP (bone morphogenetic protein) signaling pathway, was also stabilized by BTZ (Figure 4B and 4C). Therefore, application of ProTA seems to have opened a new window to understand the previously reported BTZ-induced complications in bone system. KEGG pathway analysis also revealed that BTZ treatment enriched pathways including SNARE interactions in vesicular transport and biosynthesis of unsaturated fatty acids (Table 1). Upregulation of these pathways might contribute to altered cell polarity, cellular metabolism and cell signaling through clustering receptors in lipid rafts, which are known causes and hallmarks of human cancers44,45. Altogether, application of ProTA has provided the first systematic view of the pathways enriched during BTZ action.
Table 1. Pathways enriched upon Bortezomib treatment.
| Category | Term | P Value | Genes | Fold Enrichment | FDR |
|---|---|---|---|---|---|
| BIOCARTA | TSP-1 induced apoptosis in microvascular endothelial cell | 2.86E-02 | CD36, MAPK14, FOS | 10.62 | 29.14 |
| BIOCARTA | Cytokines and inflammatory response | 8.06E-02 | CSF1, CSF2, IFNB1, IL6 | 3.81 | 63.19 |
| BIOCARTA | Bone remodeling | 9.11E-02 | IFNB1, Tnfsf11, FOS | 5.72 | 67.88 |
| KEGG_PATHWAY | SNARE interactions in vesicular transport | 2.36E-03 | GOSR1, STX11, STX12, STX16, STX18, STX19, Use1 | 4.98 | 2.76 |
| KEGG_PATHWAY | Neuroactive ligand-receptor interaction | 2.53E-02 | HTR1B, Cnr2, FPR1, GABRA3, GABRA6, GLP1R, GLP2R, GLRA3, GHSR, NPBWR2, PTH1R, P2RX1, P2RX4, P2RY8, SSTR4, S1PR4, S1PR5 | 1.8 | 26.15 |
| KEGG_PATHWAY | Biosynthesis of unsaturated fatty acids | 4.51E-02 | Acot4, ACOT7, ACOX1, Acox3 | 4.92 | 42.05 |
Protein-protein interaction network analysis to identify functional hubs and sub-networks or modules in BTZ action
Most biological processes are undertaken through complex interactions, which could be conceptualized as “node-link” in network analysis. In a such network, a few nodes that have multiple links are termed as “hubs”. Highly interconnected groups of nodes, often referred to as “sub-network” or “modules”, are critical for exerting a distinct cellular function46.
The top 250 hits in ProTA were subjected to protein-protein interaction (PPI) network analysis using STRING (Search Tool for the Retrieval of Interacting Genes/Proteins, version 9.0) database47. PPI network analysis suggests that proteins including PSMC1, SMARCA4 and PAX6 might act as hubs (nodes of over 10 linkages) in the network (Figure 4A). PPI analysis also identified significant enrichment in the following sub-networks or modules: (1) DNA-dependent transcription initiation; (2) proteasome homeostasis (originally defined as “regulation of ubiquitin-protein ligase activity involved in mitotic cell cycle” by STRING); (3) chromatin modification; (4) induction of apoptosis; (5) transmembrane receptor protein tyrosine kinase signaling pathway; and (6) regulation of fatty acid oxidation. Similar results were obtained for PPI analysis performed with the top 500 ProTA-BTZ hits, except that it additionally identified the module of “Mitochondrial ATP synthesis coupled proton transport” (Supplementary information, Figure S4). Conceivably, by increasing the stability of the proteins implicated in these sub-networks, BTZ treatment drastically changed the cellular proteolytic flux and elicited fundamental changes in a variety of cellular activities, from metabolism, gene expression, cell signaling, epigenetic modifications, to control of cell cycle and apoptosis. On one hand, BTZ might act by activating the sub-networks for “induction of apoptosis”, for example, to acutely exert its cytotoxicity against cancer cells. Meanwhile, BTZ also induced a wide-spectrum of cellular responses such as triggering more chromatin modifications, modulating transmembrane signaling and switching cell metabolism that might have profound impact on the ensuing behavior and fate of the cells, which could finally contribute to the evolution of tumor drug resistance.
As BTZ has proven efficacy in treating multiple myeloma, we next attempted to examine whether the above results that were generated with HEK293FT cells would be applicable in myeloma cells. To this end, we compared the static levels of several endogenous proteins, whose stabilities were increased upon BTZ treatment in HEK293FT cells, between BTZ-sensitive (CZ-1) and BTZ-resistant myeloma cells (CZ-1/R). Human CZ-1 cells were originally derived from a Chinese patient of multiple myeloma. The BTZ-resistant cell line, CZ-1/R, was generated through intermittently treating CZ-1 cells with incremental doses of BTZ48 and indeed showed increased cell viability compared with CZ-1 cells upon BTZ treatment at various doses (Supplementary information, Figure S5). The first subject to test was fatty acid synthase (FASN), the enzyme that catalyzes the formation of long-chain fatty acids. Upregulation of FASN expression in tumor cells was previously reported to increase the formation of lipid raft where EGFR and other growth factor receptors could cluster to potentiate the growth signals for tumor cells49. Interestingly, PPI analysis and immunoblotting assay consistently revealed that the stability of FASN protein was significantly increased upon BTZ treatment in HEK293FT cells and CZ-1 cells (Figure 4A-4C). Indeed, the basal level of endogenous FASN protein in the BTZ-resistant CZ-1/R cells was significantly higher than that in the BTZ-sensitive CZ-1 cells (Figure 4D). It is likely that stabilization of FASN by BTZ might promote cell growth and confer survival advantage to the BTZ-treated cells.
BTZ stabilized proteasome subunits PSMC1 and proteasome assembly factor PSMD10
Proteasome homeostasis is dynamic and delicately regulated upon stress50,51. Proteasomal inhibition was found to activate Nrf1-52 or Nrf2-mediated53 transcription of some proteasome subunits, which gave rise to the so-called “proteasome bounce-back response”. Overexpression of or mutations in PSMB5 gene were also reported in BTZ-resistant cancer cells3,54,55. It remained unclear whether these were the only mechanisms that regulate proteasome homeostasis and lead to compensatory increase in proteasome activity upon proteasome inhibition.
Interestingly, a proteasome subunit, PSMC1, was found to be significantly stabilized upon BTZ treatment in HEK293FT cells (Figures 4A and 5A). Similarly, PSMD10, the mammalian homolog of yeast proteasome assembly factor NAS656, was also stabilized by BTZ (Figure 5A). Chase experiment with cycloheximide (CHX), which blocks protein synthesis, further indicated that BTZ treatment did prolong the half-lives of these proteins in HEK293FT cells, mainly through inhibiting their degradation (Figure 5B). Therefore, we next compared the static levels of PSMC1 and PSMD10 between CZ-1 and CZ-1/R myeloma cells. BTZ treatment also markedly increased the expression levels of endogenous PSMC1 and PSMD10 proteins in CZ-1 cells (Figure 5C), while the basal levels of endogenous PSMC1 and PSMD10 proteins in the BTZ-resistant CZ-1/R cells were significantly higher than those in the BTZ-sensitive CZ-1 cells (Figure 5D). CHX chase experiments indicated that degradation of endogenous PSMC1 and PSMD10 in human myeloma cells was indeed inhibited in the presence of BTZ (Supplementary information, Figure S6A). Interestingly, PMSB5 was found to be stable in CZ-1 cells with or without BTZ treatment, although the protein level of PSMB5 in CZ-1/R cells was indeed higher than that in CZ-1 cells (Supplementary information, Figure S6B-S6D). As PSMC1 does not directly interact with PSMB5, PSMC1 molecules that could be co-immunoprecipitated with PSMB5 should be among those incorporated into intact proteasomes. BTZ treatment was found to only slightly stabilize free PSMC1 proteins, but significantly increased its levels in the PSMB5 co-immunoprecipitates, suggesting that more functional proteasome complexes might be assembled in cells treated with BTZ (Supplementary information, Figure S6F). Conceivably, mechanisms of both transcriptional activation52,53,57 and the stabilizing effect of BTZ might work concertedly to modulate proteasome homeostasis in these myeloma cells.
Therefore, upon BTZ-induced proteasome inhibition, cells seemed to not only acutely increase the availability of proteasome subunits through blocking the degradation of proteasome subunits such as PMSC1, but also stabilize the proteasome assembly factor, PSMD10, which would further facilitate the assembly of proteasome subunits into functional proteasomes (Figure 5E). This would functionally compensate for BTZ-induced proteasome inhibition and help cells survive the proteotoxic stress, which might provide an additional avenue for the evolution of tumor drug resistance in BTZ-based chemotherapy.
Cross analysis of BTZ-induced global changes in transcriptome and protein degradome
In the cell, regulators of transcription usually undergo rapid turnover through the UPS58,59. Indeed, proteins involved in transcription initiation were stabilized by BTZ treatment and over-represented in PPI network analysis of the ProTA-BTZ hits (Figure 4A). Therefore, upon BTZ-induced proteasome inhibition, changes in gene transcription became an indispensable part of the cellular response to BTZ treatment. In order to fully understand the mode of drug action for BTZ, microarray analysis was performed to comparatively profile mRNA levels in HEK293FT cells with or without BTZ treatment (1.0 μM for 6 h). Then, genes whose transcript levels changed over 3-folds upon BTZ treatment (fold change > 3) were subjected to GO (Supplementary information, Figure S7) or PPI network analyses (Supplementary information, Figure S8). Consistent with a previous report60, BTZ treatment was found to promote the transcription of many genes involved in apoptosis or protein folding. The top 20 hub genes from both ProTA-PPI and mRNA-PPI data sets were extracted and the highly connected hub genes were then presented (Figure 6A). These hubs include IL-6 signaling transducer (IL6ST), IL-6 receptor (IL6R), TEK, interferon-α receptor 1 (IFNAR1), IL12A, CSF2RA, CD28, mitogen-activated protein kinase 8 and 9 (MAPK8, MAPK9) and CTLA4. Remarkably, IL-6 signaling was again identified as one of the hubs, which is consistent with the results of pathway analyses of the ProTA hits (Table 1).
Figure 6.

Drug-resistant myeloma cells are re-sensitized to BTZ by targeting ProTA-identified hits. (A) Cross analysis of BTZ-induced changes in both human protein degradome and transcriptome. Presented here is a network summary of the results of PPI analyses performed on the top 20 hub genes extracted from the PPI-ProTA (Figure 4A) and the PPI-transcriptome data sets (Supplementary information, Figure S8). In order to profile the changes in the transcriptome of HEK293FT cells with or without BTZ treatment, total mRNAs were extracted and subjected to microarray analysis. (B) Protein levels of endogenous IL-6 in CZ-1 cells or CZ-1/R cells, with or without BTZ treatment (100 nM, 12 h). Data were represented as mean ± SEM (n = 4; *P < 0.02). (C) Cytotoxicity of BTZ (10 nM) was further enhanced by Merck-5 (2 μM, 24 h) that inhibits JAKs in IL-6 signaling pathway. Cell viability of CZ-1 was assessed with MTT assay. Data were represented as mean ± SEM (n = 6; **P < 0.01; ***P < 0.0001). (D, E) BTZ-resistant CZ-1/R cells were treated with BTZ (5 nM in D, 10 nM in E) alone or in combination with C75 (D, 10 μg/ml) or 17-AAG (E, 50 μM) for 24 h. Viability of the myeloma cells in each group was assessed using MTT assay. Data were represented as mean ± SEM (n = 6; *P < 0.1; n = 6; ***P < 0.0001).
As shown in Figure 6B, the protein level of intracellular IL-6 was ∼50% higher in BTZ-treated CZ-1 cells than in the untreated controls. Interestingly, CZ-1/R cells showed an IL-6 level of about 40% higher than that in CZ-1 cell, and BTZ treatment did not further increase IL-6 protein levels in the BTZ-resistant CZ-1/R cells. Merck-5, an inhibitor of JAKs (Janus tyrosine kinases) in the IL-6 signaling pathway61, significantly potentiated the cytotoxicity of BTZ to myeloma cells (Figure 6C). As activation of the IL-6 signaling pathway confers drug resistance to multiple types of cancers62, upregulation of IL-6 signaling in BTZ-treated multiple myeloma cells might also contribute to the frequently observed drug resistance among BTZ-treated patients.
The prototype of combination therapy using BTZ and drugs targeting ProTA-identified hits
As interactions between proteins and small molecules are essential parts of biological processes, chemical-protein interaction (CPI) analysis could potentially help illuminate the mechanisms underlying cellular responses or drug action63. Using STITCH2, we set to identify chemicals that may interact with the top 250 hits in our ProTA-BTZ analysis. As a result, calcium (Ca2+), coenzyme A (CoA) and adenosine diphosphate (ADP) were over-represented (Supplementary information, Figure S9). Interestingly, Ca2+ was previously shown to be a critical determinant of BTZ cytotoxicity in myeloma cell lines64. Adenine nucleotides (ATP, ADP, AMP), the primary energy currencies in the cell, also act as signaling molecules that modulate activities of ion channels or activate signaling cascades65.
CoA is a coenzyme essential for fatty acid metabolism. In the CPI map, genes including FASN, ACOT7, ACOX1, ACOX3 that are involved in the pathway of biosynthesis of unsaturated fatty acid were connected to CoA (Supplementary information, Figure S9). FASN was found to be significantly stabilized upon BTZ treatment (Figure 4B and 4C), suggesting that fatty acid synthesis might be significantly activated during BTZ action, and intervention into fatty acid synthesis might represent a novel strategy to modulate BTZ cytotoxicity against myeloma cells. Indeed, as shown in Figure 6D, combined use of BTZ and the FASN inhibitor C75 (10 μg/ml) manifested enhanced toxicity against BTZ-resistant cells (CZ-1/R). Using the combination index (CI) method66 and the associated software, CompuSyn, C75 at 10 μg/ml was found to work on the myeloma cells with moderate antagonism at 1 nM BTZ, moderate synergism at 5 nM and strong synergism at 10, 15 or 20 nM BTZ (Supplementary information, Figures S10-S11).
Meanwhile, 17-AAG is a geldanamycin analog that binds to the ATP/ADP pocket of Hsp90, which is known to play a critical role in tumorigenesis and tumor drug responses and resistance65. Our GO analysis of BTZ-induced changes at the transcriptional level indicated that genes involved in “response to unfolded protein” were significantly enriched (Supplementary information, Figure S7). Indeed, combined use of BTZ and 17-AAG (50 μM) resulted in potentiated cytotoxicity against BTZ-resistant (CZ-1/R) cells (Figure 6E). As also shown in Supplementary information, Figures S10-S11, through computing CI using the Chou-Talalay method66 and the software CompuSyn (http://www.combosyn.com/), 17-AAG at 50 μM indeed had strong synergistic effect (CI < 0.30) with BTZ at all concentrations tested.
Discussion
Recently, efforts have been devoted to identifying genetic basis for drug action, cellular response and drug resistance1,3,67. Here we present a ProTA that employed the dual fluorescent protein strategy to quantitatively index the stability or turnover rates of 15 000 human proteins. Taken into account the in vivo maturation time, quantum yields and oligomerization properties of the fluorescent proteins, monomeric EGFP and RFP (mEGFP and mRFP) were selected as fluorophores of choice (Figure 1A). In constructing the ProTA reporter, mEGFP and mRFP were linked with lysine-less ubiquitin to ensure: (1) strictly co-translational expression of the fluorescent proteins; (2) efficient generation of the fluorescent proteins as separate entities; (3) minimal conjugation on the lysine-free Ub and the mEGFP fusion proteins; and (4) preserving the natural N-termini of the human ORF products to minimally impact the localization and functionality of the N-degrons (degradation signal). Our data indicate that ProTA could faithfully assess the stability of protein fusions made in ProTA configuration in responses to either drug treatment (Figure 1D) or genetic manipulation (Figure 1E), thus testifying the broad applicability of ProTA for monitoring protein stability under diverse conditions. After constructing the reporter cell library for ProTA, we used ProTA to index stabilities of approximately15 000 human proteins in HEK293FT cells without stimulation (Figure 2 and Supplementary information, Table S2).
Given the fact that degrons of many proteins are located at the C-termini, the EGFP fusion at their C-termini like that in the ProTA system might impair the functionality of these degrons. Therefore, cautions should be taken when examining ProTA-profiled protein stabilities of individual proteins, especially those with C-terminal degrons.
BTZ is currently a frontier proteasome inhibitor for treating multiple myeloma and other human malignancies with remarkable efficacy. However, a systematic understanding of the molecular basis for cellular responses to BTZ, mode of BTZ action and tumor resistance to BTZ remained elusive. To address this unmet need, we applied ProTA to profile BTZ-induced changes in protein stabilities. However, myeloma cells, like most of the other cancer cells, are aneuploid and genetically unstable, which made them unsuitable for ProTA library construction. Therefore, ProTA analysis was instead performed with HEK293FT cells of normal karyotype with or without BTZ treatment, followed by further validation carried out in patient-derived myeloma cells.
Indeed, BTZ treatment increased stabilities of many cellular proteins (Figures 3 and 4). With GO and pathway analyses of the BTZ-stabilized proteins, we showed that major pathways such as cell apoptosis and proliferation pathways were over-represented after BTZ treatment (Table 1 and Supplementary information, Figure S3). PPI network analysis further identified many pathways or sub-networks activated upon BTZ treatment (Figure 4A). Among them, upregulation and activation of pro-apoptotic pathways or cell cycle arresting mechanisms might underlie the cytotoxicity of BTZ to cancer cells. Meanwhile, alterations in fatty acid metabolism, proteasome homeostasis, chromatin modifications and remodeling seemed to constitute another part of the cellular responses and self-adjustment that might help the cells survive BTZ-induced stress. Therefore, application of ProTA has led to the first ever network perspective (Figure 4A) of proteins and networks associated with BTZ-induced changes in protein turnover.
Remarkably, our data have demonstrated for the first time that some of the proteasome subunits (e.g., PSMC1) along with proteasome assembly factor (e.g., PSMD10) were themselves substrates of proteasome and underwent proteasome-dependent degradation (Figures 4A and 5A). This mechanism might also function in concert with the previously demonstrated transcriptional regulation52,53 to form a feedback loop regulating proteasome homeostasis (Figure 5E), which could upregulate proteasome function and functionally compensate for BTZ-induced compromise in proteolytic capacity. This may collectively reduce BTZ cytotoxicity, thus providing an additional avenue for drug resistance in BTZ-treated cancer cells. The fact that static levels of the proteasomal components were elevated in the BTZ-resistant CZ-1/R cells (Figure 5D and Supplementary information, Figure S5E) further underscored this possibility.
IL-6 signaling was also activated upon BTZ treatment (Figure 6A). Experiments with CZ-1 cells derived from multiple myeloma patients further confirmed that the static level of IL-6 was indeed upregulated upon BTZ treatment (Figure 6B). Previously, it was proposed that multiple myeloma cells could adhere to bone marrow stromal cells and trigger them to secret IL-6, a key inflammatory cytokine that mediates growth signaling in various cancer lines68,69. Upregulation of IL-6 signaling by BTZ treatment might confer growth advantage in multiple myeloma cells70. Activated IL-6 signaling, together with other mechanisms including stabilized proteins involved in cell proliferation, enhanced fatty acid synthesis (Figure 4A-4C) and increased abundance of functional proteasomes (Figure 5) might collectively contribute to the evolution of tumor drug resistance that have been frequently observed in relapsed myeloma in BTZ chemotherapy.
Finally, combined use of BTZ and drugs targeting BTZ-stabilized proteins, such as C75 and 17-AAG that inhibited FASN and Hsp90, respectively, manifested synergistic toxicity against BTZ-resistant myeloma cells (Figure 6D-6E). Therefore, by targeting the key genes or pathways that ProTA identified in BTZ action, new regimens could be rationalized to help develop novel strategies to overcome tumor drug resistance.
Through profiling the human degradome, an underexplored dimension of protein dynamics, ProTA is emerging as a novel tool to elucidate potential molecular basis for drug action, cellular responses and drug resistance. Complementing existing techniques, application of ProTA would facilitate therapeutic development targeting proteostasis for treatment of human disorders.
Materials and Methods
Reagents
All chemicals were from Sigma, if not otherwise indicated. See Supplementary information, Data S1 for all antibodies used in this study.
Construction of the parental vector, pCDH-ccdB-mEGFPfu-mRFPf
The plasmid, pAG426GAL-attR1-ccdB-attR2-EGFP, originally constructed in Dr Lindquist's lab at Massachusetts Institute of Technology71, was obtained from Addgene, Inc. EGFP was replaced with DNA fragments coding mEGFP, double FLAG epitopes, human ubiquitin with all lysines substituted with arginines, mRFP and double FLAG epitopes to generate mEGFP-(FLAG)2-UbK0-mRFP-(FLAG)2 (mEGFPfu-mRFPf). Then, the fragment containing attR1-ccdB-attR2-mEGFPfu-mRFPf was subcloned into the lentivirus vector, pCDH-CMV-MCS-EF1a-Puro (System Biosciences), using XbaI and NheI restriction sites, to obtain the parental vector, pCDH-ccdB-mEGFPfu-mRFPf, for mammalian expression.
Construction of human ORF expression library
To construct ProTA library, ORFs of 15 000 cDNAs from human ORFeome V5.172 in pDONR223 (OpenBiosystems) were cloned into the parental plasmid, pCDH-ccdB-mEGFPfu-mRFPf, using Gateway cloning technique. To generate the reporter cell library, HEK293FT cells were infected with lentiviruses carrying ORFs-mEGFPfu-mRFPf supplemented with 8 μg/ml polybrene (Sigma, USA). The coverage of the ProTA reporter cell library was further confirmed through microarray analysis and PCR reactions performed with the primer sets and genomic DNAs from randomly picked colonies of the cultured ProTA library.
Data processing in ProTA
For each ORF, the sum of the eight ratios was first normalized to 1. PSI values were then generated using the formulas and , where i denotes the fraction number of the subpopulation in FACS sorting, and Ri is the fraction of the signal present for a gene in that given subpopulation i. Data were further filtered to remove those bearing one of the following characteristics: (1) a STDEV greater than 1.5; (2) a maximal value of the Ri less than 0.3; and (3) a difference between PSI and the fraction number i with the maximal Ri greater than 1. When ProTA was applied to profile BTZ-induced change in protein stability, PSI values were obtained with all the human ORF-encoded proteins in cells treated with or without BTZ (PSIBTZ and PSI0, respectively), the increases in PSIs (ΔPSI) after BTZ treatment were calculated using the formula, ΔPSI = PSIBTZ − PSI0.
Statistical analysis
Data were analyzed by using Prism 5 (GraphPad Software, Inc). Unpaired t-test was performed using GraphPad Prism version 5.0 for Windows (GraphPad Software). Results were presented as means ± SEM.
Acknowledgments
We are grateful to Dr W Bian and her team in Cell Biology Core facility of SIBCB for their excellent support in flow cytometry analysis and cell sorting, and Drs L Lin, Y Xia and H Xiao of National Center for Microarrays for their help in microarray analysis. We are particularly grateful to Dr Hsueh-Chi Yen at Institute of Mol Biol, Academia Sinica, and Dr Ivan Dikic of Goethe University for advice, and Drs Dangsheng Li, Lijian Hui and Jiarui Wu of SIBCB, and Dr Shirley Diamond of California Institute of Technology for helpful discussion. We also thank Dr Ya-lan Wu and all other members of the Hu lab for support. This work was supported by the National Natural Science Foundation of China (31270828, 31070678, 81170491, 81072070 and 81470360), the 100 Talents award from CAS to RH and the Ministry of Science and Technology, China (2010CB912100, 2012CB910800 and 2013CB910900 to RH, and 2011CB915501 to KR). RH was also supported by a Sanofi-aventis SIBS Young Investigator award and funding from the Cancer Center of Xuhui Central Hospital (CCR2012003), Shanghai Institute of Neurosciences (SKLN-201206) and the Instrument Developing Project of the Chinese Academy of Sciences (YZ201339).
Footnotes
(Supplementary information is linked to the online version of the paper on the Cell Research website.)
Supplementary Information
Immunoblotting analysis confirmed bortezomib induced changes in stability of individual fusion proteins.
Immunoblotting analysis confirmed bortezomib-induced changes in stability of endogenous proteins.
Categorization of bortezomib-induced changes in global protein stability.
Protein-protein interaction (PPI) network analysis of bortezomib-induced changes in protein stability.
Characterization of BTZ-sensitive cells (CZ-1) or BTZ-resistant cells (CZ-1/R) from a multiple myeloma (MM) patient48.
(A) The stability of PSMC1 and PSMD10 were assayed by cycloheximide (CHX) chase in CZ-1 cells.
Categorization of bortezomib-induced changes in gene transcriptions.
Protein-protein interaction (PPI) network analysis of bortezomib-induced changes in gene transcriptions.
Chemical-protein interaction (CPI) network analysis of bortezomib induced changes in protein stability.
Combined chemotherapy using BTZ and drugs that targeted the ProTA identified key genes
C75, 17-AAG and BTZ exhibit synergistic, cytotoxic effects on the MM cell line CZ-1
Annotations for the genes whose symbols were frequently used in this work
A complete list of the ProTA-identified protein stability profiles in 293FT cells treated with or without bortezomib
Materials and Methods
References
- Diaz LA, Williams RT, Wu J, et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 2012;486:537–540. doi: 10.1038/nature11219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soon WW, Hariharan M, Snyder MP. High-throughput sequencing for biology and medicine. Mol Syst Biol. 2013;9:640. doi: 10.1038/msb.2012.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker SA, Houghtaling BR, Elemento O, Kapoor TM. Using transcriptome sequencing to identify mechanisms of drug action and resistance. Nat Chem Biol. 2012;8:235–237. doi: 10.1038/nchembio.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vabulas RM, Hartl FU. Protein synthesis upon acute nutrient restriction relies on proteasome function. Science. 2005;310:1960–1963. doi: 10.1126/science.1121925. [DOI] [PubMed] [Google Scholar]
- Finley D, Ciechanover A, Varshavsky A. Ubiquitin as a central cellular regulator. Cell. 2004;116:S29–S32. doi: 10.1016/s0092-8674(03)00971-1. [DOI] [PubMed] [Google Scholar]
- Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- Powers ET, Morimoto RI, Dillin A, Kelly JW, Balch WE. Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem. 2009;78:959–991. doi: 10.1146/annurev.biochem.052308.114844. [DOI] [PubMed] [Google Scholar]
- Haendler B, Jentsch S. The ubiquitin system in health and disease. Preface. Ernst Schering Found Symp Proc. 2008;1:V–VI. [PubMed] [Google Scholar]
- Hoeller D, Dikic I. Targeting the ubiquitin system in cancer therapy. Nature. 2009;458:438–444. doi: 10.1038/nature07960. [DOI] [PubMed] [Google Scholar]
- Ang XL, Harper JW. A road map of cellular protein homeostasis. Nat Chem Biol. 2009;5:9–11. doi: 10.1038/nchembio0109-9. [DOI] [PubMed] [Google Scholar]
- Yen HC, Xu Q, Chou DM, Zhao Z, Elledge SJ. Global protein stability profiling in mammalian cells. Science. 2008;322:918–923. doi: 10.1126/science.1160489. [DOI] [PubMed] [Google Scholar]
- Yewdell JW, Lacsina JR, Rechsteiner MC, Nicchitta CV. Out with the old, in with the new? Comparing methods for measuring protein degradation. Cell Biol Int. 2011;35:457–462. doi: 10.1042/CBI20110055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner NC, Steinbach PA, Tsien RY. A guide to choosing fluorescent proteins. Nat Methods. 2005;2:905–909. doi: 10.1038/nmeth819. [DOI] [PubMed] [Google Scholar]
- Emanuele MJ, Elia AE, Xu Q, et al. Global identification of modular cullin-RING ligase substrates. Cell. 2011;147:459–474. doi: 10.1016/j.cell.2011.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khmelinskii A, Keller PJ, Bartosik A, et al. Tandem fluorescent protein timers for in vivo analysis of protein dynamics. Nat Biotechnol. 2012;30:708–714. doi: 10.1038/nbt.2281. [DOI] [PubMed] [Google Scholar]
- Komar AA, Hatzoglou M. Cellular IRES-mediated translation: the war of ITAFs in pathophysiological states. Cell Cycle. 2011;10:229–240. doi: 10.4161/cc.10.2.14472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh CT, Garneau-Tsodikova S, Gatto GJ., Jr Protein posttranslational modifications: the chemistry of proteome diversifications. Angew Chem Int Ed Engl. 2005;44:7342–7372. doi: 10.1002/anie.200501023. [DOI] [PubMed] [Google Scholar]
- Piatkov KI, Brower CS, Varshavsky A. The N-end rule pathway counteracts cell death by destroying proapoptotic protein fragments. Proc Natl Acad Sci USA. 2012;109:E1839–E1847. doi: 10.1073/pnas.1207786109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshavsky A. The N-end rule pathway and regulation by proteolysis. Protein Sci. 2011;20:1298–1345. doi: 10.1002/pro.666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasaki T, Sriram SM, Park KS, Kwon YT. The N-end rule pathway. Annual Rev Biochem. 2012;81:261–289. doi: 10.1146/annurev-biochem-051710-093308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshavsky A. Three decades of studies to understand the functions of the ubiquitin family. Methods Mol Biol. 2012;832:1–11. doi: 10.1007/978-1-61779-474-2_1. [DOI] [PubMed] [Google Scholar]
- Verma R, Oania RS, Kolawa NJ, Deshaies RJ. Cdc48/p97 promotes degradation of aberrant nascent polypeptides bound to the ribosome. Elife. 2013;2:e00308. doi: 10.7554/eLife.00308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner GC, Varshavsky A. Detecting and measuring cotranslational protein degradation in vivo. Science. 2000;289:2117–2120. doi: 10.1126/science.289.5487.2117. [DOI] [PubMed] [Google Scholar]
- Brower CS, Piatkov KI, Varshavsky A. Neurodegeneration-associated protein fragments as short-lived substrates of the N-end rule pathway. Mol Cell. 2013;50:161–171. doi: 10.1016/j.molcel.2013.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang CS, Varshavsky A. Regulation of peptide import through phosphorylation of Ubr1, the ubiquitin ligase of the N-end rule pathway. Proc Natl Acad Sci USA. 2008;105:19188–19193. doi: 10.1073/pnas.0808891105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg AL. Development of proteasome inhibitors as research tools and cancer drugs. J Cell Biol. 2012;199:583–588. doi: 10.1083/jcb.201210077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Frezza M, Schmitt S, Kanwar J, Dou QP. Bortezomib as the first proteasome inhibitor anticancer drug: current status and future perspectives. Curr Cancer Drug Targets. 2011;11:239–253. doi: 10.2174/156800911794519752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson PG, Barlogie B, Berenson J, et al. A phase 2 study of bortezomib in relapsed, refractory myeloma. N Engl J Med. 2003;348:2609–2617. doi: 10.1056/NEJMoa030288. [DOI] [PubMed] [Google Scholar]
- Richardson PG, Weller E, Lonial S, et al. Lenalidomide, bortezomib, and dexamethasone combination therapy in patients with newly diagnosed multiple myeloma. Blood. 2010;116:679–686. doi: 10.1182/blood-2010-02-268862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruschak AM, Slassi M, Kay LE, Schimmer AD. Novel proteasome inhibitors to overcome bortezomib resistance. J Natl Cancer Inst. 2011;103:1007–1017. doi: 10.1093/jnci/djr160. [DOI] [PubMed] [Google Scholar]
- Bachmair A, Finley D, Varshavsky A. In vivo half-life of a protein is a function of its amino-terminal residue. Science. 1986;234:179–186. doi: 10.1126/science.3018930. [DOI] [PubMed] [Google Scholar]
- Varshavsky A. Ubiquitin fusion technique and related methods. Methods Enzymol. 2005;399:777–799. doi: 10.1016/S0076-6879(05)99051-4. [DOI] [PubMed] [Google Scholar]
- Shimoji T, Murakami K, Sugiyama Y, et al. Identification of annexin A1 as a novel substrate for E6AP-mediated ubiquitylation. J Cell Biochem. 2009;106:1123–1135. doi: 10.1002/jcb.22096. [DOI] [PubMed] [Google Scholar]
- Hu RG, Sheng J, Qi X, Xu Z, Takahashi TT, Varshavsky A. The N-end rule pathway as a nitric oxide sensor controlling the levels of multiple regulators. Nature. 2005;437:981–986. doi: 10.1038/nature04027. [DOI] [PubMed] [Google Scholar]
- An JY, Seo JW, Tasaki T, Lee MJ, Varshavsky A, Kwon YT. Impaired neurogenesis and cardiovascular development in mice lacking the E3 ubiquitin ligases UBR1 and UBR2 of the N-end rule pathway. Proc Natl Acad Sci USA. 2006;103:6212–6217. doi: 10.1073/pnas.0601700103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PC, Sowa ME, Gygi SP, Harper JW. Alternative ubiquitin activation/conjugation cascades interact with N-end rule ubiquitin ligases to control degradation of RGS proteins. Mol Cell. 2011;43:392–405. doi: 10.1016/j.molcel.2011.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rual JF, Hirozane-Kishikawa T, Hao T, et al. Human ORFeome version 1.1: a platform for reverse proteomics. Genome Res. 2004;14:2128–2135. doi: 10.1101/gr.2973604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis G, Sherman BT, Hosack DA, et al. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
- Thomas PD, Kejariwal A, Campbell MJ, et al. PANTHER: a browsable database of gene products organized by biological function, using curated protein family and subfamily classification. Nucleic Acids Res. 2003;31:334–341. doi: 10.1093/nar/gkg115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader GD, Cary MP, Sander C. Pathguide: a pathway resource list. Nucleic Acids Res. 2006;34:D504–D506. doi: 10.1093/nar/gkj126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lioni M, Noma K, Snyder A, et al. Bortezomib induces apoptosis in esophageal squamous cell carcinoma cells through activation of the p38 mitogen-activated protein kinase pathway. Mol Cancer Ther. 2008;7:2866–2875. doi: 10.1158/1535-7163.MCT-08-0391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallek M, Bergsagel PL, Anderson KC. Multiple myeloma: increasing evidence for a multistep transformation process. Blood. 1998;91:3–21. [PMC free article] [PubMed] [Google Scholar]
- Oyajobi BO, Garrett IR, Gupta A, et al. Stimulation of new bone formation by the proteasome inhibitor, bortezomib: implications for myeloma bone disease. Br J Haematol. 2007;139:434–438. doi: 10.1111/j.1365-2141.2007.06829.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Deberardinis RJ, Thompson CB. Cellular metabolism and disease: what do metabolic outliers teach us. Cell. 2012;148:1132–1144. doi: 10.1016/j.cell.2012.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barabasi AL, Oltvai ZN. Network biology: understanding the cell's functional organization. Nat Rev Genet. 2004;5:101–113. doi: 10.1038/nrg1272. [DOI] [PubMed] [Google Scholar]
- Szklarczyk D, Franceschini A, Kuhn M, et al. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011;39:D561–D568. doi: 10.1093/nar/gkq973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou J, Lin FY, Zhang B, Zhang LZ, Ding SQ. Establishment and biological characteristics of human multiple myeloma cell line CZ-1. Chin Med J (Engl) 2004;117:115–119. [PubMed] [Google Scholar]
- Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. 2007;7:763–777. doi: 10.1038/nrc2222. [DOI] [PubMed] [Google Scholar]
- Hanna J, Meides A, Zhang DP, Finley D. A ubiquitin stress response induces altered proteasome composition. Cell. 2007;129:747–759. doi: 10.1016/j.cell.2007.03.042. [DOI] [PubMed] [Google Scholar]
- Aiken CT, Kaake RM, Wang X, Huang L. Oxidative stress-mediated regulation of proteasome complexes. Mol Cell Proteomics. 2011;10:R110.006924. doi: 10.1074/mcp.M110.006924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radhakrishnan SK, Lee CS, Young P, Beskow A, Chan JY, Deshaies RJ. Transcription factor Nrf1 mediates the proteasome recovery pathway after proteasome inhibition in mammalian cells. Mol Cell. 2010;38:17–28. doi: 10.1016/j.molcel.2010.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak MK, Kensler TW. Induction of 26S proteasome subunit PSMB5 by the bifunctional inducer 3-methylcholanthrene through the Nrf2-ARE, but not the AhR/Arnt-XRE, pathway. Biochem Biophys Res Commun. 2006;345:1350–1357. doi: 10.1016/j.bbrc.2006.05.043. [DOI] [PubMed] [Google Scholar]
- Oerlemans R, Franke NE, Assaraf YG, et al. Molecular basis of bortezomib resistance: proteasome subunit beta5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood. 2008;112:2489–2499. doi: 10.1182/blood-2007-08-104950. [DOI] [PubMed] [Google Scholar]
- Lichter DI, Danaee H, Pickard MD, et al. Sequence analysis of beta-subunit genes of the 20S proteasome in patients with relapsed multiple myeloma treated with bortezomib or dexamethasone. Blood. 2012;120:4513–4516. doi: 10.1182/blood-2012-05-426924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomko RJ, Jr, Funakoshi M, Schneider K, Wang J, Hochstrasser M. Heterohexameric ring arrangement of the eukaryotic proteasomal ATPases: implications for proteasome structure and assembly. Mol Cell. 2010;38:393–403. doi: 10.1016/j.molcel.2010.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y. Structure, assembly and homeostatic regulation of the 26S proteasome. J Mol Cell Biol. 2010;2:308–317. doi: 10.1093/jmcb/mjq030. [DOI] [PubMed] [Google Scholar]
- Ravid T, Hochstrasser M. Diversity of degradation signals in the ubiquitin-proteasome system. Nat Rev Mol Cell Biol. 2008;9:679–690. doi: 10.1038/nrm2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng F, Wenzel S, Tansey WP. Ubiquitin and proteasomes in transcription. Annu Rev Biochem. 2012;81:177–201. doi: 10.1146/annurev-biochem-052110-120012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obeng EA, Carlson LM, Gutman DM, Harrington WJ, Jr, Lee KP, Boise LH. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood. 2006;107:4907–4916. doi: 10.1182/blood-2005-08-3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinrich PC, Behrmann I, Muller-Newen G, Schaper F, Graeve L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem J. 1998;334:297–314. doi: 10.1042/bj3340297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Z, Fenoglio S, Gao DC, et al. TGF-beta IL-6 axis mediates selective and adaptive mechanisms of resistance to molecular targeted therapy in lung cancer. Proc Natl Acad Sci USA. 2010;107:15535–15540. doi: 10.1073/pnas.1009472107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn M, Szklarczyk D, Franceschini A, et al. STITCH 2: an interaction network database for small molecules and proteins. Nucleic Acids Res. 2010;38:D552–D556. doi: 10.1093/nar/gkp937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landowski TH, Megli CJ, Nullmeyer KD, Lynch RM, Dorr RT. Mitochondrial-mediated disregulation of Ca2+ is a critical determinant of Velcade (PS-341/bortezomib) cytotoxicity in myeloma cell lines. Cancer Res. 2005;65:3828–3836. doi: 10.1158/0008-5472.CAN-04-3684. [DOI] [PubMed] [Google Scholar]
- Taipale M, Jarosz DF, Lindquist S. HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat Rev Mol Cell Biol. 2010;11:515–528. doi: 10.1038/nrm2918. [DOI] [PubMed] [Google Scholar]
- Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- Mardis ER. Next-generation DNA sequencing methods. Annu Rev Genomics Hum Genet. 2008;9:387–402. doi: 10.1146/annurev.genom.9.081307.164359. [DOI] [PubMed] [Google Scholar]
- Schlachter C, Lisdat F, Frohme M, et al. Pushing the detection limits: the evanescent field in surface plasmon resonance and analyte-induced folding observation of long human telomeric repeats. Biosens Bioelectron. 2012;31:571–574. doi: 10.1016/j.bios.2011.11.003. [DOI] [PubMed] [Google Scholar]
- Conze D, Weiss L, Regen PS, et al. Autocrine production of interleukin 6 causes multidrug resistance in breast cancer cells. Cancer Res. 2001;61:8851–8858. [PubMed] [Google Scholar]
- Chauhan D, Singh AV, Ciccarelli B, Richardson PG, Palladino MA, Anderson KC. Combination of novel proteasome inhibitor NPI-0052 and lenalidomide trigger in vitro and in vivo synergistic cytotoxicity in multiple myeloma. Blood. 2010;115:834–845. doi: 10.1182/blood-2009-03-213009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberti S, Gitler AD, Lindquist S. A suite of Gateway cloning vectors for high-throughput genetic analysis in Saccharomyces cerevisiae. Yeast. 2007;24:913–919. doi: 10.1002/yea.1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamesch P, Li N, Milstein S, et al. hORFeome v3.1: a resource of human open reading frames representing over 10 000 human genes. Genomics. 2007;89:307–315. doi: 10.1016/j.ygeno.2006.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Immunoblotting analysis confirmed bortezomib induced changes in stability of individual fusion proteins.
Immunoblotting analysis confirmed bortezomib-induced changes in stability of endogenous proteins.
Categorization of bortezomib-induced changes in global protein stability.
Protein-protein interaction (PPI) network analysis of bortezomib-induced changes in protein stability.
Characterization of BTZ-sensitive cells (CZ-1) or BTZ-resistant cells (CZ-1/R) from a multiple myeloma (MM) patient48.
(A) The stability of PSMC1 and PSMD10 were assayed by cycloheximide (CHX) chase in CZ-1 cells.
Categorization of bortezomib-induced changes in gene transcriptions.
Protein-protein interaction (PPI) network analysis of bortezomib-induced changes in gene transcriptions.
Chemical-protein interaction (CPI) network analysis of bortezomib induced changes in protein stability.
Combined chemotherapy using BTZ and drugs that targeted the ProTA identified key genes
C75, 17-AAG and BTZ exhibit synergistic, cytotoxic effects on the MM cell line CZ-1
Annotations for the genes whose symbols were frequently used in this work
A complete list of the ProTA-identified protein stability profiles in 293FT cells treated with or without bortezomib
Materials and Methods